The Harmonious Interplay of Amino Acid and Monocarboxylate Transporters Induces the Robustness of Cancer Cells


{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
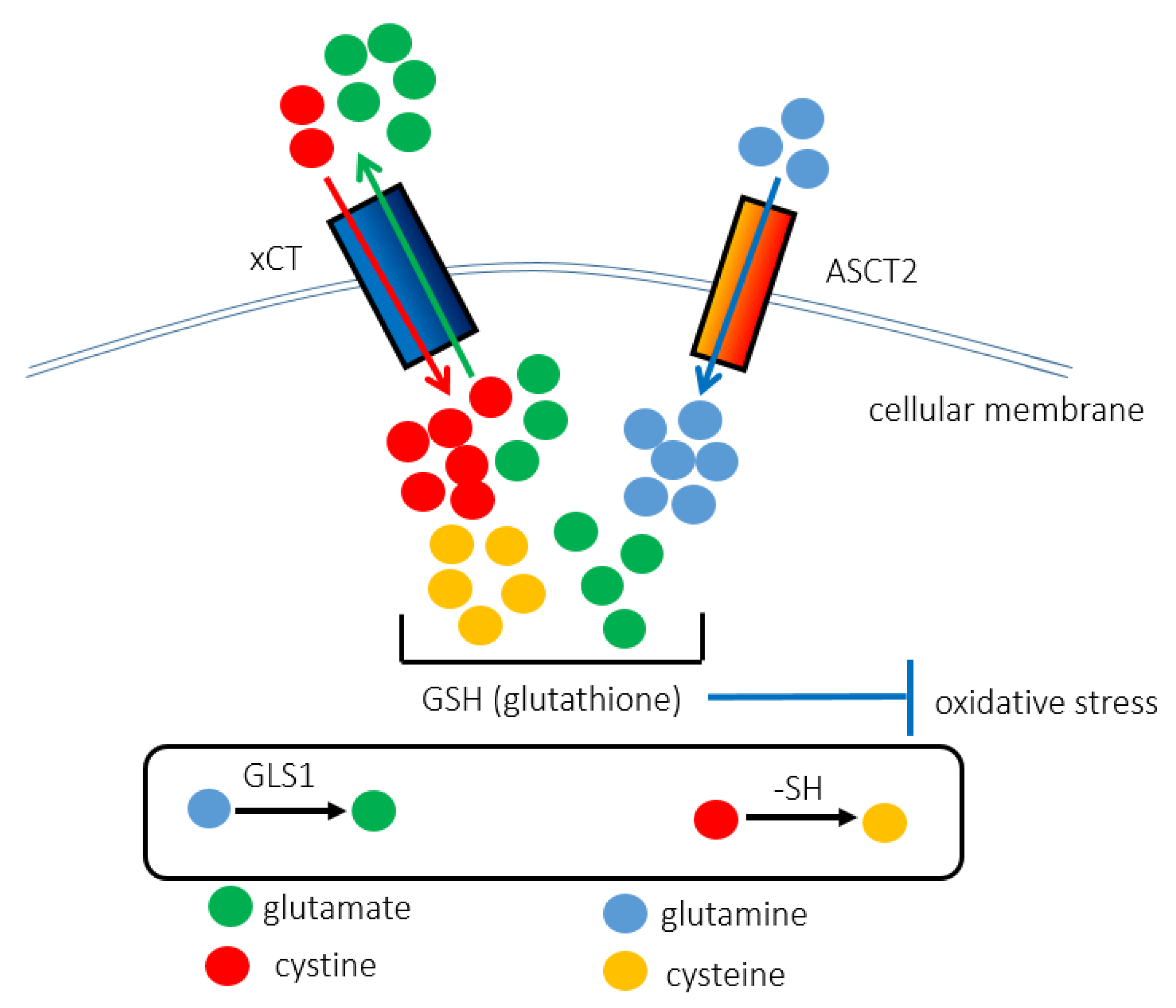
2. xCT (SLC7A11)
3. ASCT2 (SLC1A5)
4. LAT1 (SLC7A5)
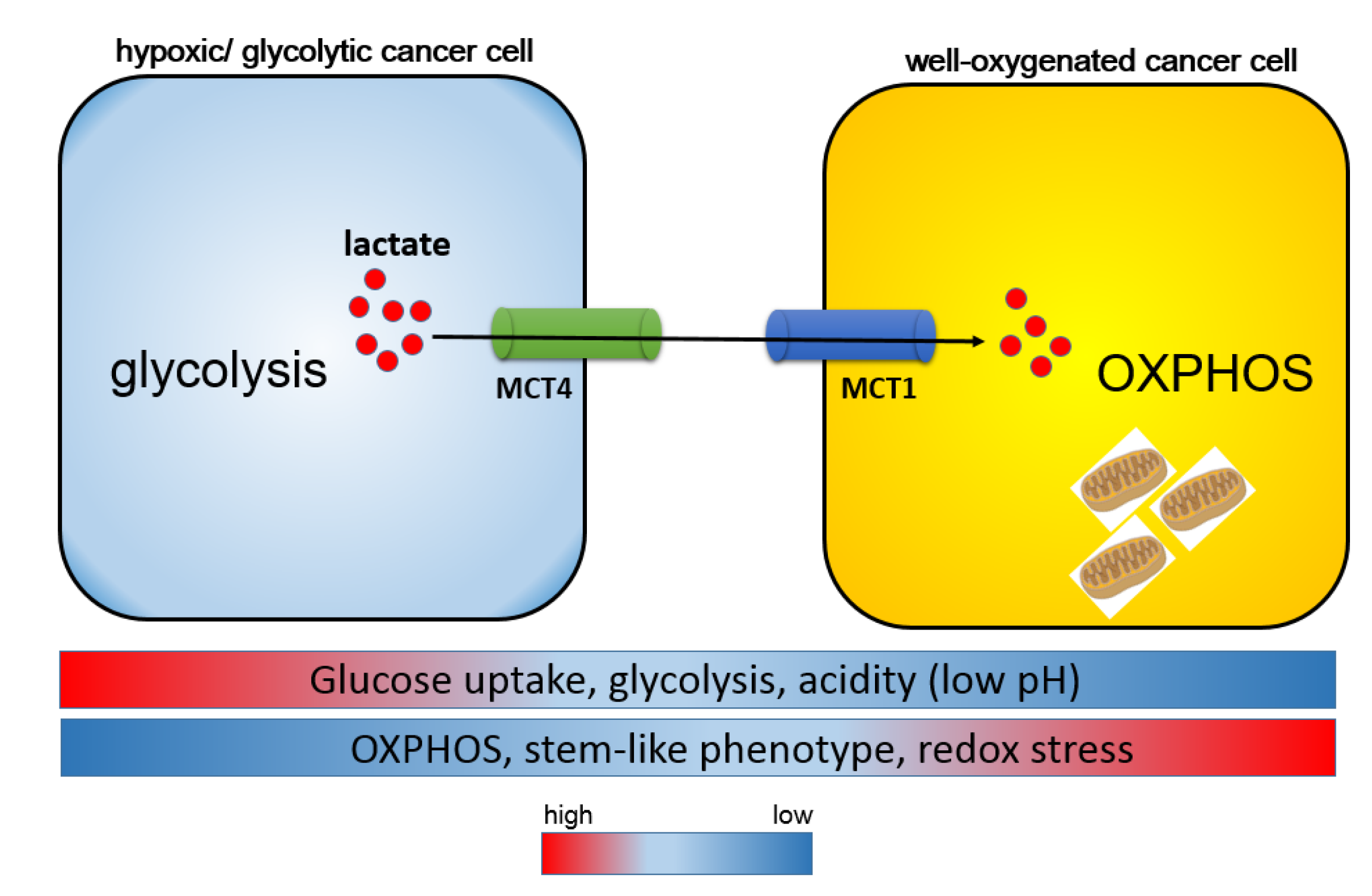
5. Monocarboxylate Transporters
6. Conclusions
Funding
Conflicts of Interest
Abbreviations
| α-KG | α-ketoglutarate |
| AMPK | AMP activated protein kinase |
| ATF4 | activating transcription factor 4 |
| BMSC | bone marrow-derived stromal cell |
| CAFs | cancer-associated fibroblasts |
| CD44v | CD44 variant |
| CD98hc | CD98 heavy chain |
| CLL | chronic lymphocytic leukemia |
| EAA | essential amino acids |
| GDH | glutamate dehydrogenase |
| HIF | hypoxia-inducible factor |
| GSH | glutathione |
| HNSCC | head and neck squamous cell carcinoma |
| MCT | monocarboxylate transporter |
| microRNA | miR |
| mTOR | the mammalian target of rapamycin |
| OXPHOS | oxidative phosphorylation |
| SSZ | sulfasalazine |
| TCA cycle | tricarboxylic acid cycle |
References
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- DeBerardinis, R.J.; Lum, J.J.; Hatzivassiliou, G.; Thompson, C.B. The Biology of Cancer: Metabolic Reprogramming Fuels Cell Growth and Proliferation. Cell Metab. 2008, 7, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Phan, L.M.; Yeung, S.J.; Lee, M.-H. Cancer metabolic reprogramming: Importance, main features, and potentials for precise targeted anti-cancer therapies. Cancer Biol. Med. 2014, 11, 1–19. [Google Scholar] [PubMed]
- Yoshida, G.J. Metabolic reprogramming: The emerging concept and associated therapeutic strategies. J. Exp. Clin. Cancer Res. 2015, 34, 111. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.-A.; Wang, C.-Y.; Hsieh, Y.-T.; Chen, Y.-J.; Wei, Y.-H. Metabolic reprogramming orchestrates cancer stem cell properties in nasopharyngeal carcinoma. Cell Cycle 2014, 14, 86–98. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, G.J.; Saya, H. Therapeutic strategies targeting cancer stem cells. Cancer Sci. 2016, 107, 5–11. [Google Scholar] [CrossRef] [PubMed]
