
MSCAT: A Machine Learning Assisted Catalog of Metabolomics Software Tools


Abstract
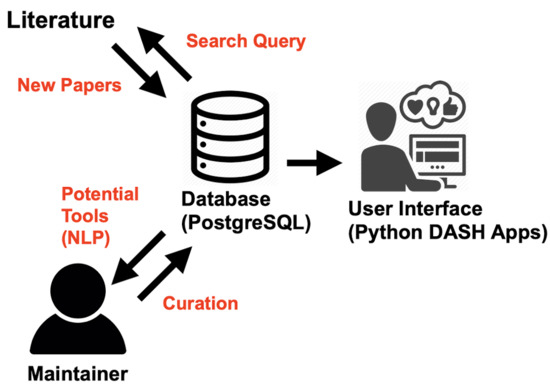
:
1. Introduction
2. Results
2.1. Categorization of Software Tools

{kind=link}
{kind=link}
{kind=link}
| Kusonmano et al. [30] | Spicer et al. [1] | Stanstrup et al. [31] | Misra et al. [18] | Chang et al. [32] |
|---|---|---|---|---|
| Data Acquisition Instrument | Pre-Processing LC-MS GC-MS NMR | Metabolite Profiling MS Data ProcessingNMR Data ProcessingUV Data Processing | Data Preprocessing | Data Preprocessing |
| Data Preprocessing Normalization Identification Binning | Annotation Identification Quantification | Metabolite Annotation Ion grouping MS/MS Structure Databases | Statistical Tools | Statistical Analysis |
| Data Analysis PCA Clustering SVM PLS | Post-Processing | Data Analysis Statistics Network Analysis Pathway Analysis | Annotation Tools | Identification |
| Data Interpretation Databases Network Visualization Pathway Analysis | Statistical Analysis | Biological Interpretation | Functional Analysis | |
| Workflow and other tools | Databases | Metabolic Modeling | ||
| Instrument-based | ||||
| Workflows |
2.2. Database Design
2.3. Literature Mining: Tool Publications
2.4. Literature Mining: Tool Name Detection
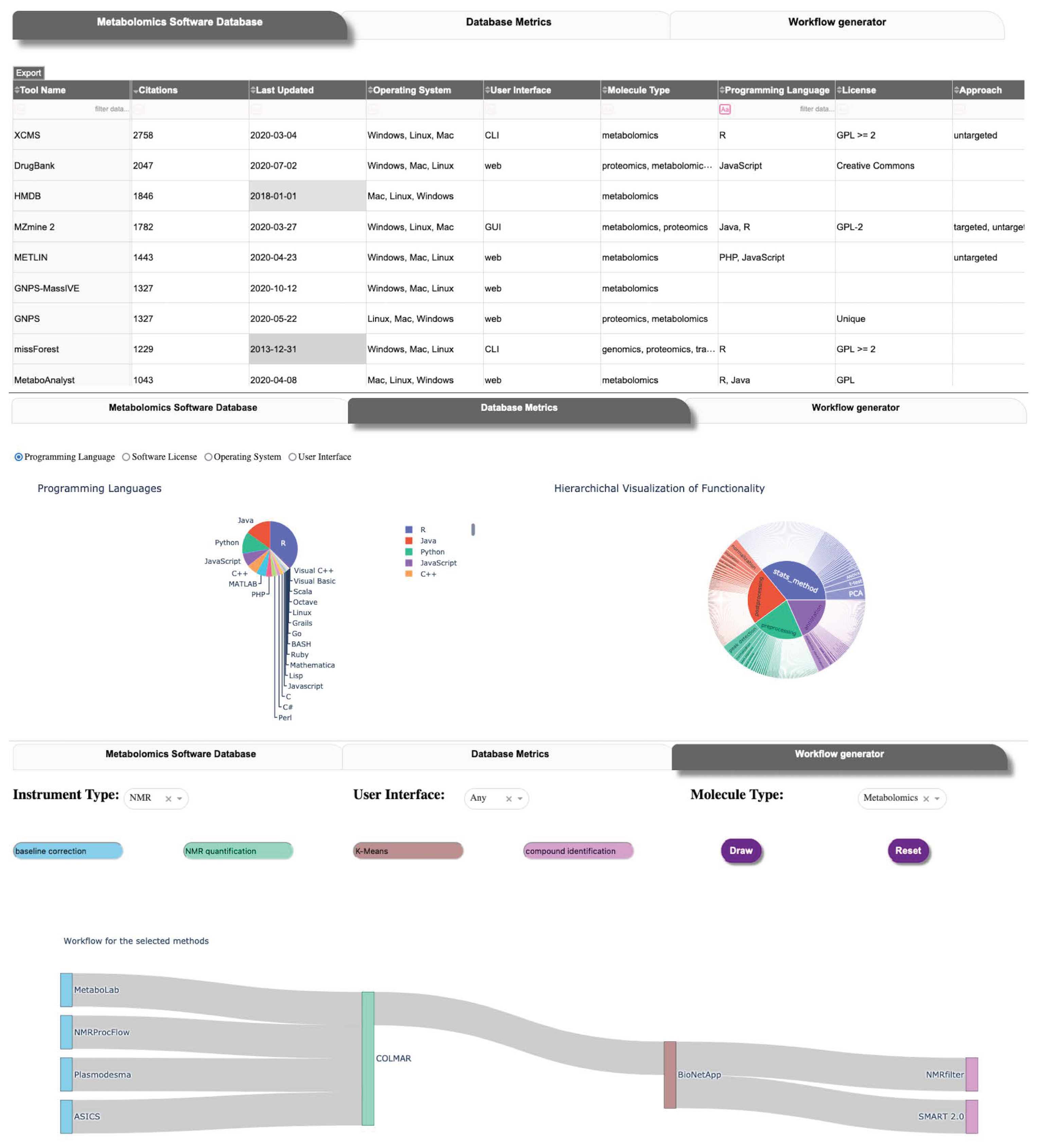
2.5. Database User Interface
2.6. Curation Workflow
3. Discussion
4. Materials and Methods
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Spicer, R.; Salek, R.M.; Moreno, P.; Canueto, D.; Steinbeck, C. Navigating freely-available software tools for metabolomics analysis. Metabolomics 2017, 13, 106. [Google Scholar] [CrossRef] [Green Version]
- Henry, V.J.; Bandrowski, A.E.; Pepin, A.S.; Gonzalez, B.J.; Desfeux, A. OMICtools: An informative directory for multi-omic data analysis. Database 2014, 2014, bau069. [Google Scholar] [CrossRef] [Green Version]
- Ellinger, J.J.; Chylla, R.A.; Ulrich, E.L.; Markley, J.L. Databases and Software for NMR-Based Metabolomics. Curr. Metab. 2013, 1, 28–40. [Google Scholar] [CrossRef] [Green Version]
