Metabotypes of Pseudomonas aeruginosa Correlate with Antibiotic Resistance, Virulence and Clinical Outcome in Cystic Fibrosis Chronic Infections

 , ,
, ,  , ,
, ,  ,
,  , ,
, ,  , ,
, ,
Abstract
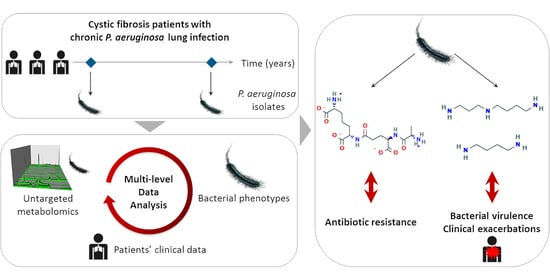
1. Introduction
2. Results
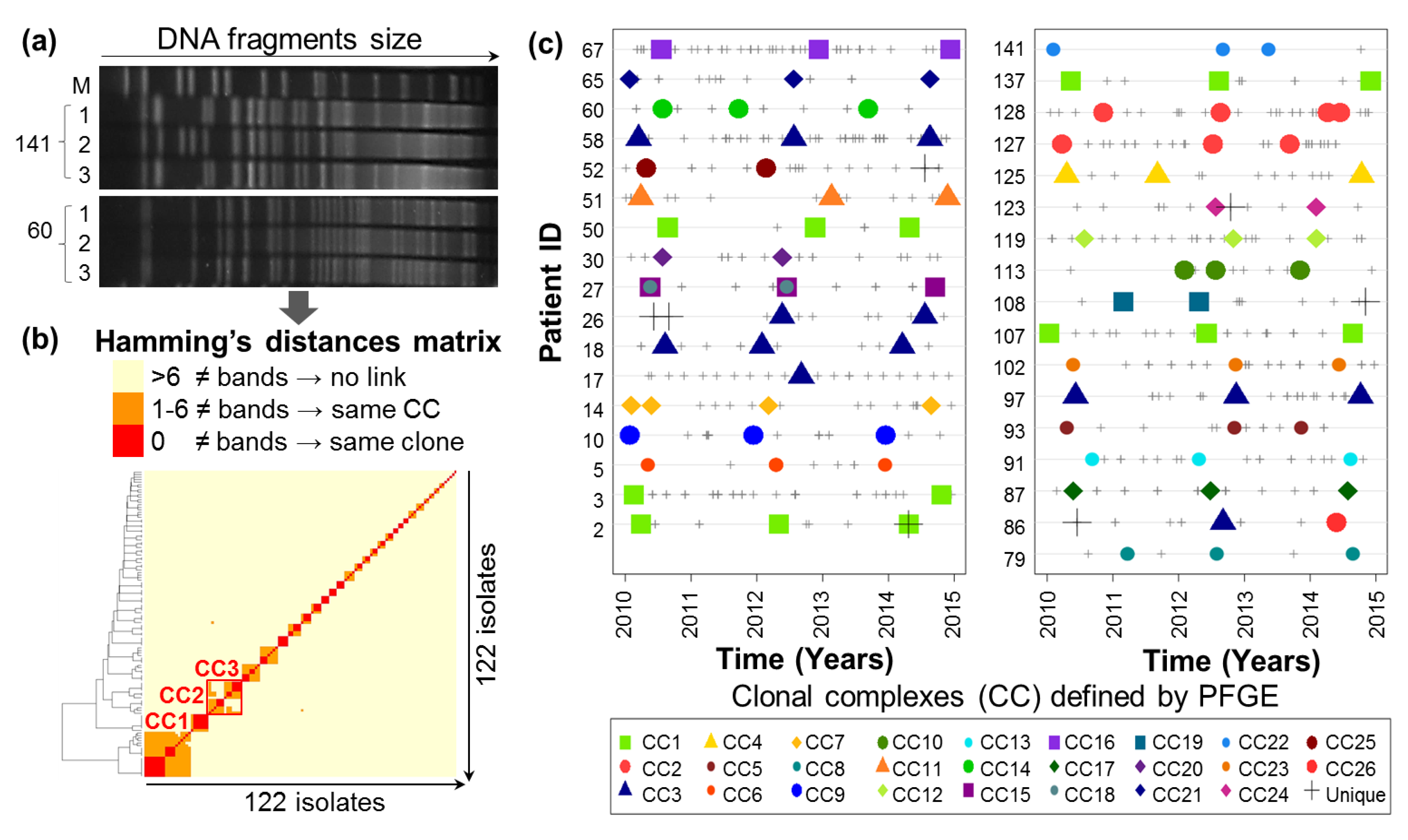
2.1. Evolutionary Relationships of P.a Clinical Isolates
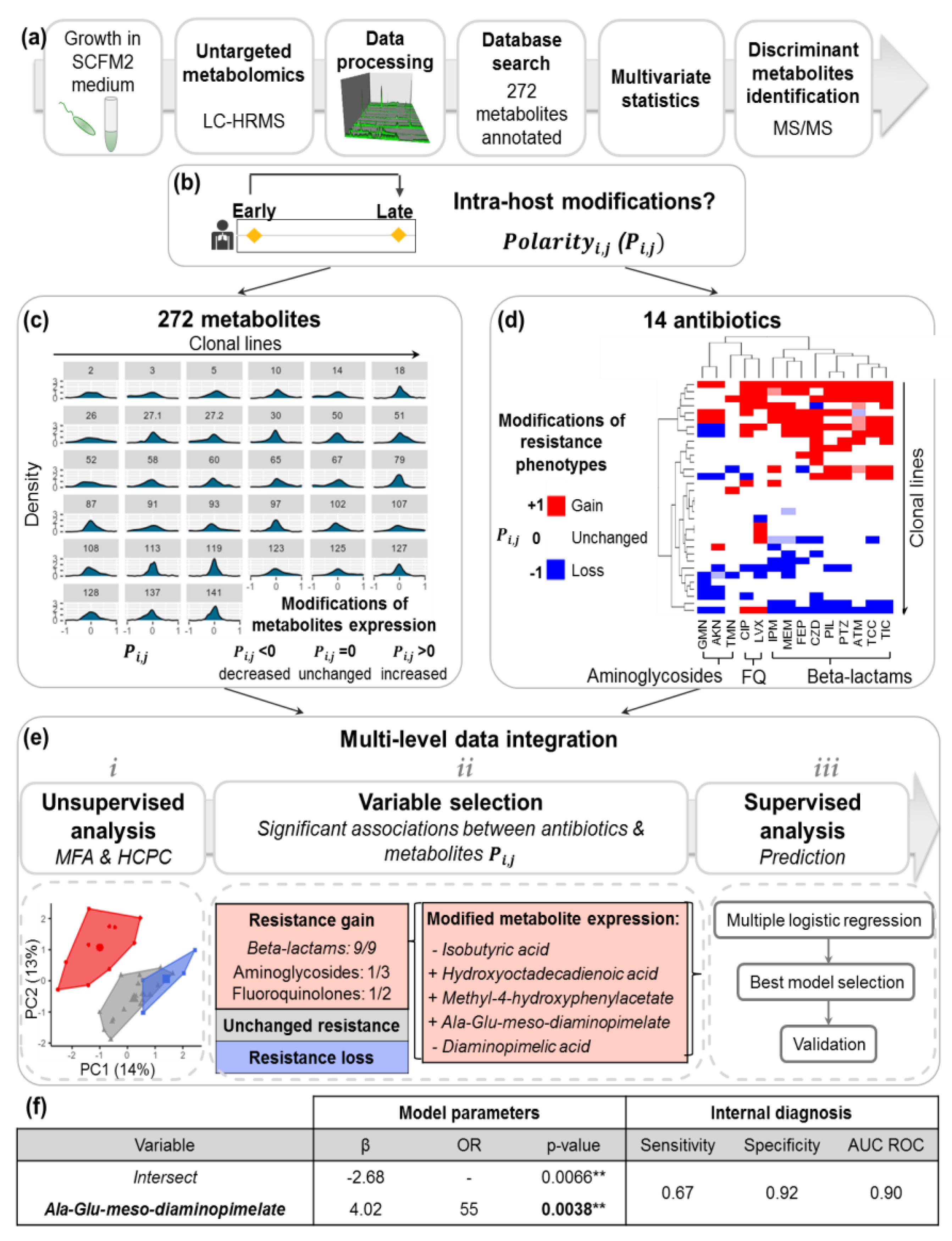
2.2. Acquisition of P.a Metabolomic Profiles by Untargeted LC-HRMS
2.3. Diversity of P.a Metabolic Evolution within CF Patients’ Lungs
2.4. Intra-Host Metabolic Adaptation Is Associated with the Acquisition of Antibiotic Resistance
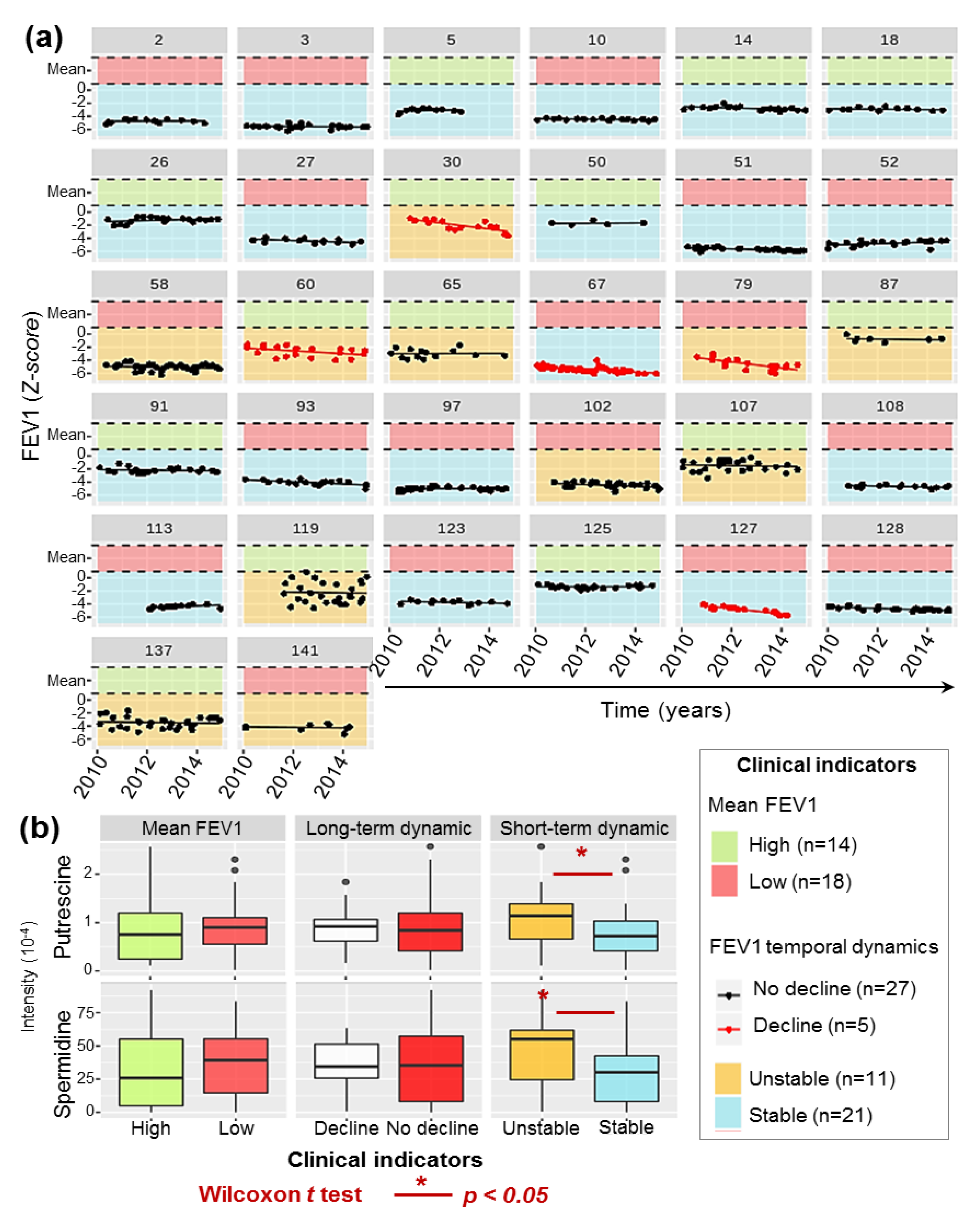
2.5. P.a Metabotypes Segregated by Differential Levels of Polyamines and Their Metabolites
2.6. Multivariate-Based Analysis of Bacterial Virulence
2.7. Polyamines Production Is Associated with the Level of P.a Virulence
2.8. High Polyamines Production By P.a Is Associated with Frequent Clinical Exacerbations
3. Discussion
4. Materials and Methods
4.1. Patients
4.1.1. Cohort Selection of Patients
4.1.2. Clinical Data and Respiratory Function Modelling
4.2. P.a Clinical Isolates
4.2.1. P.a Isolates Identification
4.2.2. Growth Conditions
4.2.3. Pulsed-Field Gel Electrophoresis Clonal Analysis
4.3. Metabolomics Analysis
4.3.1. Sample Preparation
4.3.2. Liquid Chromatography Coupled with High Resolution Mass Spectrometry (LC-HRMS) Analysis
4.3.3. LC-HRMS Data Processing
4.3.4. Metabolite Annotation
4.4. Phenotypic Assays
4.5. Polarity Degreei,j
4.6. Statistical Analyses
4.6.1. Multiscale Integration of within-Host Adaptation of Antibiotic Resistance and Metabolomics Profiles
4.6.2. Definition of Bacterial Metabotypes
4.6.3. Definition of Bacterial Level of Virulence
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Riordan, J.R.; Rommens, J.M.; Kerem, B.S.; Alon, N.O.A.; Rozmahel, R.; Grzelczak, Z.; Zielenski, J.; Lok, S.I.; Plavsic, N.; Chou, J.L.; et al. Identification of the Cystic Fibrosis Gene: Cloning and Characterization of Complementary DNA. Science 1989, 245, 1066–1073. [Google Scholar] [CrossRef] [PubMed]
- Rommens, J.M.; Iannuzzi, M.C.; Kerem, B.S.; Drumm, M.L.; Melmer, G.; Dean, M.; Rozmahel, R.; Cole, J.L.; Kennedy, D.; Hidaka, N.; et al. Identification of the Cystic Fibrosis Gene: Chromosome Walking and Jumping. Science 1989, 245, 1059–1065. [Google Scholar] [CrossRef]
- Kerem, E.; Webb, A.K. European Cystic Fibrosis Society Standards of Care: A Road Map to Improve CF Outcome. J. Cyst. Fibros. 2014, 13, 357–358. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Marshall, B.C.; Elbert, A.; Petren, K.; Rizvi, S.; Fink, A.; Ostrenga, J.; Sewall, A. Cystic Fibrosis Foundation Patient Registry 2014 Annual Data Report; Cystic Fibrosis Foundation: Bethesda, MD, USA, 2015. [Google Scholar]
- Folkesson, A.; Jelsbak, L.; Yang, L.; Johansen, H.K.; Ciofu, O.; Hoiby, N.; Molin, S. Adaptation of Pseudomonas Aeruginosa to the Cystic Fibrosis Airway: An Evolutionary Perspective. Nat. Rev. Microbiol. 2012, 10, 841–851. [Google Scholar] [CrossRef] [PubMed]