- Makena, M.R.; Ranjan, A.; Thirumala, V.; Reddy, A.P. Cancer stem cells: Road to therapeutic resistance and strategies to overcome resistance. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2020, 1866, 165339. [Google Scholar] [CrossRef]
- Yoshida, G.J. The heterogeneity of cancer stem-like cells at the invasive front. Cancer Cell Int. 2017, 17, 23. [Google Scholar] [CrossRef][Green Version]
- Frank, N.Y.; Schatton, T.; Frank, M.H. The therapeutic promise of the cancer stem cell concept. J. Clin. Investig. 2010, 120, 41–50. [Google Scholar] [CrossRef]
- Kurtova, A.V.; Xiao, J.; Mo, Q.; Pazhanisamy, S.K.; Krasnow, R.; Lerner, S.P.; Chen, F.; Roh, T.T.; Lay, E.; Ho, P.L.; et al. Blocking PGE2-induced tumour repopulation abrogates bladder cancer chemoresistance. Nat. Cell Biol. 2015, 517, 209–213. [Google Scholar] [CrossRef]
- Plaks, V.; Kong, N.; Werb, Z. The Cancer Stem Cell Niche: How Essential Is the Niche in Regulating Stemness of Tumor Cells? Cell Stem Cell 2015, 16, 225–238. [Google Scholar] [CrossRef] [PubMed]
- Cairns, R.A.; Harris, I.S.; Mak, T.W. Regulation of cancer cell metabolism. Nat. Rev. Cancer 2011, 11, 85–95. [Google Scholar] [CrossRef] [PubMed]
- Cantor, J.R.; Sabatini, D.M. Cancer Cell Metabolism: One Hallmark, Many Faces. Cancer Discov. 2012, 2, 881–898. [Google Scholar] [CrossRef] [PubMed]
- Bröer, S. Amino Acid Transporters as Targets for Cancer Therapy: Why, Where, When, and How. Int. J. Mol. Sci. 2020, 21, 6156. [Google Scholar] [CrossRef] [PubMed]
- Bhutia, Y.D.; Babu, E.; Ramachandran, S.; Ganapathy, V. Amino Acid transporters in cancer and their relevance to “glutamine addiction”: Novel targets for the design of a new class of anticancer drugs. Cancer Res. 2015, 75, 1782–1788. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.; Wang, C.; Liu, G.; Bi, C.; Wang, X.; Zhou, Q.; Jin, H. SLC7A11/xCT in cancer: Biological functions and therapeutic implications. Am J Cancer Res 2020, 10, 3106–3126. [Google Scholar]
- Huang, Y.; Dai, Z.; Barbacioru, C.; Sadée, W. Cystine-Glutamate Transporter SLC7A11 in Cancer Chemosensitivity and Chemoresistance. Cancer Res. 2005, 65, 7446–7454. [Google Scholar] [CrossRef]
- Lo, M.; Wang, Y.Z.; Gout, P.W. The x(c)- cystine/glutamate antiporter: A potential target for therapy of cancer and other diseases. J. Cell Physiol. 2008, 215, 593–602. [Google Scholar] [CrossRef]
