
Altered Ca2+ Handling and Oxidative Stress Underlie Mitochondrial Damage and Skeletal Muscle Dysfunction in Aging and Disease


{kind=link}
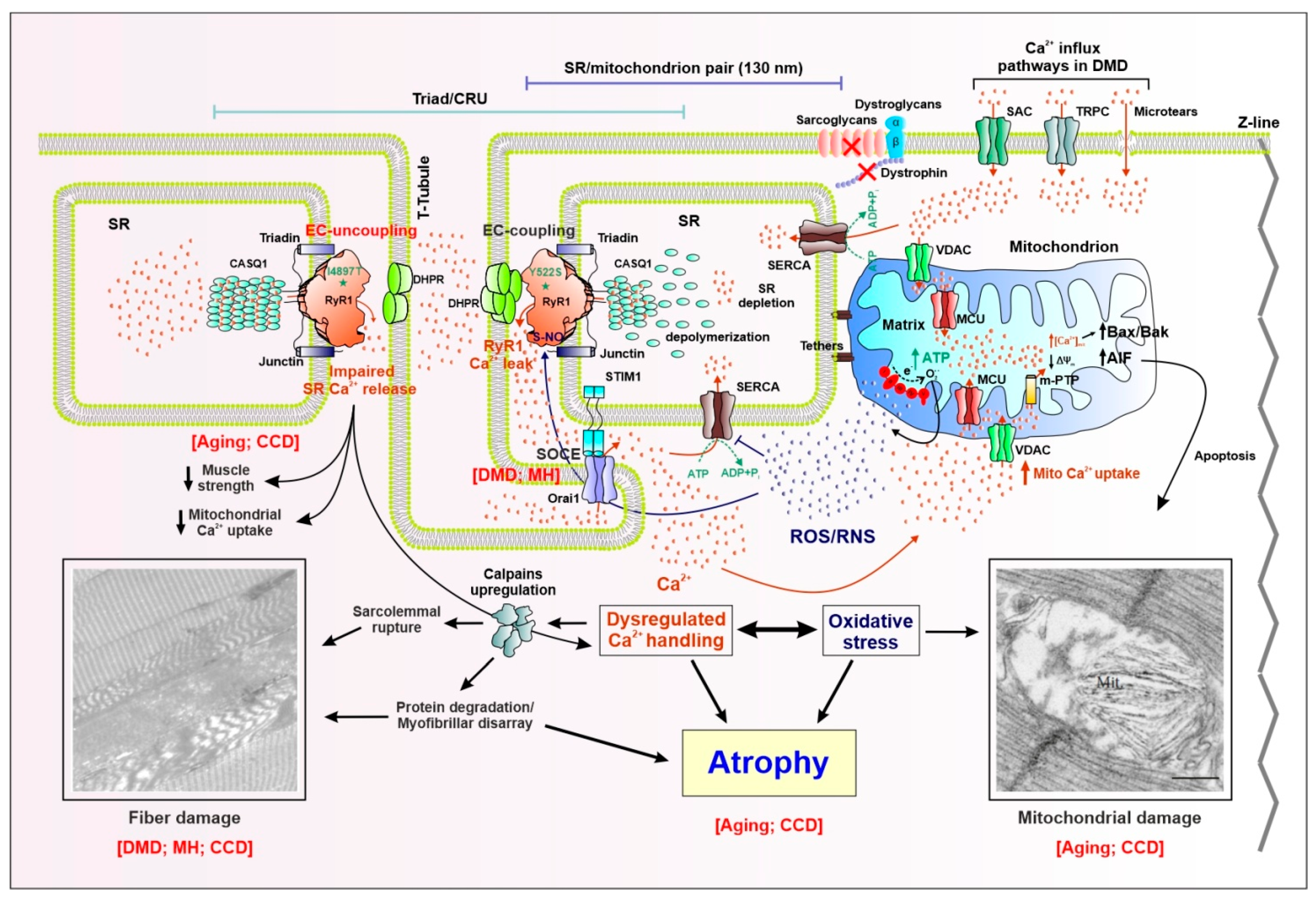
Abstract
1. Ca2+ Signaling in Skeletal Muscle
1.1. Excitation–Contraction (EC) Coupling in Skeletal Muscle
1.2. Store-Operated Ca2+ Entry in Skeletal Muscle
2. Mitochondria in Skeletal Muscle
2.1. Mitochondrial ATP Production and ROS Generation
2.2. Mitochondrial Ca2+ Uptake and Ca2+ Microdomains in Skeletal Muscle
3. Altered Ca2+ Handling and Mitochondrial ROS Production in Inherited Forms of Skeletal Muscle Disease
3.1. Muscular Dystrophy
3.2. Malignant Hyperthermia and Central Core Disease
4. Role of Altered Ca2+ Handling and Mitochondrial ROS Production in Loss of Muscle Mass and Reduced Contractility in Sarcopenia
5. Altered Mitochondrial Function and Ca2+ Homeostasis in Muscle Atrophy
6. Concluding Remarks and Future Directions
Funding
Conflicts of Interest
Abbreviations
| AIF | apoptosis inducing factor |
| ATP | adenosine triphosphate |
| Ca2+ | calcium |
| CCD | central core disease |
| CEU | Ca2+ entry unit |
| CRU | Ca2+ release unit |
| CASQ1 | calsequestrin-1 |
| DGC | dystrophin-glycoprotein complex |
| DMD | Duchenne muscular dystrophy |
| DHPR | dihydropyridine receptor |
| EC coupling | excitation-contraction coupling |
| ER | endoplasmic reticulum |
| MCU | mitochondrial Ca2+ uniporter |
| MD | musclular dystrophy |
| MH | malignant hyperthermia |
| RNS | reactive nitrogen species |
| ROS | reactive oxygen species |
| RyR1 | ryanodine receptor type-1 |
| SERCA | sarco/endoplasmic Ca2+ ATPase |
| SOCE | store-operated Ca2+ entry |
| SR | sarcoplasmic reticulum |
| STIM1 | stromal-interacting molecule 1 |
| TA | tubular aggregate |
| TAM | tubular aggregate myopathy |
| TCA | tricarboxylic acid |
| T-tubule | transverse tubule |
References
- Franzini-Armstrong, C. Structure of sarcoplasmic reticulum. Fed. Proc. 1980, 39, 2403–2409. [Google Scholar] [PubMed]
- Franzini-Armstrong, C. Freeze-fracture of frog slow tonic fibers. Structure of surface and internal membranes. Tissue Cell 1984, 16, 647–664. [Google Scholar] [CrossRef]
- Franzini-Armstrong, C.; Jorgensen, A.O. Structure and development of E-C coupling units in skeletal muscle. Annu. Rev. Physiol. 1994, 56, 509–534. [Google Scholar] [CrossRef] [PubMed]
- Schneider, M.F.; Chandler, W.K. Voltage dependent charge movement in skeletal muscle: A possible step in excitation-contraction coupling. Nature 1973, 242, 244–246. [Google Scholar] [CrossRef]
- Smith, J.S.; Imagawa, T.; Ma, J.; Fill, M.; Campbell, K.P.; Coronado, R. Purified ryanodine receptor from rabbit skeletal muscle is the calcium-release channel of sarcoplasmic reticulum. J. Gen. Physiol. 1988, 92, 1–26. [Google Scholar] [CrossRef]
- Schneider, M.F. Control of calcium release in functioning skeletal muscle fibers. Annu. Rev. Physiol. 1994, 56, 463–484. [Google Scholar] [CrossRef]
- Franzini-Armstrong, C.; Protasi, F. Ryanodine receptors of striated muscles: A complex channel capable of multiple interactions. Physiol. Rev. 1997, 77, 699–729. [Google Scholar] [CrossRef]
- Rios, E.; Brum, G. Involvement of dihydropyridine receptors in excitation-contraction coupling in skeletal muscle. Nature 1987, 325, 717–720. [Google Scholar] [CrossRef]
- Knudson, C.M.; Chaudhari, N.; Sharp, A.H.; Powell, J.A.; Beam, K.G.; Campbell, K.P. Specific absence of the α1 subunit of the dihydropyridine receptor in mice with muscular dysgenesis. J. Biol. Chem. 1989, 264, 1345–1348. [Google Scholar] [CrossRef]
- Adams, B.A.; Tanabe, T.; Mikami, A.; Numa, S.; Beam, K.G. Intramembrane charge movement restored in dysgenic skeletal muscle by injection of dihydropyridine receptor cDNAs. Nature 1990, 346, 569–572. [Google Scholar] [CrossRef]
- Tanabe, T.; Beam, K.G.; Adams, B.A.; Niidome, T.; Numa, S. Regions of the skeletal muscle dihydropyridine receptor critical for excitation-contraction coupling. Nature 1990, 346, 567–569. [Google Scholar] [CrossRef]
- Canato, M.; Scorzeto, M.; Giacomello, M.; Protasi, F.; Reggiani, C.; Stienen, G.J.M. Massive alterations of sarcoplasmic reticulum free calcium in skeletal muscle fibers lacking calsequestrin revealed by a genetically encoded probe. Proc. Natl. Acad. Sci. USA 2010, 107, 22326–22331. [Google Scholar] [CrossRef] [PubMed]
- Ziman, A.P.; Ward, C.W.; Rodney, G.G.; Lederer, W.J.; Bloch, R.J. Quantitative measurement of Ca2+ in the sarcoplasmic reticulum lumen of mammalian skeletal muscle. Biophys. J. 2010, 99, 2705–2714. [Google Scholar] [CrossRef]
- Sztretye, M.; Yi, J.; Figueroa, L.; Zhou, J.; Royer, L.; Ríos, E. D4cpv-calsequestrin: A sensitive ratiometric biosensor accurately targeted to the calcium store of skeletal muscle. J. Gen. Physiol. 2011, 138, 211–229. [Google Scholar] [CrossRef] [PubMed]
- MacLennan, D.H.; Wong, P.T. Isolation of a calcium-sequestering protein from sarcoplasmic reticulum. Proc. Natl. Acad. Sci. USA 1971, 68, 1231–1235. [Google Scholar] [CrossRef] [PubMed]
- Meissner, G. Isolation and characterization of two types of sarcoplasmic reticulum vesicles. BBA Biomembr. 1975, 389, 51–68. [Google Scholar] [CrossRef]
- Campbell, K.P.; MacLennan, D.H.; Jorgensen, A.O. Staining of the Ca2+-binding proteins, calsequestrin, calmodulin, troponin C, and S-100, with the cationic carbocyanine dye “Stains-all”. J. Biol. Chem. 1983, 258, 11267–11273. [Google Scholar] [CrossRef]
