
First Genome-Scale Metabolic Model of Dolosigranulum pigrum Confirms Multiple Auxotrophies




Abstract
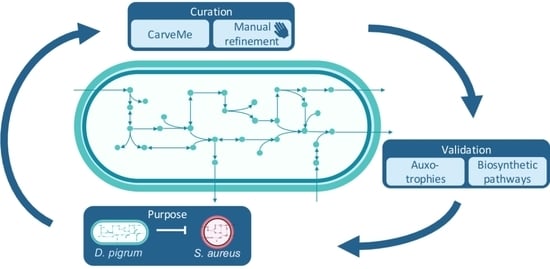
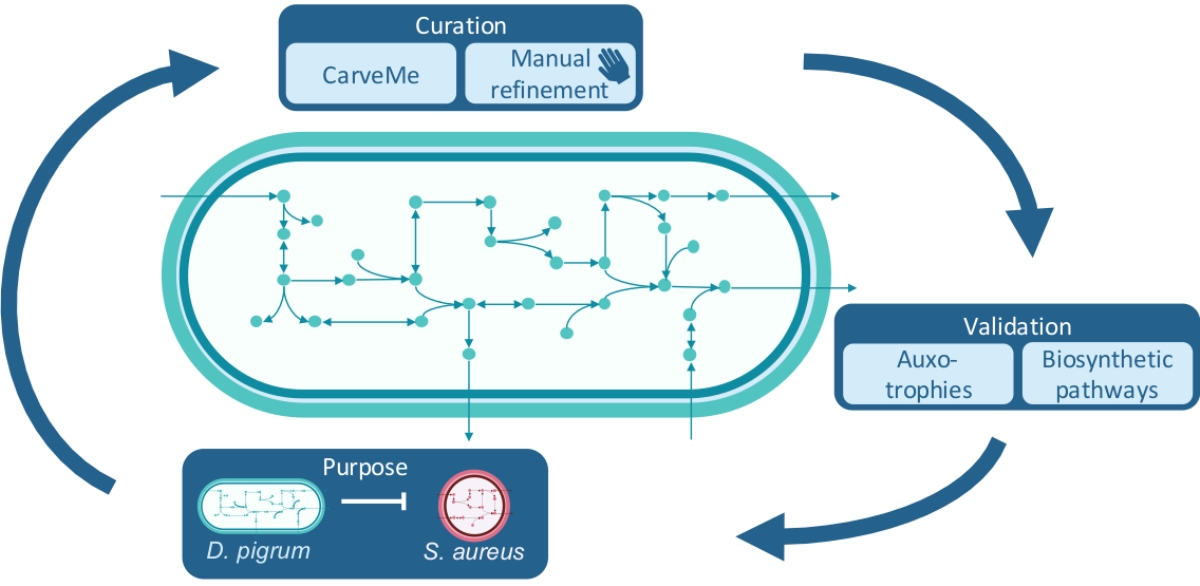
1. Introduction
2. Results
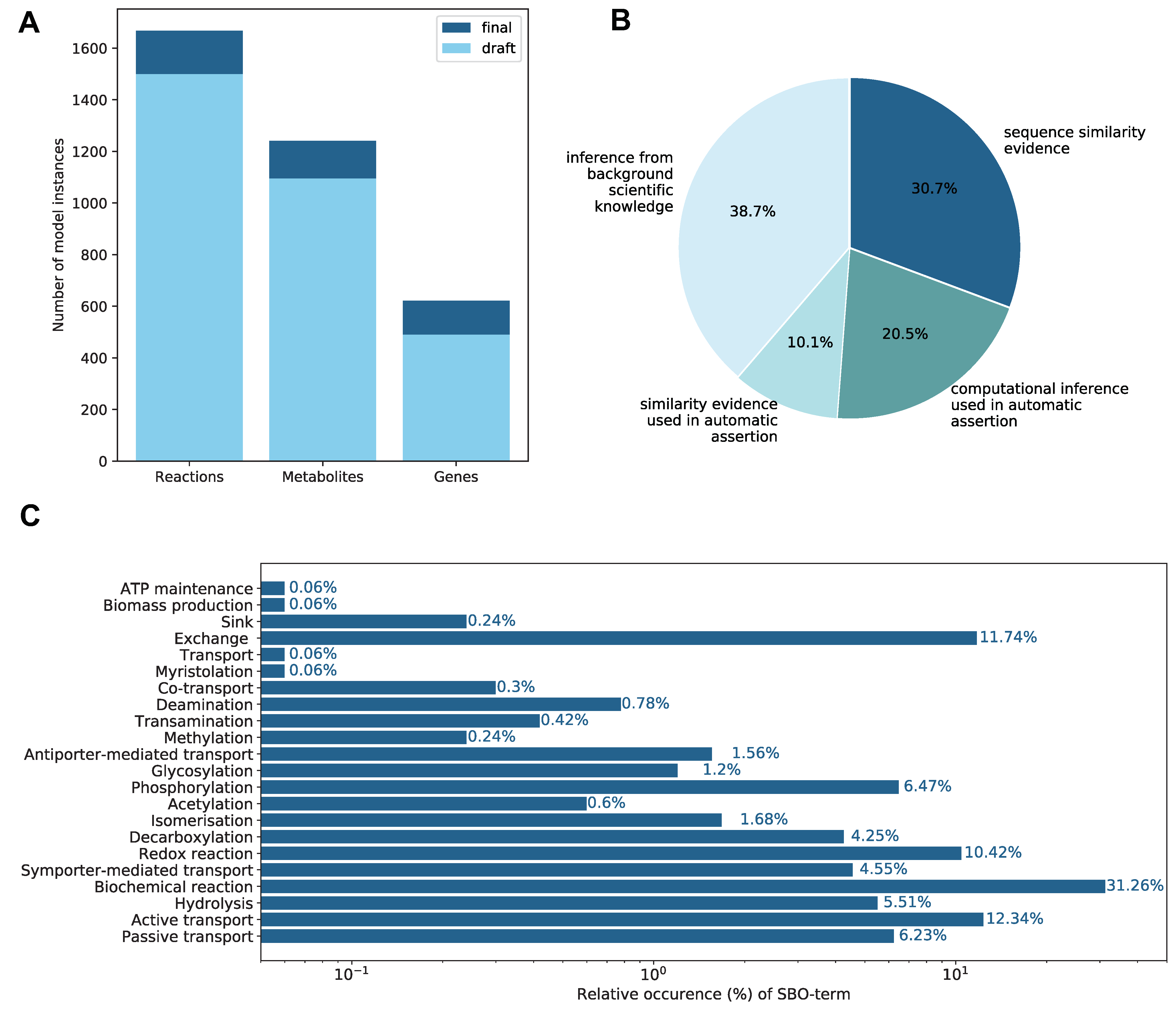
2.1. Properties of the Constructed GEM
2.1.1. Mass and Charge Imbalances
2.1.2. Annotations
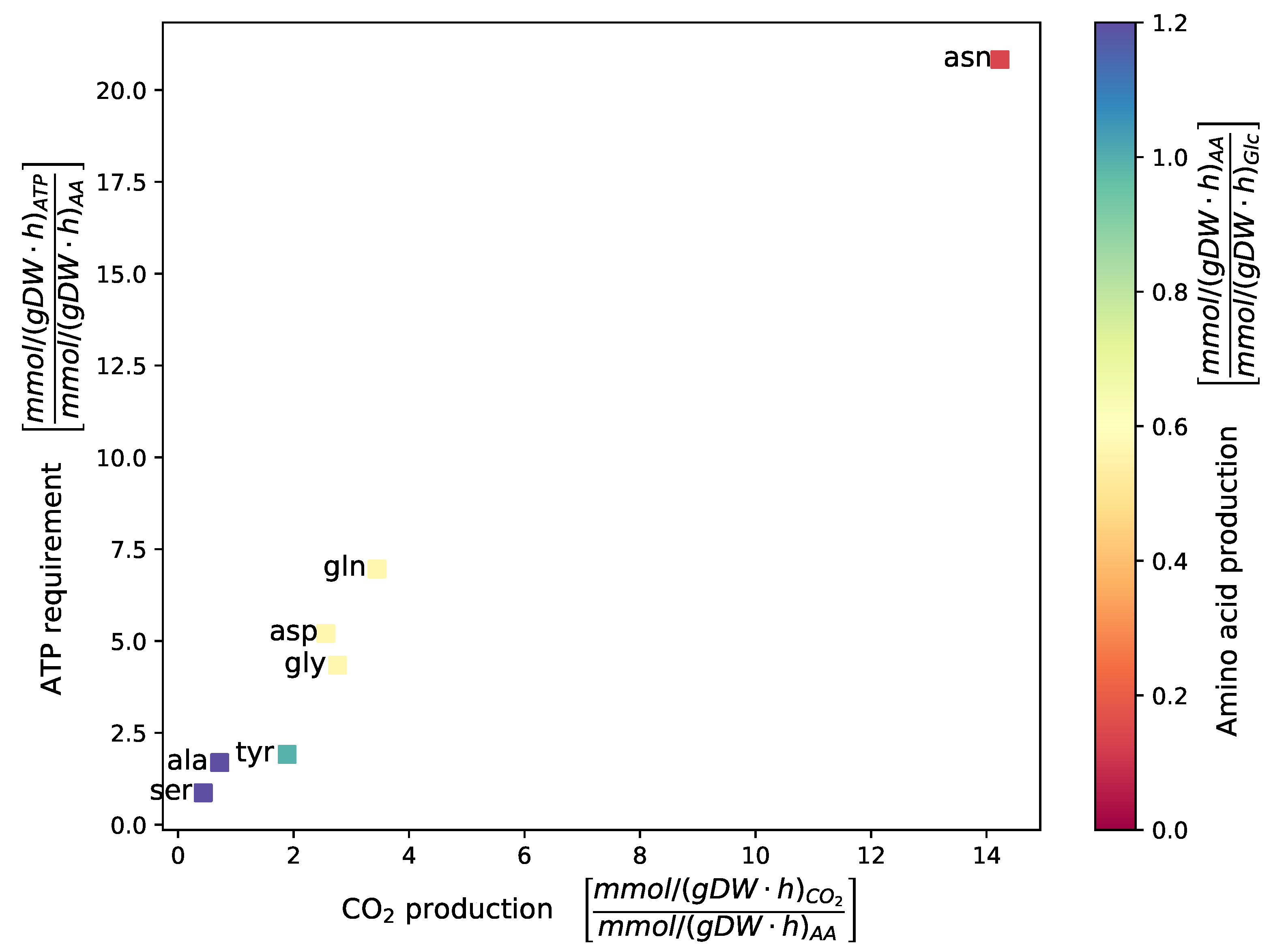
2.1.3. Biomass Objective Function
2.1.4. Subsystems and Groups
2.2. Evaluating Auxotrophies and Predicted Biosynthesis
2.2.1. Auxotrophies and Biosynthesis
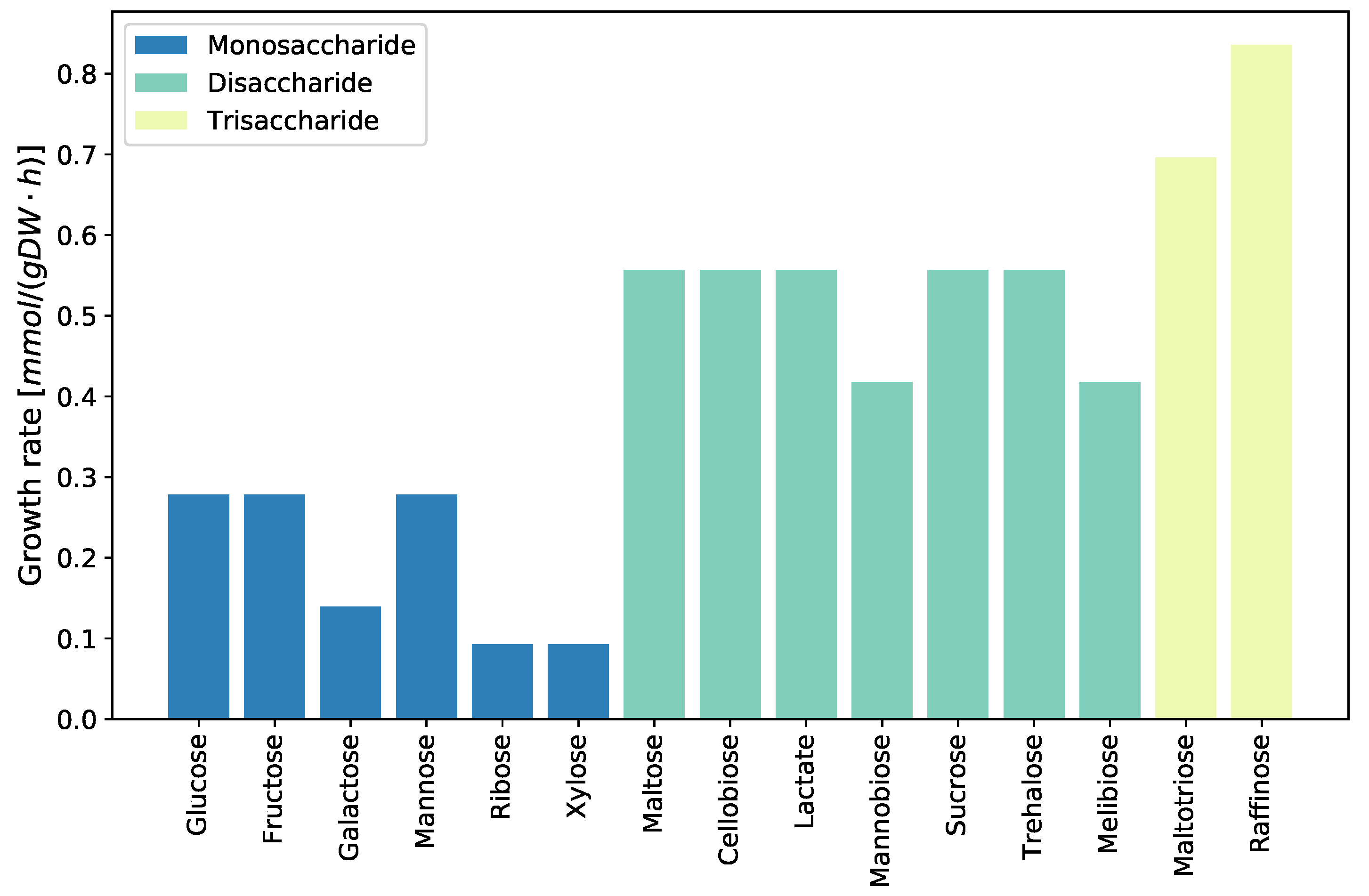
2.2.2. Carbohydrate Metabolism
2.3. Evaluating Growth Capabilities
2.3.1. Growth in SNM
2.3.2. Growth in SCFM
2.3.3. Growth in the Blood Medium
2.3.4. Growth in the Gastrointestinal Tract
2.3.5. Definition of a Minimal Medium for D. pigrum
2.3.6. Growth on Different Carbon Sources
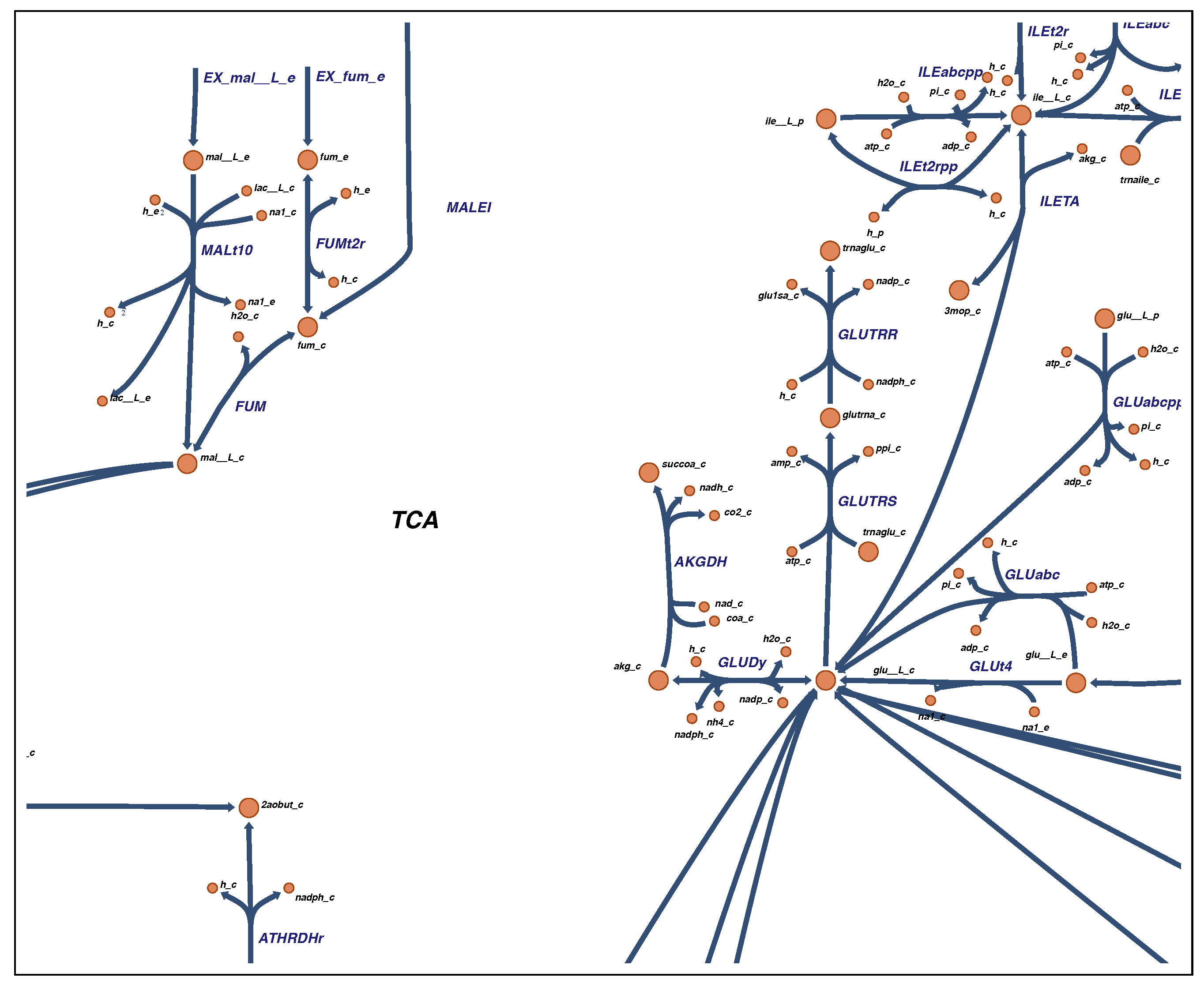
2.4. Visualization
3. Discussion
4. Materials and Methods
4.1. Building the Draft Reconstruction
4.1.1. CarveMe
4.1.2. ModelPolisher
4.1.3. Memote
4.2. Refining the Reconstruction Using Literature Evidence
4.2.1. Mass and Charge Imbalances
4.2.2. Add Gene Annotations
4.2.3. Extend Model Manually Using the KEGG Database
4.2.4. Test for Energy-Generating Cycles
4.2.5. Add More Precise SBO Terms
4.2.6. Improve Biomass Objective Function
4.2.7. Add ECO Terms
4.2.8. Remove Redundant Information
4.2.9. Add Subsystems and Groups
4.3. Evaluation and Validation of the Reconstruction
4.3.1. Evaluating Auxotrophies, Biosynthesis Capabilities, and Carbohydrate Metabolism
4.3.2. Identification of Additional Auxotrophies
4.3.3. Evaluating Growth Capabilities in Different Media
4.3.4. Defining a Minimal Medium for D. pigrum
4.3.5. Evaluating Growth Capabilities on Different Carbon Sources
4.4. Visualization
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ABC | ATP-binding cassette |
| API | application programming interface |
| ATP | adenosine triphosphate |
| BiGG | Biochemically, Genetically, and Genomically structured |
| BLAST | Basic Local Alignment Search Tool |
| BOF | biomass objective function |
| CDS | coding domain sequence |
| CF | cystic fibrosis |
| COBRA | Constraint-Based Reconstruction and Analysis |
| CTP | cytidine triphosphate |
| dATP | deoxyadenosine triphosphate |
| dCTP | deoxycytidine triphosphate |
| dGTP | deoxyguanosine triphosphate |
| dTTP | deoxythymidine triphosphate |
| EC | Enzyme Commission |
| ECF | energy-coupling factor |
| ECO | Evidence and Conclusion Ontology |
| FBA | flux balance analysis |
| fbc | flux balance constraints |
| FVA | flux variability analysis |
| GEM | genome-scale metabolic model |
| GPR | gene–protein reaction |
| GTP | guanosine triphosphate |
| HMDB | Human Metabolome Database |
| ID | identifier |
| ITP | inosine triphosphate |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| MIRIAM | Minimal Information Requested in the Annotation of Models |
| NADH | reduced nicotinamide adenine dinucleotide |
| NADPH | reduced nicotinamide adenine dinucleotide phosphat |
| NCBI | National Center for Biotechnology Information |
| REST | representational state transfer |
| SBO | Systems Biology Ontology |
| SCFM | synthetic cystic fibrosis medium |
| SNM | synthetic nasal medium |
| URT | upper respiratory tract |
| UTP | uridine triphosphate |
| TCA | tricarboxylic acid |
| VMH | Virtual Metabolic Human |
References