- Cannata, N.; Merelli, E.; Altman, R.B. Time to organize the bioinformatics resourceome. PLoS Comput. Biol. 2005, 1, e76. [Google Scholar] [CrossRef] [Green Version]
- Weber, R.J.M.; Lawson, T.N.; Salek, R.M.; Ebbels, T.M.D.; Glen, R.C.; Goodacre, R.; Griffin, J.L.; Haug, K.; Koulman, A.; Moreno, P.; et al. Computational tools and workflows in metabolomics: An international survey highlights the opportunity for harmonisation through Galaxy. Metabolomics 2017, 13, 12. [Google Scholar] [CrossRef] [Green Version]
- Johnson, C.H.; Ivanisevic, J.; Siuzdak, G. Metabolomics: Beyond biomarkers and towards mechanisms. Nat. Rev. Mol. Cell Biol. 2016, 17, 451–459. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.J.; Kim, S.H.; Kim, J.H.; Hwang, S.; Yoo, H.J. Understanding Metabolomics in Biomedical Research. Endocrinol. Metab. 2016, 31, 7–16. [Google Scholar] [CrossRef]
- Warth, B.; Spangler, S.; Fang, M.; Johnson, C.H.; Forsberg, E.M.; Granados, A.; Martin, R.L.; Domingo-Almenara, X.; Huan, T.; Rinehart, D.; et al. Exposome-Scale Investigations Guided by Global Metabolomics, Pathway Analysis, and Cognitive Computing. Anal. Chem. 2017, 89, 11505–11513. [Google Scholar] [CrossRef]
- Alonso, A.; Marsal, S.; Julia, A. Analytical methods in untargeted metabolomics: State of the art in 2015. Front. Bioeng. Biotechnol. 2015, 3, 23. [Google Scholar] [CrossRef] [Green Version]
- Bartel, J.; Krumsiek, J.; Theis, F.J. Statistical methods for the analysis of high-throughput metabolomics data. Comput. Struct. Biotechnol. J. 2013, 4, e201301009. [Google Scholar] [CrossRef] [Green Version]
- Johnson, C.H.; Ivanisevic, J.; Benton, H.P.; Siuzdak, G. Bioinformatics: The next frontier of metabolomics. Anal. Chem. 2015, 87, 147–156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uppal, K.; Walker, D.I.; Liu, K.; Li, S.; Go, Y.M.; Jones, D.P. Computational Metabolomics: A Framework for the Million Metabolome. Chem. Res. Toxicol. 2016, 29, 1956–1975. [Google Scholar] [CrossRef] [Green Version]