- Yano, K.; Zarain-Herzberg, A. Sarcoplasmic reticulum calsequestrins: Structural and functional properties. Mol. Cell. Biochem. 1994, 135, 61–70. [Google Scholar] [CrossRef]
- Ikemoto, N.; Ronjat, M.; Mészáros, L.G.; Koshita, M. Postulated Role of Calsequestrin in the Regulation of Calcium Release from Sarcoplasmic Reticulum. Biochemistry 1989, 28, 6764–6771. [Google Scholar] [CrossRef]
- Beard, N.A.; Sakowska, M.M.; Dulhunty, A.F.; Laver, D.R. Calsequestrin is an inhibitor of skeletal muscle ryanodine receptor calcium release channels. Biophys. J. 2002, 82, 310–320. [Google Scholar] [CrossRef]
- Beard, N.A.; Casarotto, M.G.; Wei, L.; Varsányi, M.; Laver, D.R.; Dulhunty, A.F. Regulation of ryanodine receptors by calsequestrin: Effect of high luminal Ca2+ phosphorylation. Biophys. J. 2005, 88, 3444–3454. [Google Scholar] [CrossRef] [PubMed]
- Sztretye, M.; Yi, J.; Figueroa, L.; Zhou, J.; Royer, L.; Allen, P.; Brum, G.; Ríos, E. Measurement of RyR permeability reveals a role of calsequestrin in termination of SR Ca 2+ release in skeletal muscle. J. Gen. Physiol. 2011, 138, 231–247. [Google Scholar] [CrossRef]
- MacLennan, D.H.; Brandl, C.J.; Korczak, B.; Green, N.M. Amino-acid sequence of a Ca2++ Mg2+-dependent ATPase from rabbit muscle sarcoplasmic reticulum, deduced from its complementary DNA sequence. Nature 1985, 316, 696–700. [Google Scholar] [CrossRef]
- Brandl, C.J.; Green, N.M.; Korczak, B.; MacLennan, D.H. Two Ca2+ ATPase genes: Homologies and mechanistic implications of deduced amino acid sequences. Cell 1986, 44, 597–607. [Google Scholar] [CrossRef]
- Hasselbach, W. Relaxation and the sarcotubular calcium pump. Fed. Proc. 1964, 23, 909–912. [Google Scholar] [PubMed]
- Armstrong, C.M.; Bezanilla, F.M.; Horowicz, P. Twitches in the presence of ethylene glycol bis(β-aminoethyl ether)-N,N′-tetraacetic acid. BBA Bioenerg. 1972, 267, 605–608. [Google Scholar] [CrossRef]
- Chandler, W.K.; Rakowski, R.F.; Schneider, M.F. Effects of glycerol treatment and maintained depolarization on charge movement in skeletal muscle. J. Physiol. 1976, 254, 285–316. [Google Scholar] [CrossRef]
- Ríos, E.; Pizarro, G.; Stefani, E. Charge movement and the nature of signal transduction in skeletal muscle excitation-contraction coupling. Annu. Rev. Physiol. 1992, 54, 109–133. [Google Scholar] [CrossRef]
- Ríos, E.; Karhanek, M.; Ma, J.; González, A. An allosteric model of the molecular interactions of excitation-contraction coupling in skeletal muscle. J. Gen. Physiol. 1993, 102, 449–481. [Google Scholar] [CrossRef]
- Hopf, F.W.; Reddy, P.; Hong, J.; Steinhardt, R.A. A capacitative calcium current in cultured skeletal muscle cells is mediated by the calcium-specific leak channel and inhibited by dihydropyridine compounds. J. Biol. Chem. 1996, 271, 22358–22367. [Google Scholar] [CrossRef]
- Kurebayashi, N.; Ogawa, Y. Depletion of Ca2+ in the sarcoplasmic reticulum stimulates Ca2+ entry into mouse skeletal muscle fibres. J. Physiol. 2001, 533, 185–199. [Google Scholar] [CrossRef]
- Putney, J.W. A model for receptor-regulated calcium entry. Cell Calcium 1986, 7, 1–12. [Google Scholar] [CrossRef]
- Parekh, A.B.; Penner, R. Store depletion and calcium influx. Physiol. Rev. 1997, 77, 901–930. [Google Scholar] [CrossRef] [PubMed]
- Parekh, A.B.; Putney, J.W. Store-operated calcium channels. Physiol. Rev. 2005, 85, 757–810. [Google Scholar] [CrossRef] [PubMed]
- Roos, J.; DiGregorio, P.J.; Yeromin, A.V.; Ohlsen, K.; Lioudyno, M.; Zhang, S.; Safrina, O.; Kozak, J.A.; Wagner, S.L.; Cahalan, M.D.; et al. STIM1, an essential and conserved component of store-operated Ca2+ channel function. J. Cell Biol. 2005, 169, 435–445. [Google Scholar] [CrossRef]
- Liou, J.; Kim, M.L.; Won, D.H.; Jones, J.T.; Myers, J.W.; Ferrell, J.E.; Meyer, T. STIM is a Ca2+ sensor essential for Ca2+-store- depletion-triggered Ca2+ influx. Curr. Biol. 2005, 15, 1235–1241. [Google Scholar] [CrossRef]
- Zhang, S.L.; Yu, Y.; Roos, J.; Kozak, J.A.; Deerinck, T.J.; Ellisman, M.H.; Stauderman, K.A.; Cahalan, M.D. STIM1 is a Ca2+ sensor that activates CRAC channels and migrates from the Ca2+ store to the plasma membrane. Nature 2005, 437, 902–905. [Google Scholar] [CrossRef]
- Feske, S.; Gwack, Y.; Prakriya, M.; Srikanth, S.; Puppel, S.H.; Tanasa, B.; Hogan, P.G.; Lewis, R.S.; Daly, M.; Rao, A. A mutation in Orai1 causes immune deficiency by abrogating CRAC channel function. Nature 2006, 441, 179–185. [Google Scholar] [CrossRef]
- Prakriya, M.; Feske, S.; Gwack, Y.; Srikanth, S.; Rao, A.; Hogan, P.G. Orai1 is an essential pore subunit of the CRAC channel. Nature 2006, 443, 230–233. [Google Scholar] [CrossRef]
- Vig, M.; Peinelt, C.; Beck, A.; Koomoa, D.L.; Rabah, D.; Koblan-Huberson, M.; Kraft, S.; Turner, H.; Fleig, A.; Penner, R.; et al. CRACM1 is a plasma membrane protein essential for store-operated Ca2+ entry. Science 2006, 312, 1220–1223. [Google Scholar] [CrossRef]
- Lyfenko, A.D.; Dirksen, R.T. Differential dependence of store-operated and excitation-coupled Ca2+ entry in skeletal muscle on STIM1 and Orai1. J. Physiol. 2008, 586, 4815–4824. [Google Scholar] [CrossRef] [PubMed]