- Lécuyer, H.; Audibert, J.; Bobigny, A.; Eckert, C.; Jannière-Nartey, C.; Buu-Hoï, A.; Mainardi, J.L.; Podglajen, I. Dolosigranulum pigrum causing nosocomial pneumonia and septicemia. J. Clin. Microbiol. 2007, 45, 3474–3475. [Google Scholar] [CrossRef]
- Escherich, T. Die Darmbakterien des Neugeborenen und Säuglings. Fortschr. Med. 1885, 3, 547–554. [Google Scholar]
- Aguirre, M.; Morrison, D.; Cookson, B.; Gay, F.; Collins, M. Phenotypic and phylogenetic characterization of some Gemella-like organisms from human infections: Description of Dolosigranulum pigrum gen. nov., sp. nov. J. Appl. Bacteriol. 1993, 75, 608–612. [Google Scholar] [CrossRef]
- Hall, G.S.; Gordon, S.; Schroeder, S.; Smith, K.; Anthony, K.; Procop, G.W. Case of synovitis potentially caused by Dolosigranulum pigrum. J. Clin. Microbiol. 2001, 39, 1202–1203. [Google Scholar] [CrossRef]
- Lin, J.C.; Hou, S.J.; Huang, L.U.; Sun, J.R.; Chang, W.K.; Lu, J.J. Acute cholecystitis accompanied by acute pancreatitis potentially caused by Dolosigranulum pigrum. J. Clin. Microbiol. 2006, 44, 2298–2299. [Google Scholar] [CrossRef]
- Hoedemaekers, A.; Schülin, T.; Tonk, B.; Melchers, W.J.; Sturm, P.D. Ventilator-associated pneumonia caused by Dolosigranulum pigrum. J. Clin. Microbiol. 2006, 44, 3461–3462. [Google Scholar] [CrossRef]
- Johnsen, B.O.; Rønning, E.J.; Onken, A.; Figved, W.; Jenum, P.A. Dolosigranulum pigrum causing biomaterial-associated arthritis. APMIS 2011, 119, 85–87. [Google Scholar] [CrossRef]
- LaClaire, L.; Facklam, R. Antimicrobial susceptibility and clinical sources of Dolosigranulum pigrum cultures. Antimicrob. Agents Chemother. 2000, 44, 2001–2003. [Google Scholar] [CrossRef]
- Lappan, R.; S Peacock, C. Corynebacterium and Dolosigranulum: Future probiotic candidates for upper respiratory tract infections. Microbiol. Aust. 2019, 40, 172–177. [Google Scholar] [CrossRef]
- Bogaert, D.; Keijser, B.; Huse, S.; Rossen, J.; Veenhoven, R.; van Gils, E.; Bruin, J.; Montijn, R.; Bonten, M.; Sanders, E. Variability and Diversity of Nasopharyngeal Microbiota in Children: A Metagenomic Analysis. PLoS ONE 2011, 6, e17035. [Google Scholar] [CrossRef]
- Laufer, A.S.; Metlay, J.P.; Gent, J.F.; Fennie, K.P.; Kong, Y.; Pettigrewa, M.M. Microbial communities of the upper respiratory tract and otitis media in children. mBio 2011, 2, 245–255. [Google Scholar] [CrossRef]
- Bomar, L.; Brugger, S.D.; Yost, B.H.; Davies, S.S.; Lemon, K.P. Corynebacterium accolens releases antipneumococcal free fatty acids from human nostril and skin surface triacylglycerols. mBio 2016, 7. [Google Scholar] [CrossRef]
- Kelly, M.S.; Surette, M.G.; Smieja, M.; Pernica, J.M.; Rossi, L.; Luinstra, K.; Steenhoff, A.P.; Feemster, K.A.; Goldfarb, D.M.; Arscott-Mills, T.; et al. The Nasopharyngeal Microbiota of Children with Respiratory Infections in Botswana. Pediatric Infect. Dis. J. 2017, 36, e211–e218. [Google Scholar] [CrossRef]
- Pettigrew, M.M.; Laufer, A.S.; Gent, J.F.; Kong, Y.; Fennie, K.P.; Metlay, J.P. Upper respiratory tract microbial communities, acute otitis media pathogens, and antibiotic use in healthy and sick children. Appl. Environ. Microbiol. 2012, 78, 6262–6270. [Google Scholar] [CrossRef]
- Lappan, R.; Imbrogno, K.; Sikazwe, C.; Anderson, D.; Mok, D.; Coates, H.; Vijayasekaran, S.; Bumbak, P.; Blyth, C.C.; Jamieson, S.E.; et al. A microbiome case-control study of recurrent acute otitis media identified potentially protective bacterial genera. BMC Microbiol. 2018, 18, 1–20. [Google Scholar] [CrossRef]