- Blazenovic, I.; Kind, T.; Ji, J.; Fiehn, O. Software Tools and Approaches for Compound Identification of LC-MS/MS Data in Metabolomics. Metabolites 2018, 8, 31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Misra, B.B. New tools and resources in metabolomics: 2016–2017. Electrophoresis 2018, 39, 909–923. [Google Scholar] [CrossRef]
- Misra, B.B. Data normalization strategies in metabolomics: Current challenges, approaches, and tools. Eur. J. Mass Spectrom. 2020, 26, 165–174. [Google Scholar] [CrossRef]
- Misra, B.B. Open-Source Software Tools, Databases, and Resources for Single-Cell and Single-Cell-Type Metabolomics. Methods Mol. Biol. 2020, 2064, 191–217. [Google Scholar] [CrossRef] [PubMed]
- Misra, B.B.; Mohapatra, S. Tools and resources for metabolomics research community: A 2017–2018 update. Electrophoresis 2019, 40, 227–246. [Google Scholar] [CrossRef]
- Misra, B.B.; van der Hooft, J.J. Updates in metabolomics tools and resources: 2014–2015. Electrophoresis 2016, 37, 86–110. [Google Scholar] [CrossRef] [Green Version]
- O’Shea, K.; Misra, B.B. Software tools, databases and resources in metabolomics: Updates from 2018 to 2019. Metabolomics 2020, 16, 36. [Google Scholar] [CrossRef]
- Misra, B.B.; Fahrmann, J.F.; Grapov, D. Review of emerging metabolomic tools and resources: 2015–2016. Electrophoresis 2017, 38, 2257–2274. [Google Scholar] [CrossRef]
- Misra, B.B. New software tools, databases, and resources in metabolomics: Updates from 2020. Metabolomics 2021, 17, 49. [Google Scholar] [CrossRef]
- Sugimoto, M.; Kawakami, M.; Robert, M.; Soga, T.; Tomita, M. Bioinformatics Tools for Mass Spectroscopy-Based Metabolomic Data Processing and Analysis. Curr. Bioinform. 2012, 7, 96–108. [Google Scholar] [CrossRef]
- Peters, K.; Bradbury, J.; Bergmann, S.; Capuccini, M.; Cascante, M.; de Atauri, P.; Ebbels, T.M.D.; Foguet, C.; Glen, R.; Gonzalez-Beltran, A.; et al. PhenoMeNal: Processing and analysis of metabolomics data in the cloud. Gigascience 2019, 8, giy149. [Google Scholar] [CrossRef] [Green Version]