- Sousa, A.M.; Pereira, M.O. Pseudomonas Aeruginosa Diversification during Infection Development in Cystic Fibrosis Lungs—A Review. Pathogens 2014, 3, 680–703. [Google Scholar] [CrossRef] [PubMed]
- Salsgiver, E.L.; Fink, A.K.; Knapp, E.A.; LiPuma, J.J.; Olivier, K.N.; Marshall, B.C.; Saiman, L. Changing Epidemiology of the Respiratory Bacteriology of Patients With Cystic Fibrosis. Chest 2016, 149, 390–400. [Google Scholar] [CrossRef]
- Aaron, S.D.; Stephenson, A.L.; Cameron, D.W.; Whitmore, G.A. A Statistical Model to Predict One-Year Risk of Death in Patients with Cystic Fibrosis. J. Clin. Epidemiol. 2015, 68, 1336–1345. [Google Scholar] [CrossRef] [PubMed]
- Konstan, M.W.; Morgan, W.J.; Butler, S.M.; Pasta, D.J.; Craib, M.L.; Silva, S.J.; Stokes, D.C.; Wohl, M.E.B.; Wagener, J.S.; Regelmann, W.E.; et al. Risk Factors for Rate of Decline in Forced Expiratory Volume in One Second in Children and Adolescents with Cystic Fibrosis. J. Pediatr. 2007, 151. [Google Scholar] [CrossRef]
- Vandevanter, D.R.; Wagener, J.S.; Pasta, D.J.; Elkin, E.; Jacobs, J.R.; Morgan, W.J.; Konstan, M.W. Pulmonary Outcome Prediction (POP) Tools for Cystic Fibrosis Patients. Pediatr. Pulmonol. 2010, 45, 1156–1166. [Google Scholar] [CrossRef][Green Version]
- Bhagirath, A.Y.; Li, Y.; Somayajula, D.; Dadashi, M.; Badr, S.; Duan, K. Cystic Fibrosis Lung Environment and Pseudomonas Aeruginosa Infection. BMC Pulm. Med. 2016, 16. [Google Scholar] [CrossRef]
- Emerson, J.; Rosenfeld, M.; McNamara, S.; Ramsey, B.; Gibson, R.L. Pseudomonas Aeruginosa and Other Predictors of Mortality and Morbidity in Young Children with Cystic Fibrosis. Pediatr. Pulmonol. 2002, 34, 91–100. [Google Scholar] [CrossRef] [PubMed]
- Malhotra, S.; Hayes, D.; Wozniak, D.J. Cystic Fibrosis and Pseudomonas Aeruginosa: The Host-Microbe Interface. Clin. Microbiol. Rev. 2019, 32. [Google Scholar] [CrossRef] [PubMed]
- Hogardt, M.; Heesemann, J. Adaptation of Pseudomonas Aeruginosa during Persistence in the Cystic Fibrosis Lung. Int. J. Med. Microbiol. 2010, 300, 557–562. [Google Scholar] [CrossRef] [PubMed]
- Smith, W.D.; Bardin, E.; Cameron, L.; Edmondson, C.L.; Farrant, K.V.; Martin, I.; Murphy, R.A.; Soren, O.; Turnbull, A.R.; Wierre-Gore, N.; et al. Current and Future Therapies for Pseudomonas Aeruginosa Infection in Patients with Cystic Fibrosis. FEMS Microbiol. Lett. 2017, 364. [Google Scholar] [CrossRef] [PubMed]
- Hauser, A.R.; Jain, M.; Bar-Meir, M.; McColley, S.A. Clinical Significance of Microbial Infection and Adaptation in Cystic Fibrosis. Clin. Microbiol. Rev. 2011, 24, 29–70. [Google Scholar] [CrossRef]
- Faure, E.; Kwong, K.; Nguyen, D. Pseudomonas Aeruginosa in Chronic Lung Infections: How to Adapt Within the Host? Front. Immunol. 2018, 9, 2416. [Google Scholar] [CrossRef]
- Behrends, V.; Ryall, B.; Zlosnik, J.E.; Speert, D.P.; Bundy, J.G.; Williams, H.D. Metabolic Adaptations of Pseudomonas Aeruginosa during Cystic Fibrosis Chronic Lung Infections. Environ. Microbiol. 2013, 15, 398–408. [Google Scholar] [CrossRef]