- Closs, E.I.; Wagner, C.A.; Palacin, M.; Endou, H.; Kanai, Y. CATs and HATs: The SLC7 family of amino acid transporters. Pflügers Arch. Eur. J. Physiol. 2004, 447, 532–542. [Google Scholar] [CrossRef]
- Sato, H.; Shiiya, A.; Kimata, M.; Maebara, K.; Tamba, M.; Sakakura, Y.; Makino, N.; Sugiyama, F.; Yagami, K.-I.; Moriguchi, T.; et al. Redox Imbalance in Cystine/Glutamate Transporter-deficient Mice. J. Biol. Chem. 2005, 280, 37423–37429. [Google Scholar] [CrossRef]
- Sasaki, H.; Sato, H.; Kuriyama-Matsumura, K.; Sato, K.; Maebara, K.; Wang, H.; Tamba, M.; Itoh, K.; Yamamoto, M.; Bannai, S. Electrophile Response Element-mediated Induction of the Cystine/Glutamate Exchange Transporter Gene Expression. J. Biol. Chem. 2002, 277, 44765–44771. [Google Scholar] [CrossRef] [PubMed]
- Ji, X.; Qian, J.; Rahman, S.M.J.; Siska, P.J.; Zou, Y.; Harris, B.K.; Hoeksema, M.D.; Trenary, I.A.; Heidi, C.; Eisenberg, R.; et al. xCT (SLC7A11)-mediated metabolic reprogramming promotes non-small cell lung cancer progression. Oncogene 2018, 37, 5007–5019. [Google Scholar] [CrossRef] [PubMed]
- Habib, E.; Linher-Melville, K.; Lin, H.-X.; Singh, G. Expression of xCT and activity of system xc− are regulated by NRF2 in human breast cancer cells in response to oxidative stress. Redox Biol. 2015, 5, 33–42. [Google Scholar] [CrossRef] [PubMed]
- Bauer, A.K.; Hill, T., 3rd; Alexander, C.M. The involvement of NRF2 in lung cancer. Oxid. Med. Cell. Longev. 2013, 2013, 746432. [Google Scholar] [CrossRef]
- Harding, H.P.; Zhang, Y.; Zeng, H.; Novoa, I.; Lu, P.D.; Calfon, M.; Sadri, N.; Yun, C.; Popko, B.; Paules, R.; et al. An Integrated Stress Response Regulates Amino Acid Metabolism and Resistance to Oxidative Stress. Mol. Cell 2003, 11, 619–633. [Google Scholar] [CrossRef]
- Lewerenz, J.; Sato, H.; Albrecht, P.; Henke, N.; Noack, R.; Methner, A.; Maher, P. Mutation of ATF4 mediates resistance of neuronal cell lines against oxidative stress by inducing xCT expression. Cell Death Differ. 2011, 19, 847–858. [Google Scholar] [CrossRef]
- Lewerenz, J.; Maher, P. Basal Levels of eIF2α Phosphorylation Determine Cellular Antioxidant Status by Regulating ATF4 and xCT Expression. J. Biol. Chem. 2008, 284, 1106–1115. [Google Scholar] [CrossRef]