- Krassowski, M.; Das, V.; Sahu, S.K.; Misra, B.B. State of the Field in Multi-Omics Research: From Computational Needs to Data Mining and Sharing. Front. Genet. 2020, 11, 610798. [Google Scholar] [CrossRef]
- Majumder, E.L.; Billings, E.M.; Benton, H.P.; Martin, R.L.; Palermo, A.; Guijas, C.; Rinschen, M.M.; Domingo-Almenara, X.; Montenegro-Burke, J.R.; Tagtow, B.A.; et al. Cognitive analysis of metabolomics data for systems biology. Nat. Protoc. 2021, 16, 1376–1418. [Google Scholar] [CrossRef]
- Lindon, J.C.; Holmes, E.; Nicholson, J.K. Metabonomics: Systems biology in pharmaceutical research and development. Curr. Opin. Mol. Ther. 2004, 6, 265–272. [Google Scholar]
- Nicholson, J.K.; Lindon, J.C. Systems biology: Metabonomics. Nature 2008, 455, 1054–1056. [Google Scholar] [CrossRef]
- Simons, M.; Misra, A.; Sriram, G. Genome-scale models of plant metabolism. Methods Mol. Biol. 2014, 1083, 213–230. [Google Scholar] [CrossRef]
- Zappia, L.; Phipson, B.; Oshlack, A. Exploring the single-cell RNA-seq analysis landscape with the scRNA-tools database. PLoS Comput. Biol. 2018, 14, e1006245. [Google Scholar] [CrossRef]
- Kusonmano, K.; Vongsangnak, W.; Chumnanpuen, P. Informatics for Metabolomics. Adv. Exp. Med. Biol 2016, 939, 91–115. [Google Scholar] [CrossRef]
- Stanstrup, J.; Broeckling, C.D.; Helmus, R.; Hoffmann, N.; Mathe, E.; Naake, T.; Nicolotti, L.; Peters, K.; Rainer, J.; Salek, R.M.; et al. The metaRbolomics Toolbox in Bioconductor and beyond. Metabolites 2019, 9, 200. [Google Scholar] [CrossRef] [Green Version]
- Chang, H.Y.; Colby, S.M.; Du, X.; Gomez, J.D.; Helf, M.J.; Kechris, K.; Kirkpatrick, C.R.; Li, S.; Patti, G.J.; Renslow, R.S.; et al. A Practical Guide to Metabolomics Software Development. Anal. Chem. 2021, 93, 1912–1923. [Google Scholar] [CrossRef]
- PostgreSQL: The World’s Most Advanced Open Source Relational Database. Available online: https://www.postgresql.org/ (accessed on 28 June 2021).