- Quinn, R.A.; Adem, S.; Mills, R.H.; Comstock, W.; Deright Goldasich, L.; Humphrey, G.; Aksenov, A.A.; Melnik, A.V.; Da Silva, R.; Ackermann, G.; et al. Neutrophilic Proteolysis in the Cystic Fibrosis Lung Correlates with a Pathogenic Microbiome. Microbiome 2019, 7. [Google Scholar] [CrossRef]
- Marvig, R.L.; Sommer, L.M.; Molin, S.; Johansen, H.K. Convergent Evolution and Adaptation of Pseudomonas Aeruginosa within Patients with Cystic Fibrosis. Nat. Genet. 2015, 47, 57–64. [Google Scholar] [CrossRef]
- Smith, E.E.; Buckley, D.G.; Wu, Z.; Saenphimmachak, C.; Hoffman, L.R.; D’Argenio, D.A.; Miller, S.I.; Ramsey, B.W.; Speert, D.P.; Moskowitz, S.M.; et al. Genetic Adaptation by Pseudomonas Aeruginosa to the Airways of Cystic Fibrosis Patients. Proc. Natl. Acad. Sci. USA 2006, 103, 8487–8492. [Google Scholar] [CrossRef]
- Damkiaer, S.; Yang, L.; Molin, S.; Jelsbak, L. Evolutionary Remodeling of Global Regulatory Networks during Long-Term Bacterial Adaptation to Human Hosts. Proc. Natl. Acad. Sci. USA 2013, 110, 7766–7771. [Google Scholar] [CrossRef] [PubMed]
- Marvig, R.L.; Dolce, D.; Sommer, L.M.; Petersen, B.; Ciofu, O.; Campana, S.; Molin, S.; Taccetti, G.; Johansen, H.K. Within-Host Microevolution of Pseudomonas Aeruginosa in Italian Cystic Fibrosis Patients. BMC Microbiol. 2015, 15, 218. [Google Scholar] [CrossRef] [PubMed]
- La Rosa, R.; Johansen, H.K.; Molin, S. Convergent Metabolic Specialization through Distinct Evolutionary Paths in Pseudomonas Aeruginosa. MBio 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Klockgether, J.; Miethke, N.; Kubesch, P.; Bohn, Y.S.; Brockhausen, I.; Cramer, N.; Eberl, L.; Greipel, J.; Herrmann, C.; Herrmann, S.; et al. Intraclonal Diversity of the Pseudomonas Aeruginosa Cystic Fibrosis Airway Isolates TBCF10839 and TBCF121838: Distinct Signatures of Transcriptome, Proteome, Metabolome, Adherence and Pathogenicity despite an Almost Identical Genome Sequence. Environ. Microbiol. 2013, 15, 191–210. [Google Scholar] [CrossRef]
- Depke, T.; Thöming, J.G.; Kordes, A.; Häussler, S.; Brönstrup, M. Untargeted LC-MS Metabolomics Differentiates Between Virulent and Avirulent Clinical Strains of Pseudomonas Aeruginosa. Biomolecules 2020, 10, 1041. [Google Scholar] [CrossRef]
- Mielko, K.A.; Jabłoński, S.J.; Milczewska, J.; Sands, D.; Łukaszewicz, M.; Młynarz, P. Metabolomic Studies of Pseudomonas Aeruginosa. World J. Microbiol. Biotechnol. 2019, 35, 178. [Google Scholar] [CrossRef]
- Behrends, V.; Bell, T.J.; Liebeke, M.; Cordes-Blauert, A.; Ashraf, S.N.; Nair, C.; Zlosnik, J.E.; Williams, H.D.; Bundy, J.G. Metabolite Profiling to Characterize Disease-Related Bacteria: Gluconate Excretion by Pseudomonas Aeruginosa Mutants and Clinical Isolates from Cystic Fibrosis Patients. J. Biol. Chem. 2013, 288, 15098–15109. [Google Scholar] [CrossRef]
- Borgos, S.E.F.; Skjåstad, R.; Tøndervik, A.; Aas, M.; Aasen, I.M.; Brunsvik, A.; Holten, T.; Iversen, O.J.; Ahlen, C.; Zahlsen, K. Rapid Metabolic Profiling of Developing Pseudomonas Aeruginosa Biofilms by High-Resolution Mass Spectrometry Fingerprinting. Ann. Microbiol. 2015, 65, 891–898. [Google Scholar] [CrossRef]