- Ishimoto, T.; Nagano, O.; Yae, T.; Tamada, M.; Motohara, T.; Oshima, H.; Oshima, M.; Ikeda, T.; Asaba, R.; Yagi, H.; et al. CD44 Variant Regulates Redox Status in Cancer Cells by Stabilizing the xCT Subunit of System xc− and Thereby Promotes Tumor Growth. Cancer Cell 2011, 19, 387–400. [Google Scholar] [CrossRef]
- Yae, T.; Tsuchihashi, K.; Ishimoto, T.; Motohara, T.; Yoshikawa, M.; Yoshida, G.J.; Wada, T.; Masuko, T.; Mogushi, K.; Tanaka, H.; et al. Alternative splicing of CD44 mRNA by ESRP1 enhances lung colonization of metastatic cancer cell. Nat. Commun. 2012, 3, 883. [Google Scholar] [CrossRef]
- Granot, Z.; Henke, E.; Comen, E.A.; King, T.A.; Norton, L.; Benezra, R. Tumor entrained neutrophils inhibit seeding in the premetastatic lung. Cancer Cell 2011, 20, 300–314. [Google Scholar] [CrossRef]
- Zhang, W.; Trachootham, D.; Liu, J.; Chen, G.; Pelicano, H.; Garcia-Prieto, C.; Lu, W.; Burger, J.A.; Croce, C.M.; Plunkett, W.; et al. Stromal control of cystine metabolism promotes cancer cell survival in chronic lymphocytic leukaemia. Nat. Cell Biol. 2012, 14, 276–286. [Google Scholar] [CrossRef] [PubMed]
- Wilde, L.; Roche, M.; Domingo-Vidal, M.; Tanson, K.; Philp, N.; Curry, J.; Martinez-Outschoorn, U.E. Metabolic coupling and the Reverse Warburg Effect in cancer: Implications for novel biomarker and anticancer agent development. Semin. Oncol. 2017, 44, 198–203. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, G.J.; Azuma, A.; Miura, Y.; Orimo, A. Activated Fibroblast Program Orchestrates Tumor Initiation and Progression; Molecular Mechanisms and the Associated Therapeutic Strategies. Int. J. Mol. Sci. 2019, 20, 2256. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, G.J. Regulation of heterogeneous cancer-associated fibroblasts: The molecular pathology of activated signaling pathways. J. Exp. Clin. Cancer Res. 2020, 39, 1–15. [Google Scholar] [CrossRef]
- Bonifácio, V.D.B.; Pereira, S.A.; Serpa, J.; Vicente, J.B. Cysteine metabolic circuitries: Druggable targets in cancer. Br. J. Cancer 2020, 1–18. [Google Scholar] [CrossRef]