- Biesbroek, G.; Bosch, A.A.; Wang, X.; Keijser, B.J.; Veenhoven, R.H.; Sanders, E.A.; Bogaert, D. The impact of breastfeeding on nasopharyngeal microbial communities in infants. Am. J. Respir. Crit. Care Med. 2014, 190, 298–308. [Google Scholar] [CrossRef]
- Biesbroek, G.; Tsivtsivadze, E.; Sanders, E.A.; Montijn, R.; Veenhoven, R.H.; Keijser, B.J.; Bogaert, D. Early respiratory microbiota composition determines bacterial succession patterns and respiratory health in children. Am. J. Respir. Crit. Care Med. 2014, 190, 1283–1292. [Google Scholar] [CrossRef]
- Bosch, A.A.; De Steenhuijsen Piters, W.A.; Van Houten, M.A.; Chu, M.L.J.; Biesbroek, G.; Kool, J.; Pernet, P.; De Groot, P.K.C.; Eijkemans, M.J.; Keijser, B.J.; et al. Maturation of the infant respiratory microbiota, environmental drivers, and health consequences. Am. J. Respir. Crit. Care Med. 2017, 196, 1582–1590. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, K.; Linnemann, R.W.; Mansbach, J.M.; Ajami, N.J.; Espinola, J.A.; Petrosino, J.F.; Piedra, P.A.; Stevenson, M.D.; Sullivan, A.F.; Thompson, A.D.; et al. Nasal Airway Microbiota Profile and Severe Bronchiolitis in Infants: A Case-control Study. Pediatric Infect. Dis. J. 2017, 36, 1044–1051. [Google Scholar] [CrossRef] [PubMed]
- Prevaes, S.M.; De Winter-De Groot, K.M.; Janssens, H.M.; De Steenhuijsen Piters, W.A.; Tramper-Stranders, G.A.; Wyllie, A.L.; Hasrat, R.; Tiddens, H.A.; Van Westreenen, M.; Van Der Ent, C.K.; et al. Development of the nasopharyngeal microbiota in infants with cystic fibrosis. Am. J. Respir. Crit. Care Med. 2016, 193, 504–515. [Google Scholar] [CrossRef] [PubMed]
- Chonmaitree, T.; Jennings, K.; Golovko, G.; Khanipov, K.; Pimenova, M.; Patel, J.A.; McCormick, D.P.; Loeffelholz, M.J.; Fofanov, Y. Nasopharyngeal microbiota in infants and changes during viral upper respiratory tract infection and acute otitis media. PLoS ONE 2017, 12, e0180630. [Google Scholar] [CrossRef]
- Lopes, S.P.; Ceri, H.; Azevedo, N.F.; Pereira, M.O. Antibiotic resistance of mixed biofilms in cystic fibrosis: Impact of emerging microorganisms on treatment of infection. Int. J. Antimicrob. Agents 2012, 40, 260–263. [Google Scholar] [CrossRef] [PubMed]
- Lopes, S.P.; Azevedo, N.F.; Pereira, M.O. Emergent Bacteria in Cystic Fibrosis: In Vitro Biofilm Formation and Resilience under Variable Oxygen Conditions. BioMed Res. Int. 2014, 2014. [Google Scholar] [CrossRef]
- Lopes, S.P.; Azevedo, N.F.; Pereira, M.O. Developing a model for cystic fibrosis sociomicrobiology based on antibiotic and environmental stress. Int. J. Med. Microbiol. 2017, 307, 460–470. [Google Scholar] [CrossRef]
- Brugger, S.D.; Eslami, S.M.; Pettigrew, M.M.; Escapa, I.F.; Henke, M.T.; Kong, Y.; Lemon, K.P. Dolosigranulum pigrum Cooperation and Competition in Human Nasal Microbiota. mSphere 2020, 5. [Google Scholar] [CrossRef] [PubMed]
- Sakr, A.; Brégeon, F.; Mège, J.L.; Rolain, J.M.; Blin, O. Staphylococcus aureus nasal colonization: An update on mechanisms, epidemiology, risk factors, and subsequent infections. Front. Microbiol. 2018, 9, 2419. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, A.; Bénard, S.; Cyr, S. Hospital Cost of Staphylococcal Infection after Cardiothoracic or Orthopedic Operations in France: A Retrospective Database Analysis. Surg. Infect. 2015, 16, 428–435. [Google Scholar] [CrossRef]