- Walter, S.; Gudowius, P.; Boßhammer, J.; Römling, U.; Weißbrodt, H.; Schürmann, W.; Von Der Hardt, H.; Tümmler, B. Epidemiology of Chronic Pseudomonas Aeruginosa Infections in the Airways of Lung Transplant Recipients with Cystic Fibrosis. Thorax 1997, 52, 318–321. [Google Scholar] [CrossRef]
- Römling, U.; Greipel, J.; Tümmler, B. Gradient of Genomic Diversity in the Pseudomonas Aeruginosa Chromosome. Mol. Microbiol. 1995, 17, 323–332. [Google Scholar] [CrossRef]
- Turner, K.H.; Wessel, A.K.; Palmer, G.C.; Murray, J.L.; Whiteley, M. Essential Genome of Pseudomonas Aeruginosa in Cystic Fibrosis Sputum. Proc. Natl. Acad. Sci. USA 2015, 112, 4110–4115. [Google Scholar] [CrossRef] [PubMed]
- Aros-Calt, S.; Castelli, F.A.; Lamourette, P.; Gervasi, G.; Junot, C.; Muller, B.H.; Fenaille, F. Metabolomic investigation of Staphylococcus aureus antibiotic susceptibility by liquid chromatography coupled to high-resolution mass spectrometry. In Methods in Molecular Biology; Humana Press Inc.: Totowa, NJ, USA, 2019; Volume 1871, pp. 279–293. [Google Scholar]
- Huang, W.; Brewer, L.K.; Jones, J.W.; Nguyen, A.T.; Marcu, A.; Wishart, D.S.; Oglesby-Sherrouse, A.G.; Kane, M.A.; Wilks, A. PAMDB: A Comprehensive Pseudomonas Aeruginosa Metabolome Database. Nucleic Acids Res. 2018, 46, D575–D580. [Google Scholar] [CrossRef] [PubMed]
- Stalon, V.; Vander Wauven, C.; Momin, P.; Legrain, C. Catabolism of Arginine, Citrulline and Ornithine by Pseudomonas and Related Bacteria. J. Gen. Microbiol. 1987, 133, 2487–2495. [Google Scholar] [CrossRef] [PubMed]
- Harun, S.N.; Hennig, S.; Wainwright, C.; Klein, K. A Systematic Review of Studies Examining the Rate of Lung Function Decline in Patients with Cystic Fibrosis. Paediatr. Respir. Rev. 2016, 20, 55–66. [Google Scholar] [CrossRef] [PubMed]
- Nkam, L.; Lambert, J.; Latouche, A.; Bellis, G.; Burgel, P.R.; Hocine, M.N. A 3-Year Prognostic Score for Adults with Cystic Fibrosis. J. Cyst. Fibros. 2017, 16, 702–708. [Google Scholar] [CrossRef]
- Jacobs, C.; Huang, L.J.; Bartowsky, E.; Normark, S.; Park, J.T. Bacterial Cell Wall Recycling Provides Cytosolic Muropeptides as Effectors for Beta-Lactamase Induction. EMBO J. 1994, 13, 4684–4694. [Google Scholar] [CrossRef]
- Torrens, G.; Pérez-Gallego, M.; Moya, B.; Munar-Bestard, M.; Zamorano, L.; Cabot, G.; Blázquez, J.; Ayala, J.A.; Oliver, A.; Juan, C. Targeting the Permeability Barrier and Peptidoglycan Recycling Pathways to Disarm Pseudomonas Aeruginosa against the Innate Immune System. PLoS ONE 2017, 12, e0181932. [Google Scholar] [CrossRef]
- Zamorano, L.; Reeve, T.M.; Juan, C.; Moyá, B.; Cabot, G.; Vocadlo, D.J.; Mark, B.L.; Oliver, A. AmpG Inactivation Restores Susceptibility of Pan-β-Lactam-Resistant Pseudomonas Aeruginosa Clinical Strains. Antimicrob. Agents Chemother. 2011, 55, 1990–1996. [Google Scholar] [CrossRef]
- Bitonti, A.J.; Kelly, S.E.; McCann, P.P. Regulation of Growth and Macromolecular Synthesis by Putrescine and Spermidine in Pseudomonas Aeruginosa. Life Sci. 1984, 34, 1513–1520. [Google Scholar] [CrossRef]
- Shah, P.; Swiatlo, E. A Multifaceted Role for Polyamines in Bacterial Pathogens. Mol. Microbiol. 2008, 68, 4–16. [Google Scholar] [CrossRef]