- Wei-Lapierre, L.; Carrell, E.M.; Boncompagni, S.; Protasi, F.; Dirksen, R.T. Orai1-dependent calcium entry promotes skeletal muscle growth and limits fatigue. Nat. Commun. 2013, 4, 1–2. [Google Scholar] [CrossRef]
- Sztretye, M.; Geyer, N.; Vincze, J.; Al-Gaadi, D.; Oláh, T.; Szentesi, P.; Kis, G.; Antal, M.; Balatoni, I.; Csernoch, L.; et al. SOCE Is Important for Maintaining Sarcoplasmic Calcium Content and Release in Skeletal Muscle Fibers. Biophys. J. 2017, 113, 2496–2507. [Google Scholar] [CrossRef]
- Boncompagni, S.; Michelucci, A.; Pietrangelo, L.; Dirksen, R.T.; Protasi, F. Exercise-dependent formation of new junctions that promote STIM1-Orai1 assembly in skeletal muscle. Sci. Rep. 2017, 7, 1–2, Erratum in 2018, 8, 17463. [Google Scholar] [CrossRef] [PubMed]
- Michelucci, A.; Boncompagni, S.; Pietrangelo, L.; García-Castañeda, M.; Takano, T.; Malik, S.; Dirksen, R.T.; Protasi, F. Transverse tubule remodeling enhances orai1-dependent Ca2+ entry in skeletal muscle. Elife 2019, 8, e47576. [Google Scholar] [CrossRef] [PubMed]
- Michelucci, A.; Boncompagni, S.; Pietrangelo, L.; Takano, T.; Protasi, F.; Dirksen, R.T. Pre-assembled Ca2+ entry units and constitutively active Ca2+ entry in skeletal muscle of calsequestrin-1 knockout mice. J. Gen. Physiol. 2020, 152, e202012617. [Google Scholar] [CrossRef] [PubMed]
- Stiber, J.; Hawkins, A.; Zhang, Z.S.; Wang, S.; Burch, J.; Graham, V.; Ward, C.C.; Seth, M.; Finch, E.; Malouf, N.; et al. STIM1 signalling controls store-operated calcium entry required for development and contractile function in skeletal muscle. Nat. Cell Biol. 2008, 10, 688–697. [Google Scholar] [CrossRef]
- Carrell, E.M.; Coppola, A.R.; McBride, H.J.; Dirksen, R.T. Orai1 enhances muscle endurance by promoting fatigue-resistant type I fiber content but not through acute store-operated Ca2+ entry. FASEB J. 2016, 30, 4109–4119. [Google Scholar] [CrossRef]
- Protasi, F.; Pietrangelo, L.; Boncompagni, S. Calcium entry units (CEUs): Perspectives in skeletal muscle function and disease. J. Muscle Res. Cell Motil. 2020. Epub ahead of print. [Google Scholar] [CrossRef]
- Launikonis, B.S.; Ríos, E. Store-operated Ca2+ entry during intracellular Ca2+ release in mammalian skeletal muscle. J. Physiol. 2007, 583, 81–97. [Google Scholar] [CrossRef]
- Edwards, J.N.; Murphy, R.M.; Cully, T.R.; von Wegner, F.; Friedrich, O.; Launikonis, B.S. Ultra-rapid activation and deactivation of store-operated Ca2+ entry in skeletal muscle. Cell Calcium 2010, 47, 458–467. [Google Scholar] [CrossRef]
- Darbellay, B.; Arnaudeau, S.; Bader, C.R.; Konig, S.; Bernheim, L. STIM1L is a new actin-binding splice variant involved in fast repetitive Ca2+ release. J. Cell Biol. 2011, 194, 335–346. [Google Scholar] [CrossRef] [PubMed]
- Koenig, X.; Choi, R.H.; Launikonis, B.S. Store-operated Ca2+ entry is activated by every action potential in skeletal muscle. Commun. Biol. 2018, 1, 31. [Google Scholar] [CrossRef]
- Darbellay, B.; Arnaudeau, S.; König, S.; Jousset, H.; Bader, C.; Demaurex, N.; Bernheim, L. STIM1- and Orai1-dependent store-operated calcium entry regulates human myoblast differentiation. J. Biol. Chem. 2009, 284, 5370–5380. [Google Scholar] [CrossRef] [PubMed]
- Kiviluoto, S.; Decuypere, J.P.; De Smedt, H.; Missiaen, L.; Parys, J.B.; Bultynck, G. STIM1 as a key regulator for Ca2+ homeostasis in skeletal-muscle development and function. Skelet. Muscle 2011, 1, 16. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Finch, E.A.; Graham, V.; Zhang, Z.-S.; Ding, J.-D.; Burch, J.; Oh-hora, M.; Rosenberg, P. STIM1-Ca2+ Signaling Is Required for the Hypertrophic Growth of Skeletal Muscle in Mice. Mol. Cell. Biol. 2012, 32, 3009–3017. [Google Scholar] [CrossRef]
- Böhm, J.; Chevessier, F.; De Paula, A.M.; Koch, C.; Attarian, S.; Feger, C.; Hantaï, D.; Laforêt, P.; Ghorab, K.; Vallat, J.M.; et al. Constitutive activation of the calcium sensor STIM1 causes tubular-aggregate myopathy. Am. J. Hum. Genet. 2013, 92, 271–278. [Google Scholar] [CrossRef]
- Endo, Y.; Noguchi, S.; Hara, Y.; Hayashi, Y.K.; Motomura, K.; Miyatake, S.; Murakami, N.; Tanaka, S.; Yamashita, S.; Kizu, R.; et al. Dominant mutations in ORAI1 cause tubular aggregate myopathy with hypocalcemia via constitutive activation of store-operated Ca2+ channels. Hum. Mol. Genet. 2015, 24, 637–648. [Google Scholar] [CrossRef]
- Edwards, J.N.; Friedrich, O.; Cully, T.R.; Von Wegner, F.; Murphy, R.M.; Launikonis, B.S. Upregulation of store-operated Ca2+ entry in dystrophic mdx mouse muscle. Am. J. Physiol. Cell Physiol. 2010, 299, C42–C50. [Google Scholar] [CrossRef]
- Zhao, X.; Moloughney, J.G.; Zhang, S.; Komazaki, S.; Weisleder, N. Orai1 Mediates Exacerbated Ca2+ Entry in Dystrophic Skeletal Muscle. PLoS ONE 2012, 7, e49862. [Google Scholar] [CrossRef]
- Cully, T.R.; Choi, R.H.; Bjorksten, A.R.; George Stephenson, D.; Murphy, R.M.; Launikonis, B.S. Junctional membrane Ca2+ dynamics in human muscle fibers are altered by malignant hyperthermia causative RyR mutation. Proc. Natl. Acad. Sci. USA 2018, 115, 8215–8220. [Google Scholar] [CrossRef]
- Duke, A.M.; Hopkins, P.M.; Calaghan, S.C.; Halsall, J.P.; Steele, D.S. Store-operated Ca2+ entry in malignant hyperthermia-susceptible human skeletal muscle. J. Biol. Chem. 2010, 285, 25645–25653. [Google Scholar] [CrossRef]