- Turner, N.A.; Sharma-Kuinkel, B.K.; Maskarinec, S.A.; Eichenberger, E.M.; Shah, P.P.; Carugati, M.; Holland, T.L.; Fowler, V.G. Methicillin-resistant Staphylococcus aureus: An overview of basic and clinical research. Nat. Rev. Microbiol. 2019, 17, 203–218. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.M.; Price, L.B.; Hungate, B.A.; Abraham, A.G.; Larsen, L.A.; Christensen, K.; Stegger, M.; Skov, R.; Andersen, P.S. Staphylococcus aureus and the ecology of the nasal microbiome. Sci. Adv. 2015, 1. [Google Scholar] [CrossRef]
- Renz, A.; Dräger, A. Curating and Comparing 114 Strain-Specific Genome-Scale Metabolic Models of Staphylococcus aureus. Preprints 2021, 2021040244. [Google Scholar] [CrossRef]
- Jansma, J.; El Aidy, S. Understanding the host-microbe interactions using metabolic modeling. Microbiome 2021, 9, 16. [Google Scholar] [CrossRef]
- Bauer, E.; Thiele, I. From Network Analysis to Functional Metabolic Modeling of the Human Gut Microbiota. mSystems 2018, 3. [Google Scholar] [CrossRef]
- Diener, C.; Gibbons, S.M.; Resendis-Antonio, O. MICOM: Metagenome-Scale Modeling To Infer Metabolic Interactions in the Gut Microbiota. mSystems 2020, 5. [Google Scholar] [CrossRef]
- Zomorrodi, A.R.; Maranas, C.D. OptCom: A Multi-Level Optimization Framework for the Metabolic Modeling and Analysis of Microbial Communities. PLoS Comput. Biol. 2012, 8, e1002363. [Google Scholar] [CrossRef]
- Bauer, E.; Zimmermann, J.; Baldini, F.; Thiele, I.; Kaleta, C. BacArena: Individual-based metabolic modeling of heterogeneous microbes in complex communities. PLoS Comput. Biol. 2017, 13, e1005544. [Google Scholar] [CrossRef]
- Carey, M.A.; Dräger, A.; Beber, M.E.; Papin, J.A.; Yurkovich, J.T. Community standards to facilitate development and address challenges in metabolic modeling. Mol. Syst. Biol. 2020, 16, e9235. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Sato, Y.; Furumichi, M.; Morishima, K.; Tanabe, M. New approach for understanding genome variations in KEGG. Nucleic Acids Res. 2019, 47, D590–D595. [Google Scholar] [CrossRef]
- Lieven, C.; Beber, M.E.; Olivier, B.G.; Bergmann, F.T.; Ataman, M.; Babaei, P.; Bartell, J.A.; Blank, L.M.; Chauhan, S.; Correia, K.; et al. MEMOTE for standardized genome-scale metabolic model testing. Nat. Biotechnol. 2020, 38, 272–276. [Google Scholar] [CrossRef]
- Monk, J.M.; Lloyd, C.J.; Brunk, E.; Mih, N.; Sastry, A.; King, Z.; Takeuchi, R.; Nomura, W.; Zhang, Z.; Mori, H.; et al. iML1515, a knowledgebase that computes Escherichia coli traits. Nat. Biotechnol. 2017, 35, 904–908. [Google Scholar] [CrossRef] [PubMed]
- Edwards, J.S.; Palsson, B.O. The Escherichia coli MG1655 in silico metabolic genotype: Its definition, characteristics, and capabilities. Proc. Natl. Acad. Sci. USA 2000, 97, 5528–5533. [Google Scholar] [CrossRef]
- Machado, D.; Andrejev, S.; Tramontano, M.; Patil, K.R. Fast automated reconstruction of genome-scale metabolic models for microbial species and communities. Nucleic Acids Res. 2018, 46, 7542–7553. [Google Scholar] [CrossRef]
- Seaver, S.M.D.; Liu, F.; Zhang, Q.; Jeffryes, J.; Faria, J.P.; Edirisinghe, J.N.; Mundy, M.; Chia, N.; Noor, E.; Beber, M.E.; et al. The ModelSEED Biochemistry Database for the integration of metabolic annotations and the reconstruction, comparison and analysis of metabolic models for plants, fungi and microbes. Nucleic Acids Res. 2021, 49, D575–D588. [Google Scholar] [CrossRef]
- Zimmermann, J.; Kaleta, C.; Waschina, S. Gapseq: Informed prediction of bacterial metabolic pathways and reconstruction of accurate metabolic models. Genome Biol. 2020. [Google Scholar] [CrossRef]