- Carriel, D.; Garcia, P.S.; Castelli, F.; Lamourette, P.; Fenaille, F.; Brochier-Armanet, C.; Elsen, S.; Gutsche, I. A Novel Subfamily of Bacterial AAT-Fold Basic Amino Acid Decarboxylases and Functional Characterization of Its First Representative: Pseudomonas Aeruginosa LdcA. Genome Biol. Evol. 2018, 10, 3058–3075. [Google Scholar] [CrossRef] [PubMed]
- Michael, A.J. Polyamine Function in Archaea and Bacteria. J. Biol. Chem. 2018, 293, 18693–18701. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.D.; Itoh, Y.; Nakada, Y.; Jiang, Y. Functional Analysis and Regulation of the Divergent SpuABCDEFGH-SpuI Operons for Polyamine Uptake and Utilization in Pseudomonas Aeruginosa PAO1. J. Bacteriol. 2002, 184, 3765–3773. [Google Scholar] [CrossRef] [PubMed]
- Yeung, A.T.Y.; Bains, M.; Hancock, R.E.W. The Sensor Kinase CbrA Is a Global Regulator That Modulates Metabolism, Virulence, and Antibiotic Resistance in Pseudomonas Aeruginosa. J. Bacteriol. 2011, 193, 918–931. [Google Scholar] [CrossRef]
- Zhou, L.; Wang, J.; Zhang, L.H. Modulation of Bacterial Type III Secretion System by a Spermidine Transporter Dependent Signaling Pathway. PLoS ONE 2007, 2. [Google Scholar] [CrossRef]
- Twomey, K.B.; Alston, M.; An, S.-Q.; O’Connell, O.J.; McCarthy, Y.; Swarbreck, D.; Febrer, M.; Dow, J.M.; Plant, B.J.; Ryan, R.P. Microbiota and Metabolite Profiling Reveal Specific Alterations in Bacterial Community Structure and Environment in the Cystic Fibrosis Airway during Exacerbation. PLoS ONE 2013, 8, e82432. [Google Scholar] [CrossRef]
- Schneider, B.L.; Reitzer, L. Pathway and Enzyme Redundancy in Putrescine Catabolism in Escherichia Coli. J. Bacteriol. 2012, 194, 4080–4088. [Google Scholar] [CrossRef]
- Le Gouëllec, A.; Moyne, O.; Meynet, E.; Toussaint, B.; Fauvelle, F. High-Resolution Magic Angle Spinning NMR-Based Metabolomics Revealing Metabolic Changes in Lung of Mice Infected with P. Aeruginosa Consistent with the Degree of Disease Severity. J. Proteome Res. 2018, 17, 3409–3417. [Google Scholar] [CrossRef]
- Whiteson, K.; Agrawal, S.; Agrawal, A. Differential Responses of Human Dendritic Cells to Metabolites from the Oral/Airway Microbiome. Clin. Exp. Immunol. 2017, 188, 371–379. [Google Scholar] [CrossRef]
- Wang, J.; Wang, J.; Zhang, L.H. Immunological Blocking of Spermidine-Mediated Host–Pathogen Communication Provides Effective Control against Pseudomonas Aeruginosa Infection. Microb. Biotechnol. 2020, 13, 87–96. [Google Scholar] [CrossRef]
- Nguyen, A.T.; Oglesby-Sherrouse, A.G. Interactions between Pseudomonas Aeruginosa and Staphylococcus Aureus during Co-Cultivations and Polymicrobial Infections. Appl. Microbiol. Biotechnol. 2016, 100, 6141–6148. [Google Scholar] [CrossRef] [PubMed]
- Quinn, R.A.; Whiteson, K.; Lim, Y.W.; Salamon, P.; Bailey, B.; Mienardi, S.; Sanchez, S.E.; Blake, D.; Conrad, D.; Rohwer, F. A Winogradsky-Based Culture System Shows an Association between Microbial Fermentation and Cystic Fibrosis Exacerbation. ISME J. 2015, 9, 1024–1038. [Google Scholar] [CrossRef] [PubMed]