- Thornton, A.M.; Zhao, X.; Weisleder, N.; Brotto, L.S.; Bougoin, S.; Nosek, T.M.; Reid, M.; Hardin, B.; Pan, Z.; Ma, J.; et al. Store-Operated Ca2+ Entry (SOCE) contributes to normal skeletal muscle contractility in young but not in aged skeletal muscle. Aging (Albany. N. Y.) 2011, 3, 621–634. [Google Scholar] [CrossRef]
- Boncompagni, S.; Pecorai, C.; Michelucci, A.; Pietrangelo, L.; Protasi, F. Long-Term Exercise Reduces Formation of Tubular Aggregates and Promotes Maintenance of Ca2+ Entry Units in Aged Muscle. Front. Physiol. 2021, 11, 601057. [Google Scholar] [CrossRef] [PubMed]
- Allen, D.G.; Lamb, G.D.; Westerblad, H. Skeletal muscle fatigue: Cellular mechanisms. Physiol. Rev. 2008, 88, 287–332. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, P. Coupling of phosphorylation to electron and hydrogen transfer by a chemi-osmotic type of mechanism. Nature 1961, 191, 144–148. [Google Scholar] [CrossRef]
- Brookes, P.S.; Yoon, Y.; Robotham, J.L.; Anders, M.W.; Sheu, S.S. Calcium, ATP, and ROS: A mitochondrial love-hate triangle. Am. J. Physiol. Cell Physiol. 2004, 287, C817–C833. [Google Scholar] [CrossRef] [PubMed]
- Madeira, V.M.C. Overview of mitochondrial bioenergetics. Methods Mol. Biol. 2018, 1782, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Finkel, T. Signal transduction by reactive oxygen species. J. Cell Biol. 2011, 194, 7–15. [Google Scholar] [CrossRef]
- Schieber, M.; Chandel, N.S. ROS function in redox signaling and oxidative stress. Curr. Biol. 2014, 24, R453. [Google Scholar] [CrossRef]
- Deluca, H.F.; Engstrom, G.W. Calcium uptake by rat kidney mitochondria. Proc. Natl. Acad. Sci. USA 1961, 47, 1744–1750. [Google Scholar] [CrossRef] [PubMed]
- Scarpa, A.; Graziotti, P. Mechanisms for intracellular calcium regulation in heart: I. stopped-flow measurements of ca++ uptake by cardiac mitochondria. J. Gen. Physiol. 1973, 62, 756–772. [Google Scholar] [CrossRef] [PubMed]
- Carafoli, E.; Crompton, M. The regulation of intracellular calcium by mitochondria. Ann. N. Y. Acad. Sci. 1978, 307, 269–284. [Google Scholar] [CrossRef] [PubMed]
- Sembrowich, W.L.; Quintinskie, J.J.; Li, G. Calcium uptake in mitochondria from different skeletal muscle types. J. Appl. Physiol. 1985, 59, 137–141. [Google Scholar] [CrossRef] [PubMed]
- Rizzuto, R.; Simpson, A.W.M.; Brini, M.; Pozzan, T. Rapid changes of mitochondrial Ca2+ revealed by specifically targeted recombinant aequorin. Nature 1992, 358, 325–327. [Google Scholar] [CrossRef] [PubMed]
- Rudolf, R.; Mongillo, M.; Magalhäes, P.J.; Pozzan, T. In vivo monitoring of Ca2+ uptake into mitochondria of mouse skeletal muscle during contraction. J. Cell Biol. 2004, 166, 527–536. [Google Scholar] [CrossRef] [PubMed]
- Rossi, A.E.; Boncompagni, S.; Wei, L.; Protasi, F.; Dirksen, R.T. Differential impact of mitochondrial positioning on mitochondrial Ca2+ uptake and ca 2+ spark suppression in skeletal muscle. Am. J. Physiol. Cell Physiol. 2011, 301, C1128–C1139. [Google Scholar] [CrossRef] [PubMed]
- De Stefani, D.; Raffaello, A.; Teardo, E.; Szabó, I.; Rizzuto, R. A forty-kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature 2011, 476, 336–340. [Google Scholar] [CrossRef]
- Baughman, J.M.; Perocchi, F.; Girgis, H.S.; Plovanich, M.; Belcher-Timme, C.A.; Sancak, Y.; Bao, X.R.; Strittmatter, L.; Goldberger, O.; Bogorad, R.L.; et al. Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature 2011, 476, 341–345. [Google Scholar] [CrossRef]
- Rossi, A.E.; Boncompagni, S.; Dirksen, R.T. Sarcoplasmic reticulum-mitochondrial symbiosis: Bidirectional signaling in skeletal muscle. Exerc. Sport Sci. Rev. 2009, 37, 29–35. [Google Scholar] [CrossRef]
- Favaro, G.; Romanello, V.; Varanita, T.; Andrea Desbats, M.; Morbidoni, V.; Tezze, C.; Albiero, M.; Canato, M.; Gherardi, G.; De Stefani, D.; et al. DRP1-mediated mitochondrial shape controls calcium homeostasis and muscle mass. Nat. Commun. 2019, 10, 2576. [Google Scholar] [CrossRef] [PubMed]
- Logan, C.V.; Szabadkai, G.; Sharpe, J.A.; Parry, D.A.; Torelli, S.; Childs, A.M.; Kriek, M.; Phadke, R.; Johnson, C.A.; Roberts, N.Y.; et al. Loss-of-function mutations in MICU1 cause a brain and muscle disorder linked to primary alterations in mitochondrial calcium signaling. Nat. Genet. 2014, 46, 188–193. [Google Scholar] [CrossRef] [PubMed]
- Lewis-Smith, D.; Kamer, K.J.; Griffin, H.; Childs, A.M.; Pysden, K.; Titov, D.; Duff, J.; Pyle, A.; Taylor, R.W.; Yu-Wai-Man, P.; et al. Homozygous deletion in MICU1 presenting with fatigue and lethargy in childhood. Neurol. Genet. 2016, 2, e59. [Google Scholar] [CrossRef]
- Musa, S.; Eyaid, W.; Kamer, K.; Ali, R.; Al-Mureikhi, M.; Shahbeck, N.; Al Mesaifri, F.; Makhseed, N.; Mohamed, Z.; Alshehhi, W.A.; et al. A middle eastern founder mutation expands the genotypic and phenotypic spectrum of mitochondrial MICU1 deficiency: A report of 13 patients. JIMD Rep. 2019, 43, 79–83. [Google Scholar] [CrossRef]
- Rizzuto, R.; Brini, M.; Murgia, M.; Pozzan, T. Microdomains with high Ca2+ close to IP3-sensitive channels that are sensed by neighboring mitochondria. Science 1993, 262, 744–747. [Google Scholar] [CrossRef]
- Ogata, T.; Yamasaki, Y. Scanning electron-microscopic studies on the three-dimensional structure of mitochondria in the mammalian red, white and intermediate muscle fibers. Cell Tissue Res. 1985, 241, 251–256. [Google Scholar] [CrossRef]