- Stonebraker, M.; Rowe, L.A. The design of Postgres. ACM Sigmod Rec. 1986, 15, 340–355. [Google Scholar] [CrossRef]
- Kent, W. A simple guide to five normal forms in relational database theory. Commun. ACM 1983, 26, 120–125. [Google Scholar] [CrossRef] [Green Version]
- Katz, D.S.; Gruenpeter, M.; Honeyman, T. Taking a fresh look at FAIR for research software. Patterns 2021, 2, 100222. [Google Scholar] [CrossRef]
- Wilkinson, M.D.; Dumontier, M.; Aalbersberg, I.J.; Appleton, G.; Axton, M.; Baak, A.; Blomberg, N.; Boiten, J.-W.; da Silva Santos, L.B.; Bourne, P.E. The FAIR Guiding Principles for scientific data management and stewardship. Sci. Data 2016, 3, 160018. [Google Scholar] [CrossRef] [Green Version]
- Lample, G.; Ballesteros, M.; Subramanian, S.; Kawakami, K.; Dyer, C. Neural architectures for named entity recognition. arXiv 2016, arXiv:1603.01360. [Google Scholar]
- Li, J.; Sun, A.; Han, J.; Li, C. A survey on deep learning for named entity recognition. IEEE Trans. Knowl. Data Eng. 2020. [Google Scholar] [CrossRef]
- Bird, S. NLTK: The natural language toolkit. In Proceedings of the COLING/ACL 2006 Interactive Presentation Sessions, Sydney, Australia, 17–18 July 2006; pp. 69–72. [Google Scholar]
- Kocaman, V.; Talby, D. Spark NLP: Natural Language Understanding at Scale. Softw. Impacts 2021, 8, 100058. [Google Scholar] [CrossRef]
- Peters, M.E.; Neumann, M.; Iyyer, M.; Gardner, M.; Clark, C.; Lee, K.; Zettlemoyer, L. Deep contextualized word representations. arXiv 2018, arXiv:1802.05365. [Google Scholar]
- Huang, Z.; Xu, W.; Yu, K. Bidirectional LSTM-CRF models for sequence tagging. arXiv 2015, arXiv:1508.01991. [Google Scholar]
- Zheng, S.; Jayasumana, S.; Romera-Paredes, B.; Vineet, V.; Su, Z.; Du, D.; Huang, C.; Torr, P.H.S. Conditional Random Fields as Recurrent Neural Networks. In Proceedings of the 2015 IEEE International Conference on Computer Vision (ICCV), Las Condes, Chile, 7–13 December 2015; pp. 1529–1537. [Google Scholar]
- Riehmann, P.; Hanfler, M.; Froehlich, B. Interactive sankey diagrams. In Proceedings of the IEEE Symposium on Information Visualization, Minneapolis, MN, USA, 23–25 October 2005; pp. 233–240. [Google Scholar]
- Chamberlain, S.; Zhu, H.; Jahn, N.; Boettiger, C.; Ram, K. rcrossref: Client for Various ‘CrossRef’ ‘APIs’, 1.1.0. 2020. Available online: https://docs.ropensci.org/rcrossref/ (accessed on 20 September 2021).
- Sud, M.; Fahy, E.; Cotter, D.; Azam, K.; Vadivelu, I.; Burant, C.; Edison, A.; Fiehn, O.; Higashi, R.; Nair, K.S.; et al. Metabolomics Workbench: An international repository for metabolomics data and metadata, metabolite standards, protocols, tutorials and training, and analysis tools. Nucleic Acids Res. 2016, 44, D463–D470. [Google Scholar] [CrossRef] [Green Version]
- Bhamber, R.S.; Jankevics, A.; Deutsch, E.W.; Jones, A.R.; Dowsey, A.W. mzMLb: A Future-Proof Raw Mass Spectrometry Data Format Based on Standards-Compliant mzML and Optimized for Speed and Storage Requirements. J. Proteome Res. 2021, 20, 172–183. [Google Scholar] [CrossRef]
- Larralde, M.; Lawson, T.N.; Weber, R.J.; Moreno, P.; Haug, K.; Rocca-Serra, P.; Viant, M.R.; Steinbeck, C.; Salek, R.M. mzML2ISA & nmrML2ISA: Generating enriched ISA-Tab metadata files from metabolomics XML data. Bioinformatics 2017, 33, 2598–2600. [Google Scholar]
- Martens, L.; Chambers, M.; Sturm, M.; Kessner, D.; Levander, F.; Shofstahl, J.; Tang, W.H.; Römpp, A.; Neumann, S.; Pizarro, A.D. mzML—a community standard for mass spectrometry data. Mol. Cell. Proteom. 2011, 10, R110.000133. [Google Scholar] [CrossRef] [Green Version]
- Saripalle, R.; Runyan, C.; Russell, M. Using HL7 FHIR to achieve interoperability in patient health record. J. Biomed. Inform. 2019, 94, 103188. [Google Scholar] [CrossRef]
- Garijo, D.; Ratnakar, V.; Gil, Y.; Khider, D. The Software Description Ontology. Available online: https://w3id.org/okn/o/sd/1.9.0 (accessed on 28 June 2021).