- Bansal, A.; Simon, M.C. Glutathione metabolism in cancer progression and treatment resistance. J. Cell Biol. 2018, 217, 2291–2298. [Google Scholar] [CrossRef]
- McBean, G.J.; Flynn, J. Molecular mechanisms of cystine transport. Biochem. Soc. Trans. 2001, 29, 717–722. [Google Scholar] [CrossRef]
- Yoshikawa, M.; Tsuchihashi, K.; Ishimoto, T.; Yae, T.; Motohara, T.; Sugihara, E.; Onishi, N.; Masuko, T.; Yoshizawa, K.; Kawashiri, S.; et al. xCT Inhibition Depletes CD44v-Expressing Tumor Cells That Are Resistant to EGFR-Targeted Therapy in Head and Neck Squamous Cell Carcinoma. Cancer Res. 2013, 73, 1855–1866. [Google Scholar] [CrossRef]
- Mou, Y.; Wang, J.; Wu, J.; He, D.; Zhang, C.; Duan, C.; Li, B. Ferroptosis, a new form of cell death: Opportunities and challenges in cancer. J. Hematol. Oncol. 2019, 12, 1–16. [Google Scholar] [CrossRef]
- Wise, D.R.; Thompson, C.B. Glutamine addiction: A new therapeutic target in cancer. Trends Biochem. Sci. 2010, 35, 427–433. [Google Scholar] [CrossRef]
- Hassanein, M.; Hoeksema, M.D.; Shiota, M.; Qian, J.; Harris, B.K.; Chen, H.; Clark, J.E.; Alborn, W.E.; Eisenberg, R.; Massion, P.P. SLC1A5 Mediates Glutamine Transport Required for Lung Cancer Cell Growth and Survival. Clin. Cancer Res. 2013, 19, 560–570. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Li, W.; Ling, Z.; Hu, Q.; Fan, Z.; Cheng, B.; Tao, X. ASCT2 overexpression is associated with poor survival of OSCC patients and ASCT2 knockdown inhibited growth of glutamine-addicted OSCC cells. Cancer Med. 2020, 9, 3489–3499. [Google Scholar] [CrossRef] [PubMed]
- Oppedisano, F.; Pochini, L.; Galluccio, M.; Indiveri, C. The glutamine/amino acid transporter (ASCT2) reconstituted in liposomes: Transport mechanism, regulation by ATP and characterization of the glutamine/glutamate antiport. Biochim. Biophys. Acta (BBA) Biomembr. 2007, 1768, 291–298. [Google Scholar] [CrossRef] [PubMed]
- Scalise, M.; Pochini, L.; Console, L.; Losso, M.A.; Indiveri, C. The Human SLC1A5 (ASCT2) Amino Acid Transporter: From Function to Structure and Role in Cell Biology. Front. Cell Dev. Biol. 2018, 6, 96. [Google Scholar] [CrossRef] [PubMed]
- Koppula, P.; Zhang, Y.; Zhuang, L.; Gan, B. Amino acid transporter SLC7A11/xCT at the crossroads of regulating redox homeostasis and nutrient dependency of cancer. Cancer Commun (Lond) 2018, 38, 12. [Google Scholar] [CrossRef]