- Arkin, A.P.; Cottingham, R.W.; Henry, C.S.; Harris, N.L.; Stevens, R.L.; Maslov, S.; Dehal, P.; Ware, D.; Perez, F.; Canon, S.; et al. KBase: The United States department of energy systems biology knowledgebase. Nat. Biotechnol. 2018, 36, 566–569. [Google Scholar] [CrossRef]
- Norsigian, C.J.; Pusarla, N.; McConn, J.L.; Yurkovich, J.T.; Dräger, A.; Palsson, B.O.; King, Z. BiGG Models 2020: Multi-strain genome-scale models and expansion across the phylogenetic tree. Nucleic Acids Res. 2019. [Google Scholar] [CrossRef]
- Römer, M.; Eichner, J.; Dräger, A.; Wrzodek, C.; Wrzodek, F.; Zell, A. ZBIT Bioinformatics Toolbox: A Web-Platform for Systems Biology and Expression Data Analysis. PLoS ONE 2016, 11, e0149263. [Google Scholar] [CrossRef] [PubMed]
- Fritzemeier, C.J.; Hartleb, D.; Szappanos, B.; Papp, B.; Lercher, M.J. Erroneous energy-generating cycles in published genome scale metabolic networks: Identification and removal. PLoS Comput. Biol. 2017, 13, e1005494. [Google Scholar] [CrossRef] [PubMed]
- Moretti, S.; Tran, V.D.T.; Mehl, F.; Ibberson, M.; Pagni, M. MetaNetX/MNXref: Unified namespace for metabolites and biochemical reactions in the context of metabolic models. Nucleic Acids Res. 2021, 49, D570–D574. [Google Scholar] [CrossRef]
- Bateman, A.; Martin, M.J.; O’Donovan, C.; Magrane, M.; Apweiler, R.; Alpi, E.; Antunes, R.; Arganiska, J.; Bely, B.; Bingley, M.; et al. UniProt: A hub for protein information. Nucleic Acids Res. 2015, 43, D204–D212. [Google Scholar] [CrossRef]
- Morgat, A.; Lombardot, T.; Axelsen, K.B.; Aimo, L.; Niknejad, A.; Hyka-Nouspikel, N.; Coudert, E.; Pozzato, M.; Pagni, M.; Moretti, S.; et al. Updates in Rhea – an expert curated resource of biochemical reactions. Nucleic Acids Res. 2017, 45, D415–D418. [Google Scholar] [CrossRef] [PubMed]
- Karp, P.D.; Billington, R.; Caspi, R.; Fulcher, C.A.; Latendresse, M.; Kothari, A.; Keseler, I.M.; Krummenacker, M.; Midford, P.E.; Ong, Q.; et al. The BioCyc collection of microbial genomes and metabolic pathways. Brief. Bioinform. 2019, 20, 1085–1093. [Google Scholar] [CrossRef]
- Wishart, D.S.; Feunang, Y.D.; Marcu, A.; Guo, A.C.; Liang, K.; Vázquez-Fresno, R.; Sajed, T.; Johnson, D.; Li, C.; Karu, N.; et al. HMDB 4.0: The human metabolome database for 2018. Nucleic Acids Res. 2018, 46, D608–D617. [Google Scholar] [CrossRef] [PubMed]
- Sud, M.; Fahy, E.; Cotter, D.; Brown, A.; Dennis, E.A.; Glass, C.K.; Merrill, A.H.; Murphy, R.C.; Raetz, C.R.; Russell, D.W.; et al. LMSD: LIPID MAPS structure database. Nucleic Acids Res. 2007, 35. [Google Scholar] [CrossRef]
- Geer, L.Y.; Marchler-Bauer, A.; Geer, R.C.; Han, L.; He, J.; He, S.; Liu, C.; Shi, W.; Bryant, S.H. The NCBI BioSystems database. Nucleic Acids Res. 2010, 38, D492–D496. [Google Scholar] [CrossRef] [PubMed]
- Courtot, M.; Juty, N.; Knüpfer, C.; Waltemath, D.; Zhukova, A.; Dräger, A.; Dumontier, M.; Finney, A.; Golebiewski, M.; Hastings, J.; et al. Controlled vocabularies and semantics in systems biology. Mol. Syst. Biol. 2011, 7, 543. [Google Scholar] [CrossRef] [PubMed]
- Giglio, M.; Tauber, R.; Nadendla, S.; Munro, J.; Olley, D.; Ball, S.; Mitraka, E.; Schriml, L.M.; Gaudet, P.; Hobbs, E.T.; et al. ECO, the Evidence & Conclusion Ontology: Community standard for evidence information. Nucleic Acids Res. 2019, 47, D1186–D1194. [Google Scholar] [CrossRef]
- Fritze, E. Automating the Assignment of SBO-Terms. Bachelor’s Thesis, University of Tübingen, Tübingen, Germany, 2020. [Google Scholar]