- Pressler, T.; Bohmova, C.; Conway, S.; Dumcius, S.; Hjelte, L.; Høiby, N.; Kollberg, H.; Tümmler, B.; Vavrova, V. Chronic Pseudomonas Aeruginosa Infection Definition: EuroCareCF Working Group Report. J. Cyst. Fibros. 2011, 10. [Google Scholar] [CrossRef]
- Quanjer, P.H.; Stanojevic, S.; Cole, T.J.; Baur, X.; Hall, G.L.; Culver, B.H.; Enright, P.L.; Hankinson, J.L.; Ip, M.S.; Zheng, J.; et al. Multi-Ethnic Reference Values for Spirometry for the 3-95-Yr Age Range: The Global Lung Function 2012 Equations. Eur. Respir. J. 2012, 40, 1324–1343. [Google Scholar] [CrossRef] [PubMed]
- Bourlet, T.; Courcol, R.; Hermann, J.-L.; Lachaud, L.; Lamy, B.; Laudat, P.; Pangon, B.; Rémic, H.P.-L. Référentiel en Microbiologie Médicale; Vivactis Plus Ed; SFM: Paris, France, 2007. [Google Scholar]
- Romling, U.; Wingender, J.; Muller, H.; Tummler, B. A Major Pseudomonas Aeruginosa Clone Common to Patients and Aquatic Habitats. Appl. Environ. Microbiol. 1994, 60, 1734–1738. [Google Scholar] [CrossRef] [PubMed]
- Lavenir, R.; Sanroma, M.; Gibert, S.; Crouzet, O.; Laurent, F.; Kravtsoff, J.; Mazoyer, M.A.; Cournoyer, B. Spatio-Temporal Analysis of Infra-Specific Genetic Variations among a Pseudomonas Aeruginosa Water Network Hospital Population: Invasion and Selection of Clonal Complexes. J. Appl. Microbiol. 2008, 105, 1491–1501. [Google Scholar] [CrossRef] [PubMed]
- R core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2013. [Google Scholar]
- Aros-Calt, S.; Muller, B.H.; Boudah, S.; Ducruix, C.; Gervasi, G.; Junot, C.; Fenaille, F. Annotation of the Staphylococcus Aureus Metabolome Using Liquid Chromatography Coupled to High-Resolution Mass Spectrometry and Application to the Study of Methicillin Resistance. J. Proteome Res. 2015, 14, 4863–4875. [Google Scholar] [CrossRef]
- Smith, C.A.; Want, E.J.; O’Maille, G.; Abagyan, R.; Siuzdak, G. XCMS: Processing Mass Spectrometry Data for Metabolite Profiling Using Nonlinear Peak Alignment, Matching, and Identification. Anal. Chem. 2006, 78, 779–787. [Google Scholar] [CrossRef]
- Kuhl, C.; Tautenhahn, R.; Böttcher, C.; Larson, T.R.; Neumann, S. CAMERA: An Integrated Strategy for Compound Spectra Extraction and Annotation of Liquid Chromatography/Mass Spectrometry Data Sets. Anal. Chem. 2012, 84, 283–289. [Google Scholar] [CrossRef]
- Giacomoni, F.; Le Corguillé, G.; Monsoor, M.; Landi, M.; Pericard, P.; Pétéra, M.; Duperier, C.; Tremblay-Franco, M.; Martin, J.F.; Jacob, D.; et al. Workflow4Metabolomics: A Collaborative Research Infrastructure for Computational Metabolomics. Bioinformatics 2015, 31, 1493–1495. [Google Scholar] [CrossRef]
- Guitton, Y.; Tremblay-Franco, M.; Le Corguillé, G.; Martin, J.F.; Pétéra, M.; Roger-Mele, P.; Delabrière, A.; Goulitquer, S.; Monsoor, M.; Duperier, C.; et al. Create, Run, Share, Publish, and Reference Your LC–MS, FIA–MS, GC–MS, and NMR Data Analysis Workflows with the Workflow4Metabolomics 3.0 Galaxy Online Infrastructure for Metabolomics. Int. J. Biochem. Cell Biol. 2017, 93, 89–101. [Google Scholar] [CrossRef]
- Dieterle, F.; Ross, A.; Schlotterbeck, G.; Senn, H. Probabilistic Quotient Normalization as Robust Method to Account for Dilution of Complex Biological Mixtures. Application In1H NMR Metabonomics. Anal. Chem. 2006, 78, 4281–4290. [Google Scholar] [CrossRef] [PubMed]