- Timmerman, L.A.; Holton, T.; Yuneva, M.; Louie, R.J.; Padro, M.; Daemen, A.; Hu, M.; Chan, D.A.; Ethier, S.P.; van Veer, L.J.; et al. Glutamine sensitivity analysis identifies the xCT antiporter as a common triple-negative breast tumor therapeutic target. Cancer Cell 2013, 24, 450–465. [Google Scholar] [CrossRef]
- Cao, M.D.; Lamichhane, S.; Lundgren, S.; Bofin, A.M.; Fjøsne, H.; Giskeødegård, G.F.; Bathen, T.F. Metabolic characterization of triple negative breast cancer. BMC Cancer 2014, 14, 941. [Google Scholar] [CrossRef]
- McGivan, J.D.; Bungard, C.I. The transport of glutamine into mammalian cells. Front Biosci. 2007, 12, 874–882. [Google Scholar] [CrossRef]
- Eagle, H. Nutrition Needs of Mammalian Cells in Tissue Culture. Science 2006, 122, 501–504. [Google Scholar] [CrossRef]
- Fuchs, B.C.; Bode, B.P. Amino acid transporters ASCT2 and LAT1 in cancer: Partners in crime? Semin. Cancer Biol. 2005, 15, 254–266. [Google Scholar] [CrossRef]
- Moreadith, R.W.; Lehninger, A.L. The pathways of glutamate and glutamine oxidation by tumor cell mitochondria. Role of mitochondrial NAD(P)+-dependent malic enzyme. J. Biol. Chem. 1984, 259, 6215–6221. [Google Scholar] [PubMed]
- Nagarajan, A.; Malvi, P.; Wajapeyee, N. Oncogene-Directed Alterations in Cancer Cell Metabolism. Trends Cancer 2016, 2, 365–377. [Google Scholar] [CrossRef] [PubMed]
- Miller, D.M.; Thomas, S.D.; Islam, A.; Muench, D.; Sedoris, K. c-Myc and Cancer Metabolism. Clin. Cancer Res. 2012, 18, 5546–5553. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Venneti, S.; Nagrath, D. Glutaminolysis: A Hallmark of Cancer Metabolism. Annu. Rev. Biomed. Eng. 2017, 19, 163–194. [Google Scholar] [CrossRef] [PubMed]
- Medina, M.A. Glutamine and cancer. J. Nutr. 2001, 131, 2539S–2542S. [Google Scholar] [CrossRef] [PubMed]
- Csibi, A.; Fendt, S.-M.; Li, C.; Poulogiannis, G.; Choo, A.Y.; Chapski, D.J.; Jeong, S.M.; Dempsey, J.M.; Parkhitko, A.; Morrison, T.; et al. The mTORC1 Pathway Stimulates Glutamine Metabolism and Cell Proliferation by Repressing SIRT4. Cell 2013, 153, 840–854. [Google Scholar] [CrossRef] [PubMed]