- Carvalho, L.A.M.C.; Garijo, D.; Medeiros, C.B.; Gil, Y. Semantic Software Metadata for Workflow Exploration and Evolution. In Proceedings of the 2018 IEEE 14th International Conference on e-Science (e-Science), Amsterdam, The Netherlands, 29 Octtober–1 November 2018; pp. 431–441. [Google Scholar]
- Fantino, D. easyPubMed. 2019. Available online: https://rdrr.io/cran/easyPubMed/ (accessed on 20 September 2021).
- Chiu, J.P.; Nichols, E. Named entity recognition with bidirectional LSTM-CNNs. Trans. Assoc. Comput. Linguist. 2016, 4, 357–370. [Google Scholar] [CrossRef]
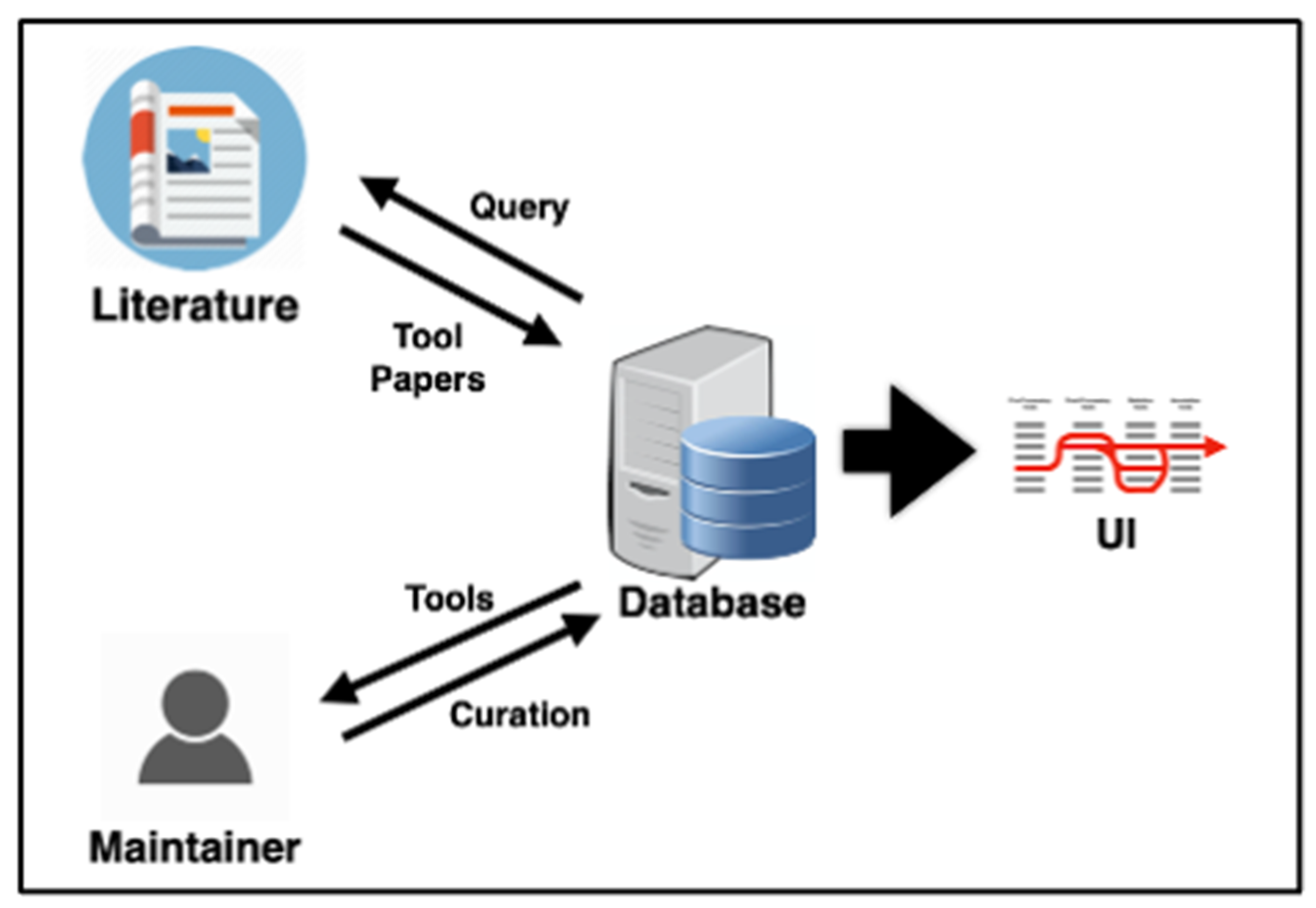
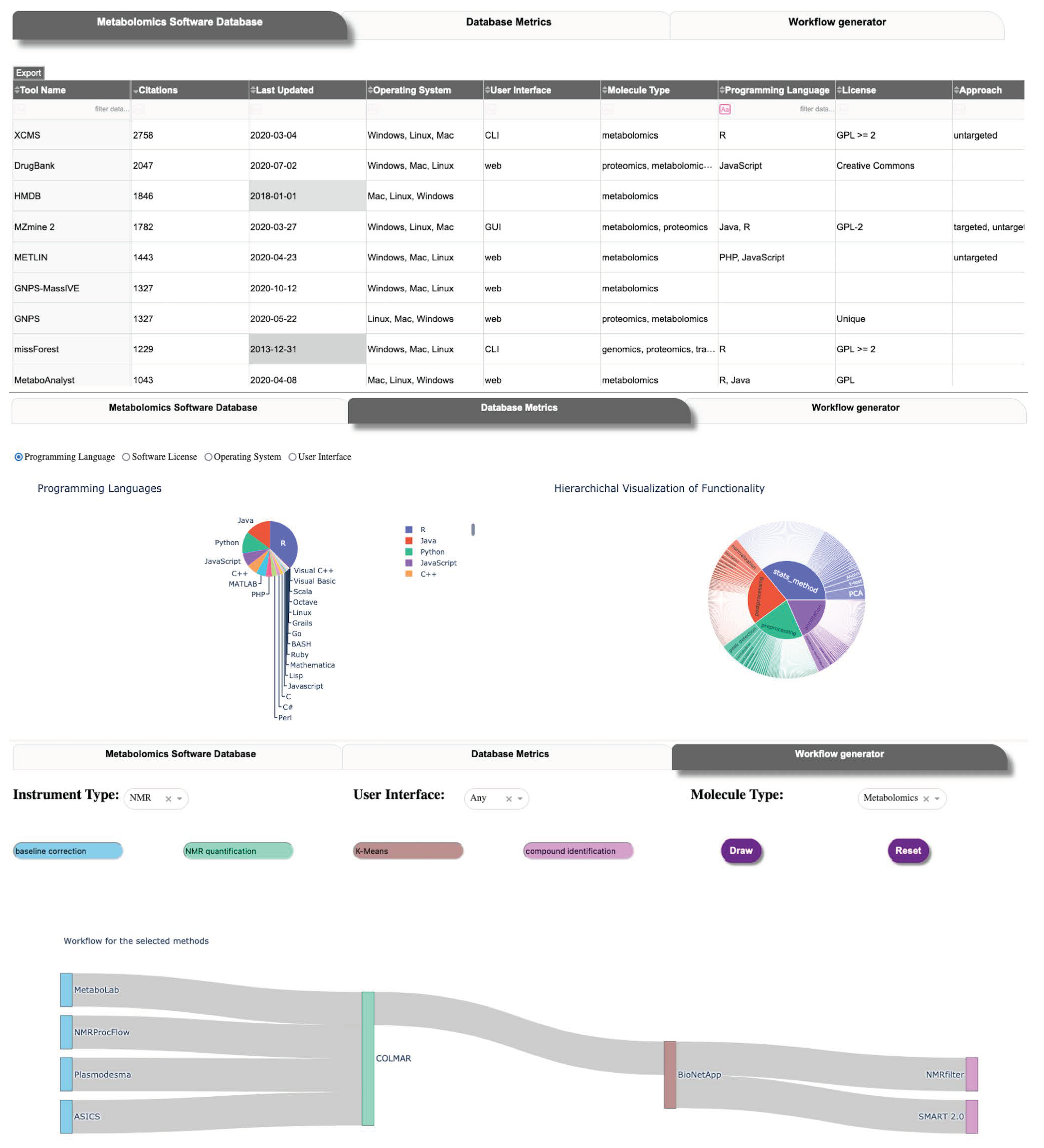
| Database Table Name | Description |
|---|---|
| Main | Tool name, release year, link to website, date of last.update, etc |
| Functional Characteristics | |
| Molecule Type | Molecule types the tool works with |
| Approach | Metabolomics approach (targeted, untargeted) if defined |
| Instrument Data | Type of instrument data the tool uses, if defined |
| Functionality | Functionality categorie(s) of the tool |
| Annotation | Annotation methods the tool provides |
| Pre-processing | Pre-processing methods the tool provides |