- Ogata, T.; Yamasaki, Y. High -resolution scanning electron-microscopic studies on the three-dimensional structure of mitochondria and sarcoplasmic reticulum in the different twitch muscle fibers of the frog. Cell Tissue Res. 1987, 250, 489–497. [Google Scholar] [CrossRef]
- Ogata, T.; Yamasaki, Y.; Shimada, Y.; Forbes, M.S.; Hikida, R.S. Ultra-high resolution scanning electron microscopic studies on the sarcoplasmic reticulum and mitochondria in various muscles: A review. Scanning Microsc. 1993, 7, 145–156. [Google Scholar] [PubMed]
- Ogata, T.; Yamasaki, Y. Ultra-high-resolution scanning electron microscopy of mitochondria and sarcoplasmic reticulum arrangement in human red, white, and intermediate muscle fibers. Anat. Rec. 1997, 248, 214–223. [Google Scholar] [CrossRef]
- Vendelin, M.; Béraud, N.; Guerrero, K.; Andrienko, T.; Kuznetsov, A.V.; Olivares, J.; Kay, L.; Saks, V.A. Mitochondrial regular arrangement in muscle cells: A “crystal-like” pattern. Am. J. Physiol. Cell Physiol. 2005, 288, C757–C767. [Google Scholar] [CrossRef] [PubMed]
- Boncompagni, S.; Rossi, A.E.; Micaroni, M.; Beznoussenko, G.V.; Polishchuk, R.S.; Dirksen, R.T.; Protasi, F. Mitochondria are linked to calcium stores in striated muscle by developmentally regulated tethering structures. Mol. Biol. Cell. 2009, 20, 1058–1067. [Google Scholar] [CrossRef]
- Rizzuto, R.; De Stefani, D.; Raffaello, A.; Mammucari, C. Mitochondria as sensors and regulators of calcium signalling. Nat. Rev. Mol. Cell Biol. 2012, 13, 566–578. [Google Scholar] [CrossRef]
- Pietrangelo, L.; D’Incecco, A.; Ainbinder, A.; Michelucci, A.; Kern, H.; Dirksen, R.T.; Boncompagni, S.; Protasi, F. Age-dependent uncoupling of mitochondria from Ca2+ release units in skeletal muscle. Oncotarget 2015, 6, 35358–35371. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, E.P.; Brown, R.H.; Kunkel, L.M. Dystrophin: The protein product of the duchenne muscular dystrophy locus. Cell 1987, 51, 919–928. [Google Scholar] [CrossRef]
- Whitehead, N.P.; Pham, C.; Gervasio, O.L.; Allen, D.G. N-Acetylcysteine ameliorates skeletal muscle pathophysiology in mdx mice. J. Physiol. 2008, 586, 2003–2014. [Google Scholar] [CrossRef] [PubMed]
- Spencer, M.J.; Tidball, J.G. Calpain translocation during muscle fiber necrosis and regeneration in dystrophin-deficient mice. Exp. Cell Res. 1996, 226, 264–272. [Google Scholar] [CrossRef]
- Whitehead, N.P.; Yeung, E.W.; Allen, D.G. Muscle damage in mdx (dystrophic) mice: Role of calcium and reactive oxygen species. Clin. Exp. Pharmacol. Physiol. 2006, 33, 657–662. [Google Scholar] [CrossRef]
- Vandebrouck, C.; Martin, D.; Van Schoor, M.C.; Debaix, H.; Gailly, P. Involvement of TRPC in the abnormal calcium influx observed in dystrophic (mdx) mouse skeletal muscle fibers. J. Cell Biol. 2002, 158, 1089–1096. [Google Scholar] [CrossRef] [PubMed]
- Millay, D.P.; Goonasekera, S.A.; Sargent, M.A.; Maillet, M.; Aronow, B.J.; Molkentin, J.D. Calcium influx is sufficient to induce muscular dystrophy through a TRPC-dependent mechanism. Proc. Natl. Acad. Sci. USA 2009, 106, 19023–19028. [Google Scholar] [CrossRef] [PubMed]
- Gailly, P. TRP channels in normal and dystrophic skeletal muscle. Curr. Opin. Pharmacol. 2012, 12, 326–334. [Google Scholar] [CrossRef] [PubMed]
- Goonasekera, S.A.; Davis, J.; Kwong, J.Q.; Accornero, F.; Wei-LaPierre, L.; Sargent, M.A.; Dirksen, R.T.; Molkentin, J.D. Enhanced Ca2+ influx from STIM1-Orai1 induces muscle pathology in mouse models of muscular dystrophy. Hum. Mol. Genet. 2014, 23, 3706–3715. [Google Scholar] [CrossRef] [PubMed]
- Bellinger, A.M.; Reiken, S.; Carlson, C.; Mongillo, M.; Liu, X.; Rothman, L.; Matecki, S.; Lacampagne, A.; Marks, A.R. Hypernitrosylated ryanodine receptor calcium release channels are leaky in dystrophic muscle. Nat. Med. 2009, 15, 325–330. [Google Scholar] [CrossRef] [PubMed]
- Andersson, D.C.; Meli, A.C.; Reiken, S.; Betzenhauser, M.J.; Umanskaya, A.; Shiomi, T.; D’Armiento, J.; Marks, A.R. Leaky ryanodine receptors in β-sarcoglycan deficient mice: A potential common defect in muscular dystrophy. Skelet. Muscle 2012, 2, 9. [Google Scholar] [CrossRef] [PubMed]
- Michelucci, A.; Paolini, C.; Canato, M.; Wei-Lapierre, L.; Pietrangelo, L.; De Marco, A.; Reggiani, C.; Dirksen, R.T.; Protasi, F. Antioxidants protect calsequestrin-1 knockout mice from halothane- and heat-induced sudden death. Anesthesiology 2015, 123, 603–617. [Google Scholar] [CrossRef]
- Michelucci, A.; De Marco, A.; Guarnier, F.A.; Protasi, F.; Boncompagni, S. Antioxidant treatment reduces formation of structural cores and improves muscle function in RYR1Y522S/WT mice. Oxid. Med. Cell. Longev. 2017, 2017, 6792694. [Google Scholar] [CrossRef] [PubMed]
- Paolini, C.; Quarta, M.; Wei-LaPierre, L.; Michelucci, A.; Nori, A.; Reggiani, C.; Dirksen, R.T.; Protasi, F. Oxidative stress, mitochondrial damage, and cores in muscle from calsequestrin-1 knockout mice. Skelet. Muscle 2015, 5, 10. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, H.; Pollock, N.; Schiemann, A.; Bulger, T.; Stowell, K. Malignant hyperthermia: A review. Orphanet J. Rare Dis. 2015, 10, 1–19. [Google Scholar] [CrossRef]
- Shy, G.M.; Magee, K.R. A new congenital non-progressive myopathy. Brain 1956, 79, 610–621. [Google Scholar] [CrossRef]
- Jungbluth, H. Central core disease. Orphanet J. Rare Dis. 2007, 2, 25. [Google Scholar] [CrossRef]