- Boudah, S.; Olivier, M.F.; Aros-Calt, S.; Oliveira, L.; Fenaille, F.; Tabet, J.C.; Junot, C. Annotation of the Human Serum Metabolome by Coupling Three Liquid Chromatography Methods to High-Resolution Mass Spectrometry. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2014, 966, 34–47. [Google Scholar] [CrossRef] [PubMed]
- Roux, A.; Xu, Y.; Heilier, J.F.; Olivier, M.F.; Ezan, E.; Tabet, J.C.; Junot, C. Annotation of the Human Adult Urinary Metabolome and Metabolite Identification Using Ultra High Performance Liquid Chromatography Coupled to a Linear Quadrupole Ion Trap-Orbitrap Mass Spectrometer. Anal. Chem. 2012, 84, 6429–6437. [Google Scholar] [CrossRef] [PubMed]
- Ogata, H.; Goto, S.; Sato, K.; Fujibuchi, W.; Bono, H.; Kanehisa, M. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 1999, 27, 29–34. [Google Scholar] [CrossRef] [PubMed]
- Wishart, D.S.; Tzur, D.; Knox, C.; Eisner, R.; Guo, A.C.; Young, N.; Cheng, D.; Jewell, K.; Arndt, D.; Sawhney, S.; et al. HMDB: The Human Metabolome Database. Nucleic Acids Res. 2007, 35. [Google Scholar] [CrossRef]
- Smith, C.A.; O’Maille, G.; Want, E.J.; Qin, C.; Trauger, S.A.; Brandon, T.R.; Custodio, D.E.; Abagyan, R.; Siuzdak, G. METLIN: A Metabolite Mass Spectral Database. Ther. Drug Monit. 2005, 27, 747–751. [Google Scholar] [CrossRef]
- Sumner, L.W.; Amberg, A.; Barrett, D.; Beale, M.H.; Beger, R.; Daykin, C.A.; Fan, T.W.M.; Fiehn, O.; Goodacre, R.; Griffin, J.L.; et al. Proposed Minimum Reporting Standards for Chemical Analysis: Chemical Analysis Working Group (CAWG) Metabolomics Standards Initiative (MSI). Metabolomics 2007, 3, 211–221. [Google Scholar] [CrossRef]
- Basso, P.; Wallet, P.; Elsen, S.; Soleilhac, E.; Henry, T.; Faudry, E.; Attree, I. Multiple Pseudomonas Species Secrete Exolysin-like Toxins and Provoke Caspase-1-Dependent Macrophage Death. Environ. Microbiol. 2017, 19, 4045–4064. [Google Scholar] [CrossRef]
- Ngo, T.D.; Plé, S.; Thomas, A.; Barette, C.; Fortuné, A.; Bouzidi, Y.; Fauvarque, M.O.; Pereira De Freitas, R.; Francisco Hilário, F.; Attreé, I.; et al. Chimeric Protein-Protein Interface Inhibitors Allow Efficient Inhibition of Type III Secretion Machinery and Pseudomonas Aeruginosa Virulence. ACS Infect. Dis. 2019, 5, 1843–1854. [Google Scholar] [CrossRef]
- The European Committee on Antimicrobial Susceptibility Testing. Breakpoint Tables for Interpretation of MICs and Zone Diameters.Version 7.1. 2017, pp. 1–20. Available online: http://www.eucast.org (accessed on 15 January 2021).
- Husson, F.; Lê, S.; Pagès, J. Exploratory Multivariate Analysis by Example Using R; CRC Press: Boca Raton, FL, USA, 2010; ISBN 9781439835814. [Google Scholar]
- Lê, S.; Josse, J.; Husson, F. FactoMine R: An R Package for Multivariate Analysis. J. Stat. Softw. 2008, 25, 1–18. [Google Scholar] [CrossRef]
- Kassambara, A.; Mundt, F.; Kassambara, A.; Mundt, F. Factoextra: Extract and Visualize the Results of Multivariate Data Analyses. BugReports. 2017, pp. 1–76. Available online: http//www.sthda.com/english/rpkgs/factoextra (accessed on 15 January 2021).