- Lachance, J.C.; Lloyd, C.J.; Monk, J.M.; Yang, L.; Sastry, A.V.; Seif, Y.; Palsson, B.O.; Rodrigue, S.; Feist, A.M.; King, Z.A.; et al. BOFdat: Generating biomass objective functions for genome-scale metabolic models from experimental data. PLoS Comput. Biol. 2019, 15, e1006971. [Google Scholar] [CrossRef]
- Keating, S.M.; Waltemath, D.; König, M.; Zhang, F.; Dräger, A.; Chaouiya, C.; Bergmann, F.T.; Finney, A.; Gillespie, C.S.; Helikar, T.; et al. SBML Level 3: An extensible format for the exchange and reuse of biological models. Mol. Syst. Biol. 2020, 16, e9110. [Google Scholar] [CrossRef]
- Thiele, I.; Palsson, B. A protocol for generating a high-quality genome-scale metabolic reconstruction. Nat. Protoc. 2010, 5, 93–121. [Google Scholar] [CrossRef]
- Ebrahim, A.; Lerman, J.A.; Palsson, B.O.; Hyduke, D.R. COBRApy: COnstraints-Based Reconstruction and Analysis for Python. BMC Syst. Biol. 2013, 7, 74. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- King, Z.A.; Dräger, A.; Ebrahim, A.; Sonnenschein, N.; Lewis, N.E.; Palsson, B.O. Escher: A web application for building, sharing, and embedding data-rich visualizations of biological pathways. PLoS Comput. Biol. 2015, 11, e1004321. [Google Scholar] [CrossRef]
- Varki, A.; Schauer, R. Chapter 14. Sialic acids. In Essentials of Glycobiology, 2nd ed.; Varki, A., Cummings, R.D., Esko, J.D., Freeze, H.H., Stanley, P., Bertozzi, C.R., Hart, G.W., Etzle, M.E., Eds.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2009; Chapter 14. [Google Scholar]
- Krismer, B.; Liebeke, M.; Janek, D.; Nega, M.; Rautenberg, M.; Hornig, G.; Unger, C.; Weidenmaier, C.; Lalk, M.; Peschel, A. Nutrient limitation governs Staphylococcus aureus metabolism and niche adaptation in the human nose. PLoS Pathog. 2014, 10, e1003862. [Google Scholar] [CrossRef] [PubMed]
- Palmer, K.L.; Aye, L.M.; Whiteley, M. Nutritional cues control Pseudomonas aeruginosa multicellular behavior in cystic fibrosis sputum. J. Bacteriol. 2007, 189, 8079–8087. [Google Scholar] [CrossRef] [PubMed]
- Swainston, N.; Smallbone, K.; Hefzi, H.; Dobson, P.D.; Brewer, J.; Hanscho, M.; Zielinski, D.C.; Ang, K.S.; Gardiner, N.J.; Gutierrez, J.M.; et al. Recon 2.2: From reconstruction to model of human metabolism. Metabolomics 2016, 12, 109. [Google Scholar] [CrossRef] [PubMed]
- Bernardes, J.P.; Mishra, N.; Tran, F.; Bahmer, T.; Best, L.; Blase, J.I.; Bordoni, D.; Franzenburg, J.; Geisen, U.; Josephs-Spaulding, J.; et al. Longitudinal Multi-omics Analyses Identify Responses of Megakaryocytes, Erythroid Cells, and Plasmablasts as Hallmarks of Severe COVID-19. Immunity 2020, 53, 1296–1314.e9. [Google Scholar] [CrossRef] [PubMed]
- Elmadfa, I. Österreichischer Ernährungsbericht 2012; Institut für Ernährungswissenschaften Universität Wien im Auftrag des Bundesministeriums für Gesundheit: Wien, Austria, 2012; p. 424. [Google Scholar]
- Magnúsdóttir, S.; Heinken, A.; Kutt, L.; Ravcheev, D.A.; Bauer, E.; Noronha, A.; Greenhalgh, K.; Jäger, C.; Baginska, J.; Wilmes, P.; et al. Generation of genome-scale metabolic reconstructions for 773 members of the human gut microbiota. Nat. Biotechnol. 2016, 35, 81–89. [Google Scholar] [CrossRef]
- Lee, H.H.; Ostrov, N.; Wong, B.G.; Gold, M.A.; Khalil, A.S.; Church, G.M. Functional genomics of the rapidly replicating bacterium Vibrio natriegens by CRISPRi. Nat. Microbiol. 2019, 4, 1105–1113. [Google Scholar] [CrossRef]
- Krismer, B.; Weidenmaier, C.; Zipperer, A.; Peschel, A. The commensal lifestyle of Staphylococcus aureus and its interactions with the nasal microbiota. Nat. Rev. Microbiol. 2017, 15, 675–687. [Google Scholar] [CrossRef]