- Quane, K.A.; Healy, J.M.S.; Keating, K.E.; Manning, B.M.; Couch, F.J.; Palmucci, L.M.; Doriguzzi, C.; Fagerlund, T.H.; Berg, K.; Ording, H.; et al. Mutations in the ryanodine receptor gene in central core disease and malignant hyperthermia. Nat. Genet. 1993, 5, 51–55. [Google Scholar] [CrossRef] [PubMed]
- Denborough, M.A.; Dennett, X.; Anderson, R.M. Central-Core Disease and Malignant Hyperpyrexia. Br. Med. J. 1973, 1, 272–273. [Google Scholar] [CrossRef][Green Version]
- Dubowitz, V.; Everson Pearse, A.G. OXidative enzymes and phosphorylase in central-core disease of muscle. Lancet 1960, 276, 23–24. [Google Scholar] [CrossRef]
- Eng, G.D.; Epstein, B.S.; Engel, W.K.; McKay, D.W.; McKay, R. Malignant Hyperthermia and Central Core Disease in a Child with Congenital Dislocating Hips: Case Presentation and Review. Arch. Neurol. 1978, 35, 189–197. [Google Scholar] [CrossRef] [PubMed]
- Frank, J.P.; Harati, Y.; Butler, I.J.; Nelson, T.E.; Scott, C.I. Central core disease and malignant hyperthermia syndrome. Ann. Neurol. 1980, 7, 11–17. [Google Scholar] [CrossRef] [PubMed]
- Shuaib, A.; Paasuke, R.T.; Brownell, A.K.W. Central core disease: Clinical features in 13 patients. Medicine 1987, 66, 389–396. [Google Scholar] [CrossRef]
- Galli, L.; Orrico, A.; Lorenzini, S.; Censini, S.; Falciani, M.; Covacci, A.; Tegazzin, V.; Sorrentino, V. Frequency and localization of mutations in the 106 exons of the RYR1 gene in 50 individuals with malignant hyperthermia. Hum. Mutat. 2006, 27, 830. [Google Scholar] [CrossRef]
- Robinson, R.; Carpenter, D.; Shaw, M.A.; Halsall, J.; Hopkins, P. Mutations in RYR1 in malignant hypertheraiia and central core disease. Hum. Mutat. 2006, 27, 977–989. [Google Scholar] [CrossRef] [PubMed]
- Canato, M.; Capitanio, P.; Cancellara, L.; Leanza, L.; Raffaello, A.; Reane, D.V.; Marcucci, L.; Michelucci, A.; Protasi, F.; Reggiani, C. Excessive Accumulation of Ca2+ in Mitochondria of Y522S-RYR1 Knock-in Mice: A Link Between Leak From the Sarcoplasmic Reticulum and Altered Redox State. Front. Physiol. 2019, 10, 1142. [Google Scholar] [CrossRef] [PubMed]
- Durham, W.J.; Aracena-Parks, P.; Long, C.; Rossi, A.E.; Goonasekera, S.A.; Boncompagni, S.; Galvan, D.L.; Gilman, C.P.; Baker, M.R.; Shirokova, N.; et al. RyR1 S-Nitrosylation Underlies Environmental Heat Stroke and Sudden Death in Y522S RyR1 Knockin Mice. Cell 2008, 133, 53–65. [Google Scholar] [CrossRef]
- Chelu, M.G.; Goonasekera, S.A.; Durham, W.J.; Tang, W.; Lueck, J.D.; Riehl, J.; Pessah, I.N.; Zhang, P.; Bhattacharjee, M.B.; Dirksen, R.T.; et al. Heat- and anesthesia-induced malignant hyperthermia in an RyR1 knock-in mouse. FASEB J. 2006, 20, 329–330. [Google Scholar] [CrossRef] [PubMed]
- Michelucci, A.; Paolini, C.; Boncompagni, S.; Canato, M.; Reggiani, C.; Protasi, F. Strenuous exercise triggers a life-threatening response in mice susceptible to malignant hyperthermia. FASEB J. 2017, 31, 3649–3662. [Google Scholar] [CrossRef] [PubMed]
- Boncompagni, S.; Rossi, A.E.; Micaroni, M.; Hamilton, S.L.; Dirksen, R.T.; Franzini-Armstrong, C.; Protasi, F. Characterization and temporal development of cores in a mouse model of malignant hyperthermia. Proc. Natl. Acad. Sci. USA 2009, 106, 21996–22001. [Google Scholar] [CrossRef] [PubMed]
- Loke, J.; MacLennan, D.H. Malignant hyperthermia and central core disease: Disorders of Ca2+ release channels. Am. J. Med. 1998, 104, 470–486. [Google Scholar] [CrossRef]
- Avila, G.; O’Brien, J.J.; Dirksen, R.T. Excitation-Contraction uncoupling by a human central core disease mutation in the ryanodine receptor. Proc. Natl. Acad. Sci. USA 2001, 98, 4215–4220. [Google Scholar] [CrossRef] [PubMed]
- Avila, G.; O’Connell, K.M.S.; Dirksen, R.T. The pore region of the skeletal muscle ryanodine receptor is a primary locus for excitation-contraction uncoupling in central core disease. J. Gen. Physiol. 2003, 121, 277–286. [Google Scholar] [CrossRef] [PubMed]
- Boncompagni, S.; Loy, R.E.; Dirksen, R.T.; Franzini-Armstrong, C. The I4895T mutation in the type 1 ryanodine receptor induces fiber-type specific alterations in skeletal muscle that mimic premature aging. Aging Cell 2010, 9, 958–970. [Google Scholar] [CrossRef] [PubMed]
- Michelucci, A.; García-Castañeda, M.; Boncompagni, S.; Dirksen, R.T. Role of STIM1/ORAI1-mediated store-operated Ca2+ entry in skeletal muscle physiology and disease. Cell Calcium 2018, 76, 101–115. [Google Scholar] [CrossRef] [PubMed]
- Manno, C.; Figueroa, L.; Royer, L.; Pouvreau, S.; Lee, C.S.; Volpe, P.; Nori, A.; Zhou, J.; Meissner, G.; Hamilton, S.L.; et al. Altered Ca2+ concentration, permeability and buffering in the myofibre Ca2+ store of a mouse model of malignant hyperthermia. J. Physiol. 2013, 591, 4439–4457. [Google Scholar] [CrossRef] [PubMed]
- Yarotskyy, V.; Protasi, F.; Dirksen, R.T. Accelerated Activation of SOCE Current in Myotubes from Two Mouse Models of Anesthetic- and Heat-Induced Sudden Death. PLoS ONE 2013, 8, e77633. [Google Scholar] [CrossRef]
- Evans, W.J. What is sarcopenia? J. Gerontol. Ser. A Biol. Sci. Med Sci. 1995, 50, 5–8. [Google Scholar] [CrossRef]
- Roubenoff, R.; Hughes, V.A. Sarcopenia: Current concepts. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2000, 55, M716–M724. [Google Scholar] [CrossRef] [PubMed]