- Kassambara, A. Practical Guide to Principal Component Methods in R; edition 1, STHDA 2017. Available online: http://www.sthda.com/english/wiki/practical-guide-to-principal-component-methods-in-r (accessed on 15 January 2021).
- MacQueen, J. Some methods for classification and analysis of multivariate observations. In Proceedings of the 5th Berkeley Symposium on Mathematical Statistics and Probability; University of California Press: Berkeley, CA, USA, 1967; Volume 1, pp. 281–297. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Metabotype | Metabolite | Relative Abundance | p-Value (One-Way ANOVA) | Identification Status (*) |
|---|---|---|---|---|
| 1 | Spermidine | − | 2.8 × 10−11 | a, c, d |
| Cytosine | − | 5.2 × 10−10 | a, b, d | |
| Putrescine | − | 1.3 × 10−0.9 | a, c, d | |
| Adenosine monophosphate (AMP) | − | 3.6 × 10−0.9 | a, b, d | |
| Uridine diphosphate (UDP)-Galactose (UDP-Glucose) | − | 4.6 × 10−0.9 | a, b, d | |
| Cytidine diphosphate (CDP) | − | 5.9 × 10−0.9 | a | |
| Adenosine diphosphate (ADP) | − | 8.1 × 10−0.9 | a, b, d | |
| Guanosine | − | 1.9 × 10−0.8 | a, c, d | |
| N2-Succinyl-L-ornithine | − | 2.6 × 10−0.8 | a | |
| UDP-N-acetylgalactosamine (UDP-N-acetylglucosamine) | − | 4.1 × 10−0.8 | a, b, d | |
| 2 | Guanine | + | 1.4 × 10−0.6 | a, b, d |
| UDP-N-acetylgalactosamine (UDP-N-acetylglucosamine) | + | 1.8 × 10−0.6 | a, b, d | |
| 12-Hydroxydodecanoic acid | + | 2.6 × 10−0.6 | a, b, d | |
| Guanosine monophosphate | + | 4.5 × 10−0.6 | a, c, d | |
| Pentoses phosphate | + | 1.7 × 10−0.5 | a, b, d | |
| N2-Succinyl-L-ornithine | + | 2.3 × 10−0.5 | a | |
| Glucosamine 6-phosphate (Galactosamine 6-phosphate) | + | 2.4 × 10−0.5 | a, b, d | |
| Cytosine | + | 3.7 × 10−0.5 | a, b, d | |
| Guanosine | + | 5.2 × 10−0.5 | a, c, d | |
| UDP-Galactose (UDP-Glucose) | + | 1.5 × 10−0.4 | a, b, d | |
| 3 | 1-Hydroxy-2-nonyl-4(1H)-quinolinone | + | 7.5 × 10−0.8 | a |
| Palmitoleic acid | + | 2.5 × 10−0.7 | a | |
| Glycerylphosphorylethanolamine/ sn-glycero-3- phosphoethanolamine | + | 3.0 × 10−0.7 | a, f | |
| AMP | + | 2.2 × 10−0.6 | a, b, d | |
| N2-Succinyl-L-glutamic acid 5-semialdehyde | − | 3.3 × 10−0.6 | a | |
| Heptadecenoic acid | + | 3.6 × 10−0.6 | a | |
| N-Acetylornithine | + | 5.4 × 10−0.6 | a, c, d | |
| Tetradecanoyl-phosphate (n-C14:0) | + | 1.0 × 10−0.5 | a | |
| Indoleglycerol phosphate | + | 1.2 × 10−0.5 | a | |
| Glycerol | + | 1.2 × 10−0.5 | a |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moyne, O.; Castelli, F.; Bicout, D.J.; Boccard, J.; Camara, B.; Cournoyer, B.; Faudry, E.; Terrier, S.; Hannani, D.; Huot-Marchand, S.; et al. Metabotypes of Pseudomonas aeruginosa Correlate with Antibiotic Resistance, Virulence and Clinical Outcome in Cystic Fibrosis Chronic Infections. Metabolites 2021, 11, 63. https://doi.org/10.3390/metabo11020063
Moyne O, Castelli F, Bicout DJ, Boccard J, Camara B, Cournoyer B, Faudry E, Terrier S, Hannani D, Huot-Marchand S, et al. Metabotypes of Pseudomonas aeruginosa Correlate with Antibiotic Resistance, Virulence and Clinical Outcome in Cystic Fibrosis Chronic Infections. Metabolites. 2021; 11(2):63. https://doi.org/10.3390/metabo11020063
Chicago/Turabian StyleMoyne, Oriane, Florence Castelli, Dominique J. Bicout, Julien Boccard, Boubou Camara, Benoit Cournoyer, Eric Faudry, Samuel Terrier, Dalil Hannani, Sarah Huot-Marchand, and et al. 2021. "Metabotypes of Pseudomonas aeruginosa Correlate with Antibiotic Resistance, Virulence and Clinical Outcome in Cystic Fibrosis Chronic Infections" Metabolites 11, no. 2: 63. https://doi.org/10.3390/metabo11020063
APA StyleMoyne, O., Castelli, F., Bicout, D. J., Boccard, J., Camara, B., Cournoyer, B., Faudry, E., Terrier, S., Hannani, D., Huot-Marchand, S., Léger, C., Maurin, M., Ngo, T.-D., Plazy, C., Quinn, R. A., Attree, I., Fenaille, F., Toussaint, B., & Le Gouëllec, A. (2021). Metabotypes of Pseudomonas aeruginosa Correlate with Antibiotic Resistance, Virulence and Clinical Outcome in Cystic Fibrosis Chronic Infections. Metabolites, 11(2), 63. https://doi.org/10.3390/metabo11020063