| Post-processing | Post-processing methods the tool provides |
| Statistical Analysis | Statistical methods the tool provides |
| Software Characteristics | |
| File formats | The file formats the tool works with |
| Operating system | Tool’s operating system compatibility |
| Programming language | Tool’s available programming languages |
| Software license | Tool’s software licenses |
| User interface | Tool’s available User interfaces |
| Containers | Is the tool available as a container (URL) |
| Citations | Aggregate number of citations of tool publications |
| Publications | Publications associated with the tool |
| Concept | Metabolomics | Software |
|---|---|---|
| Keywords | Metabolomics Metabolonics Metabonomics | Programming Language |
| Controlled Vocabulary (Mesh) | Metabolomics | Software |
| Concept | Metabolomics | Software |
|---|---|---|
| Keywords | Metabolomics Metabonomics Metabolonics Metabolite Multiomic Mixomic Metabolome | Algorithm Toolkit Code Software (framework/pipeline/tool/package/suite/workflow) Open source Source code Web application Command line Programming language Github Gitlab Sourceforge Bioconductor Bio(python/java/ruby) |
| Controlled Vocabulary (MeSH) | Metabolomics Metabolome | Computing Methodologies |
| Token | POS | Sentence ID | Label |
|---|---|---|---|
| MetaComp | NN | 1.0 | T |
| comprehensive | JJ | 1.0 | O |
| analysis | NN | 1.0 | O |
| software | NN | 1.0 | O |
| comparative | JJ | 1.0 | O |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dekermanjian, J.; Labeikovsky, W.; Ghosh, D.; Kechris, K. MSCAT: A Machine Learning Assisted Catalog of Metabolomics Software Tools. Metabolites 2021, 11, 678. https://doi.org/10.3390/metabo11100678
Dekermanjian J, Labeikovsky W, Ghosh D, Kechris K. MSCAT: A Machine Learning Assisted Catalog of Metabolomics Software Tools. Metabolites. 2021; 11(10):678. https://doi.org/10.3390/metabo11100678
Chicago/Turabian StyleDekermanjian, Jonathan, Wladimir Labeikovsky, Debashis Ghosh, and Katerina Kechris. 2021. "MSCAT: A Machine Learning Assisted Catalog of Metabolomics Software Tools" Metabolites 11, no. 10: 678. https://doi.org/10.3390/metabo11100678
APA StyleDekermanjian, J., Labeikovsky, W., Ghosh, D., & Kechris, K. (2021). MSCAT: A Machine Learning Assisted Catalog of Metabolomics Software Tools. Metabolites, 11(10), 678. https://doi.org/10.3390/metabo11100678