- Brugger, S.D.; Eslami, S.M.; Pettigrew, M.M.; Escapa, I.F.; Henke, M.M.; Kong, Y.; Lemon, K.P. Dolosigranulum pigrum cooperation and competition in human nasal microbiota. bioRxiv 2019, 678698. [Google Scholar] [CrossRef]
- Juty, N.; Le Novère, N.; Laibe, C. Identifiers.org and MIRIAM Registry: Community resources to provide persistent identification. Nucleic Acids Res. 2012, 40, D580–D586. [Google Scholar] [CrossRef]
- Kelly, D.J.; Hughes, N.J. The Citric Acid Cycle and Fatty Acid Biosynthesis. In Helicobacter Pylori; ASM Press: Washington, DC, USA, 2014; pp. 135–146. [Google Scholar] [CrossRef]
- Huynen, M.A.; Dandekar, T.; Bork, P. Variation and evolution of the citric-acid cycle: A genomic perspective. Trends Microbiol. 1999, 7, 281–291. [Google Scholar] [CrossRef]
- Cordwell, S.J. Microbial genomes and ’missing’ enzymes: Redefining biochemical pathways. Arch. Microbiol. 1999, 172, 269–279. [Google Scholar] [CrossRef]
- Tabor, C.W.; Tabor, H. Polyamines in microorganisms. Microbiol. Rev. 1985, 49, 81–99. [Google Scholar] [CrossRef]
- Du, Q.; Wang, H.; Xie, J. Thiamin (vitamin B1) biosynthesis and regulation: A rich source of antimicrobial drug targets? Int. J. Biol. Sci. 2011, 7, 41–52. [Google Scholar] [CrossRef]
- Green, J.M.; Nichols, B.P. p-Aminobenzoate biosynthesis in Escherichia coli: Purification of aminodeoxychorismate lyase and cloning of pabC. J. Biol. Chem. 1991, 266, 12971–12975. [Google Scholar] [CrossRef]
- Rodionov, D.A.; Hebbeln, P.; Eudes, A.; Ter Beek, J.; Rodionova, I.A.; Erkens, G.B.; Slotboom, D.J.; Gelfand, M.S.; Osterman, A.L.; Hanson, A.D.; et al. A novel class of modular transporters for vitamins in prokaryotes. J. Bacteriol. 2009, 91, 42–51. [Google Scholar] [CrossRef] [PubMed]
- Sokolovskaya, O.M.; Shelton, A.N.; Taga, M.E. Sharing vitamins: Cobamides unveil microbial interactions. Science 2020, 369. [Google Scholar] [CrossRef]
- Kitts, P.A.; Church, D.M.; Oise Thibaud-Nissen, F.; Choi, J.; Hem, V.; Sapojnikov, V.; Smith, R.G.; Tatusova, T.; Xiang, C.; Zherikov, A.; et al. Assembly: A resource for assembled genomes at NCBI. Nucleic Acids Res. 2015, 44, 73–80. [Google Scholar] [CrossRef]
- Hucka, M.; Bergmann, F.T.; Dräger, A.; Hoops, S.; Keating, S.M.; Le Novère, N.; Myers, C.J.; Olivier, B.G.; Sahle, S.; Schaff, J.C.; et al. Systems Biology Markup Language (SBML) Level 3 Version 1 Core. J. Integr. Bioinform. 2018, 15, 1. [Google Scholar] [CrossRef]
- Olivier, B.G.; Bergmann, F.T. SBML Level 3 Package: Flux Balance Constraints version 2. J. Integr. Bioinform. 2018, 15, 20170082. [Google Scholar] [CrossRef] [PubMed]
- Bornstein, B.J.; Keating, S.M.; Jouraku, A.; Hucka, M. LibSBML: An API Library for SBML. Bioinformatics 2008, 24, 880–881. [Google Scholar] [CrossRef]
- Benson, D.A.; Karsch-Mizrachi, I.; Lipman, D.J.; Ostell, J.; Sayers, E.W. GenBank. Nucleic Acids Res. 2011, 39, D32–D37. [Google Scholar] [CrossRef]
- Orth, J.D.; Conrad, T.M.; Na, J.; Lerman, J.A.; Nam, H.; Feist, A.M.; Palsson, B. A comprehensive genome-scale reconstruction of Escherichia coli metabolism-2011. Mol. Syst. Biol. 2011, 7, 535. [Google Scholar] [CrossRef] [PubMed]
- Xavier, J.C.; Patil, K.R.; Rocha, I. Integration of Biomass Formulations of Genome-Scale Metabolic Models with Experimental Data Reveals Universally Essential Cofactors in Prokaryotes. Metab. Eng. 2017, 39, 200–208. [Google Scholar] [CrossRef]
- Hucka, M.; Smith, L.P. SBML Level 3 package: Groups, Version 1 Release 1. J. Integr. Bioinform. 2016, 13, 1. [Google Scholar] [CrossRef]