- Lexell, J. Human aging, muscle mass, and fiber type composition. J. Gerontol. Ser. A Biol. Sci. Med Sci. 1995, 50, 11–16. [Google Scholar]
- Frontera, W.R.; Suh, D.; Krivickas, L.S.; Hughes, V.A.; Goldstein, R.; Roubenoff, R. Skeletal muscle fiber quality in older men and women. Am. J. Physiol. Cell Physiol. 2000, 279, C611–C618. [Google Scholar] [CrossRef]
- Liu, W.; Klose, A.; Forman, S.; Paris, N.D.; Wei-LaPierre, L.; Cortés-Lopéz, M.; Tan, A.; Flaherty, M.; Miura, P.; Dirksen, R.T.; et al. Loss of adult skeletal muscle stem cells drives age-related neuromuscular junction degeneration. Elife 2017, 6, e26464. [Google Scholar] [CrossRef]
- Faulkner, J.A.; Larkin, L.M.; Claflin, D.R.; Brooks, S.V. Age-related changes in the structure and function of skeletal muscles. Clin. Exp. Pharmacol. Physiol. 2007, 34, 1091–1096. [Google Scholar] [CrossRef]
- Delbono, O.; O’Rourke, K.S.; Ettinger, W.H. Excitation-calcium release uncoupling in aged single human skeletal muscle fibers. J. Membr. Biol. 1995, 148, 211–222. [Google Scholar] [CrossRef]
- Renganathan, M.; Messi, M.L.; Delbono, O. Dihydropyridine receptor-ryanodine receptor uncoupling in aged skeletal muscle. J. Membr. Biol. 1997, 157, 247–253. [Google Scholar] [CrossRef]
- Weisleder, N.; Brotto, M.; Komazaki, S.; Pan, Z.; Zhao, X.; Nosek, T.; Parness, J.; Takeshima, H.; Ma, J. Muscle aging is associated with compromised Ca2+ spark signaling and segregated intracellular Ca2+ release. J. Cell Biol. 2006, 174, 639–645. [Google Scholar] [CrossRef]
- Weisleder, N.; Ma, J. Altered Ca2+ sparks in aging skeletal and cardiac muscle. Ageing Res. Rev. 2008, 7, 177–188. [Google Scholar] [CrossRef] [PubMed]
- Boncompagni, S.; D’Amelio, L.; Fulle, S.; Fanò, G.; Protasi, F. Progressive disorganization of the excitation-contraction coupling apparatus in aging human skeletal muscle as revealed by electron microscopy: A possible role in the decline of muscle performance. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2006, 61, 995–1008. [Google Scholar] [CrossRef]
- Zampieri, S.; Pietrangelo, L.; Loefler, S.; Fruhmann, H.; Vogelauer, M.; Burggraf, S.; Pond, A.; Grim-Stieger, M.; Cvecka, J.; Sedliak, M.; et al. Lifelong physical exercise delays age-associated skeletal muscle decline. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2015, 70, 163–173. [Google Scholar] [CrossRef]
- Pietrangelo, L.; Michelucci, A.; Ambrogini, P.; Sartini, S.; Guarnier, F.A.; Fusella, A.; Zamparo, I.; Mammucari, C.; Protasi, F.; Boncompagni, S. Muscle activity prevents the uncoupling of mitochondria from Ca 2+ Release Units induced by ageing and disuse. Arch. Biochem. Biophys. 2019, 663, 22–33. [Google Scholar] [CrossRef]
- Andersson, D.C.; Betzenhauser, M.J.; Reiken, S.; Meli, A.C.; Umanskaya, A.; Xie, W.; Shiomi, T.; Zalk, R.; Lacampagne, A.; Marks, A.R. Ryanodine receptor oxidation causes intracellular calcium leak and muscle weakness in aging. Cell Metab. 2011, 14, 196–207. [Google Scholar] [CrossRef]
- Pan, Z.; Yang, D.; Nagaraj, R.Y.; Nosek, T.A.; Nishi, M.; Takeshima, H.; Cheng, H.; Ma, J. Dysfunction of store-operated calcium channel in muscle cells lacking mg29. Nat. Cell Biol. 2002, 4, 379–383. [Google Scholar] [CrossRef]
- Zhao, X.; Weisleder, N.; Thornton, A.; Oppong, Y.; Campbell, R.; Ma, J.; Brotto, M. Compromised store-operated Ca2+ entry in aged skeletal muscle. Aging Cell 2008, 7, 561–568. [Google Scholar] [CrossRef]
- Payne, A.M.; Jimenez-Moreno, R.; Wang, Z.M.; Messi, M.L.; Delbono, O. Role of Ca2+, membrane excitability, and Ca2+ stores in failing muscle contraction with aging. Exp. Gerontol. 2009, 44, 261–273. [Google Scholar] [CrossRef][Green Version]
- Edwards, J.N.; Blackmore, D.G.; Gilbert, D.F.; Murphy, R.M.; Launikonis, B.S. Store-operated calcium entry remains fully functional in aged mouse skeletal muscle despite a decline in STIM1 protein expression. Aging Cell 2011, 10, 675–685. [Google Scholar] [CrossRef] [PubMed]
- Powers, S.K.; Kavazis, A.N.; McClung, J.M. Oxidative stress and disuse muscle atrophy. J. Appl. Physiol. 2007, 102, 2389–2397. [Google Scholar] [CrossRef] [PubMed]
- Booth, F.W.; Seider, M.J. Early change in skeletal muscle protein synthesis after limb immobilization of rats. J. Appl. Physiol. Respir. Environ. Exerc. Physiol. 1979, 47, 974–977. [Google Scholar] [CrossRef] [PubMed]
- Talbert, E.E.; Smuder, A.J.; Min, K.; Kwon, O.S.; Powers, S.K. Calpain and caspase-3 play required roles in immobilization-induced limb muscle atrophy. J. Appl. Physiol. 2013, 114, 1482–1489. [Google Scholar] [CrossRef] [PubMed]
- Goll, D.E.; Thompson, V.F.; Li, H.; Wei, W.; Cong, J. The calpain system. Physiol. Rev. 2003, 83, 731–801. [Google Scholar] [CrossRef] [PubMed]
- Weiss, N.; Andrianjafiniony, T.; Dupré-Aucouturier, S.; Pouvreau, S.; Desplanches, D.; Jacquemond, V. Altered myoplasmic Ca2+ handling in rat fast-twitch skeletal muscle fibres during disuse atrophy. Pflug. Arch. Eur. J. Physiol. 2010, 459, 631–644. [Google Scholar] [CrossRef] [PubMed]
- Ingalls, C.P.; Warren, G.L.; Armstrong, R.B. Intracellular Ca2+ transients in mouse soleus muscle after hindlimb unloading and reloading. J. Appl. Physiol. 1999, 87, 386–390. [Google Scholar] [CrossRef]
- Mirza, K.A.; Tisdale, M.J. Role of Ca2+ in proteolysis-inducing factor (PIF)-induced atrophy of skeletal muscle. Cell. Signal. 2012, 24, 2118–2122. [Google Scholar] [CrossRef] [PubMed]