- Renz, A.; Mostolizadeh, R.; Dräger, A. Clinical Applications of Metabolic Models in SBML Format. In Systems Medicine; Wolkenhauer, O., Ed.; Academic Press: Oxford, UK, 2020; Volume 3, pp. 362–371. [Google Scholar] [CrossRef]
- Kluyver, T.; Ragan-Kelley, B.; Pérez, F.; Granger, B.; Bussonnier, M.; Frederic, J.; Kelley, K.; Hamrick, J.; Grout, J.; Corlay, S.; et al. Jupyter Notebooks-a publishing format for reproducible computational workflows. In Positioning and Power in Academic Publishing: Players, Agents and Agendas; Loizides, F., Scmidt, B., Eds.; IOS Press: Amsterdam, The Netherlands, 2016; pp. 87–90. [Google Scholar]
- Malik-Sheriff, R.S.; Glont, M.; Nguyen, T.V.N.; Tiwari, K.; Roberts, M.G.; Xavier, A.; Vu, M.T.; Men, J.; Maire, M.; Kananathan, S.; et al. BioModels—15 years of sharing computational models in life science. Nucleic Acids Res. 2020, 48, D407–D415. [Google Scholar] [CrossRef]
- Bergmann, F.T.; Adams, R.; Moodie, S.; Cooper, J.; Glont, M.; Golebiewski, M.; Hucka, M.; Laibe, C.; Miller, A.K.; Nickerson, D.P.; et al. COMBINE archive and OMEX format: One file to share all information to reproduce a modeling project. BMC Bioinform. 2014, 15, 369. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Methionine | Arginine | Glutamine | Putrescine | Spermidine | Biotin | Niacin | |
|---|---|---|---|---|---|---|---|
| Auxotrophy | ✔ | ✔ | ✔ | ✔ | ✔ | ✔ | |
| Biosynthesis | ✔ | ||||||
| Reported reactions | ✔ | ✔ | ✔ | ||||
| Transporter | ✔ | ✔ | ✔ | ✔ | ✔ |
| Amino Acids | Trace Minerals | Other Molecules |
|---|---|---|
| l-leucine | Cl- (chloride) | d-glucose |
| l-threonine | K+ (potassium) | 4-aminobenzoate |
| l-arginine | Ca2+ (calcium) | riboflavin |
| l-lysine | Mg2+ (magnesium) | thiamine |
| l-proline | Mn2+ (manganese) | niacin |
| l-glutamate | Co2+ (cobalt) | meso-2,6-diaminoheptanedioate |
| l-histidine | Zn2+ (zinc) | O2 (oxygen) |
| l-isoleucine | Cu2+ (copper) | |
| l-methionine | Fe2+ (iron II) | |
| l-tryptophane | Na+ (sodium) | |
| l-valine | Ni2+ (nickel) | |
| l-cysteine | SO42- (sulfate) | |
| l-phenylalanine | HPO42- (phosphate) |
| ECO Term | Term Name | Assignment |
|---|---|---|
| ECO:0000001 | inference from background scientific knowledge | no GPR |
| ECO:0000251 | similarity evidence used in automatic assertion | GPR but no hit in UniProt |
| ECO:0000363 | computational inference used in automatic assertion | UniProt: ‘Predicted’ |
| ECO:0000044 | sequence similarity evidence | UniProt: ‘Inferred from homology’ |
| ECO:0000009 | transcript expression evidence | UniProt: ‘Evidence at transcript level’ |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Renz, A.; Widerspick, L.; Dräger, A. First Genome-Scale Metabolic Model of Dolosigranulum pigrum Confirms Multiple Auxotrophies. Metabolites 2021, 11, 232. https://doi.org/10.3390/metabo11040232
Renz A, Widerspick L, Dräger A. First Genome-Scale Metabolic Model of Dolosigranulum pigrum Confirms Multiple Auxotrophies. Metabolites. 2021; 11(4):232. https://doi.org/10.3390/metabo11040232
Chicago/Turabian StyleRenz, Alina, Lina Widerspick, and Andreas Dräger. 2021. "First Genome-Scale Metabolic Model of Dolosigranulum pigrum Confirms Multiple Auxotrophies" Metabolites 11, no. 4: 232. https://doi.org/10.3390/metabo11040232
APA StyleRenz, A., Widerspick, L., & Dräger, A. (2021). First Genome-Scale Metabolic Model of Dolosigranulum pigrum Confirms Multiple Auxotrophies. Metabolites, 11(4), 232. https://doi.org/10.3390/metabo11040232