- Eley, H.L.; Russell, S.T.; Tisdale, M.J. Mechanism of activation of dsRNA-dependent protein kinase (PKR) in muscle atrophy. Cell. Signal. 2010, 22, 783–790. [Google Scholar] [CrossRef]
- Powers, S.K.; Smuder, A.J.; Judge, A.R. Oxidative stress and disuse muscle atrophy: Cause or consequence? Curr. Opin. Clin. Nutr. Metab. Care 2012, 15, 240–245. [Google Scholar] [CrossRef]
- Matecki, S.; Dridi, H.; Jung, B.; Saint, N.; Reiken, S.R.; Scheuermann, V.; Mrozek, S.; Santulli, G.; Umanskaya, A.; Petrof, B.J.; et al. Leaky ryanodine receptors contribute to diaphragmatic weakness during mechanical ventilation. Proc. Natl. Acad. Sci. USA 2016, 113, 9069–9074. [Google Scholar] [CrossRef]
- Agrawal, A.; Rathor, R.; Kumar, R.; Suryakumar, G.; Singh, S.N.; Kumar, B. Redox modification of ryanodine receptor contributes to impaired Ca2+ homeostasis and exacerbates muscle atrophy under high altitude. Free. Radic. Biol. Med. 2020, 160, 643–656. [Google Scholar] [CrossRef]
- Qaisar, R.; Bhaskaran, S.; Ranjit, R.; Sataranatarajan, K.; Premkumar, P.; Huseman, K.; Van Remmen, H. Restoration of SERCA ATPase prevents oxidative stress-related muscle atrophy and weakness. Redox Biol. 2019, 20, 68–74. [Google Scholar] [CrossRef] [PubMed]
- Qaisar, R.; Qayum, M.; Muhammad, T. Reduced sarcoplasmic reticulum Ca2+ ATPase activity underlies skeletal muscle wasting in asthma. Life Sci. 2021, 273, 119296. [Google Scholar] [CrossRef]
- Tomiya, S.; Tamura, Y.; Kouzaki, K.; Kotani, T.; Wakabayashi, Y.; Noda, M.; Nakazato, K. Cast immobilization of hindlimb upregulates sarcolipin expression in atrophied skeletal muscles and increases thermogenesis in C57BL/6J mice. Am. J. Physiol.Regul. Integr. Comp. Physiol. 2019, 317, R649–R661. [Google Scholar] [CrossRef] [PubMed]
- Kavazis, A.N.; Talbert, E.E.; Smuder, A.J.; Hudson, M.B.; Nelson, W.B.; Powers, S.K. Mechanical ventilation induces diaphragmatic mitochondrial dysfunction and increased oxidant production. Free Radic. Biol. Med. 2009, 46, 842–850. [Google Scholar] [CrossRef] [PubMed]
- Muller, F.L.; Song, W.; Jang, Y.C.; Liu, Y.; Sabia, M.; Richardson, A.; Van Remmen, H. Denervation-induced skeletal muscle atrophy is associated with increased mitochondrial ROS production. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2007, 293, R1159–R1168. [Google Scholar] [CrossRef]
- Abadi, A.; Glover, E.I.; Isfort, R.J.; Raha, S.; Safdar, A.; Yasuda, N.; Kaczor, J.J.; Melov, S.; Hubbard, A.; Qu, X.; et al. Limb immobilization induces a coordinate down-regulation of mitochondrial and other metabolic pathways in men and women. PLoS ONE 2009, 4, e6518. [Google Scholar] [CrossRef]
- Picard, M.; Azuelos, I.; Jung, B.; Giordano, C.; Matecki, S.; Hussain, S.; White, K.; Li, T.; Liang, F.; Benedetti, A.; et al. Mechanical ventilation triggers abnormal mitochondrial dynamics and morphology in the diaphragm. J. Appl. Physiol. 2015, 118, 1161–1171. [Google Scholar] [CrossRef]
- Min, K.; Smuder, A.J.; Kwon, O.S.; Kavazis, A.N.; Szeto, H.H.; Powers, S.K. Mitochondrial-targeted antioxidants protect skeletal muscle against immobilization-induced muscle atrophy. J. Appl. Physiol. 2011, 111, 1459–1466. [Google Scholar] [CrossRef]
- Adhihetty, P.J.; Ljubicic, V.; Menzies, K.J.; Hood, D.A. Differential susceptibility of subsarcolemmal and intermyofibrillar mitochondria to apoptotic stimuli. Am. J. Physiol. Cell Physiol. 2005, 289, C994–C1001. [Google Scholar] [CrossRef] [PubMed]
- O’Leary, M.F.N.; Vainshtein, A.; Carter, H.N.; Zhang, Y.; Hood, D.A. Denervation-induced mitochondrial dysfunction and autophagy in skeletal muscle of apoptosis-deficient animals. Am. J. Physiol. Cell Physiol. 2012, 303, C447–C454. [Google Scholar] [CrossRef]
- Karam, C.; Yi, J.; Xiao, Y.; Dhakal, K.; Zhang, L.; Li, X.; Manno, C.; Xu, J.; Li, K.; Cheng, H.; et al. Absence of physiological Ca2+ transients is an initial trigger for mitochondrial dysfunction in skeletal muscle following denervation. Skelet. Muscle 2017, 7, 6. [Google Scholar] [CrossRef] [PubMed]
- Krysiewicz, S.; Mellinger, B.C. The role of imaging in the diagnostic evaluation of impotence. Am. J. Roentgenol. 1989, 153, 1133–1139. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Michelucci, A.; Liang, C.; Protasi, F.; Dirksen, R.T. Altered Ca2+ Handling and Oxidative Stress Underlie Mitochondrial Damage and Skeletal Muscle Dysfunction in Aging and Disease. Metabolites 2021, 11, 424. https://doi.org/10.3390/metabo11070424
Michelucci A, Liang C, Protasi F, Dirksen RT. Altered Ca2+ Handling and Oxidative Stress Underlie Mitochondrial Damage and Skeletal Muscle Dysfunction in Aging and Disease. Metabolites. 2021; 11(7):424. https://doi.org/10.3390/metabo11070424
Chicago/Turabian StyleMichelucci, Antonio, Chen Liang, Feliciano Protasi, and Robert T. Dirksen. 2021. "Altered Ca2+ Handling and Oxidative Stress Underlie Mitochondrial Damage and Skeletal Muscle Dysfunction in Aging and Disease" Metabolites 11, no. 7: 424. https://doi.org/10.3390/metabo11070424
APA StyleMichelucci, A., Liang, C., Protasi, F., & Dirksen, R. T. (2021). Altered Ca2+ Handling and Oxidative Stress Underlie Mitochondrial Damage and Skeletal Muscle Dysfunction in Aging and Disease. Metabolites, 11(7), 424. https://doi.org/10.3390/metabo11070424