- Wise, D.R.; DeBerardinis, R.J.; Mancuso, A.; Sayed, N.; Zhang, X.-Y.; Pfeiffer, H.K.; Nissim, I.; Daikhin, E.; Yudkoff, M.; McMahon, S.B.; et al. Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proc. Natl. Acad. Sci. USA 2008, 105, 18782–18787. [Google Scholar] [CrossRef]
- Esslinger, C.S.; Cybulski, K.A.; Rhoderick, J.F. Ngamma-aryl glutamine analogues as probes of the ASCT2 neutral amino acid transporter binding site. Bioorg. Med. Chem. 2005, 13, 1111–1118. [Google Scholar] [CrossRef]
- Nicklin, P.; Bergman, P.; Zhang, B.; Triantafellow, E.; Wang, H.; Nyfeler, B.; Yang, H.; Hild, M.; Kung, C.; Wilson, C.; et al. Bidirectional Transport of Amino Acids Regulates mTOR and Autophagy. Cell 2009, 136, 521–534. [Google Scholar] [CrossRef]
- Duran, R.V.; Oppliger, W.; Robitaille, A.M.; Heiserich, L.; Skendaj, R.; Gottlieb, E.; Hall, M.N. Glutaminolysis activates Rag-mTORC1 signaling. Mol. Cell 2012, 47, 349–358. [Google Scholar] [CrossRef]
- Okazaki, S.; Umene, K.; Yamasaki, J.; Suina, K.; Otsuki, Y.; Yoshikawa, M.; Minami, Y.; Masuko, T.; Kawaguchi, S.; Nakayama, H.; et al. Glutaminolysis-related genes determine sensitivity to xCT-targeted therapy in head and neck squamous cell carcinoma. Cancer Sci. 2019, 110, 3453–3463. [Google Scholar] [CrossRef] [PubMed]
- Toyoda, M.; Kaira, K.; Ohshima, Y.; Ishioka, N.S.; Shino, M.; Sakakura, K.; Takayasu, Y.; Takahashi, K.; Tominaga, H.; Oriuchi, N.; et al. Prognostic significance of amino-acid transporter expression (LAT1, ASCT2, and xCT) in surgically resected tongue cancer. Br. J. Cancer 2014, 110, 2506–2513. [Google Scholar] [CrossRef] [PubMed]
- Han, S.; Huang, T.; Li, W.; Wang, X.; Wu, X.; Liu, S.; Yang, W.; Shi, Q.; Li, H.; Hou, F. Prognostic Value of CD44 and Its Isoforms in Advanced Cancer: A Systematic Meta-Analysis With Trial Sequential Analysis. Front. Oncol. 2019, 9, 39. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Holst, J. L-type amino acid transport and cancer: Targeting the mTORC1 pathway to inhibit neoplasia. Am. J. Cancer Res. 2015, 5, 1281–1294. [Google Scholar]
- Bodoor, K.; Almomani, R.; Alqudah, M.; Haddad, Y.; Samouri, W. LAT1 (SLC7A5) Overexpression in Negative Her2 Group of Breast Cancer: A Potential Therapy Target. Asian Pac. J. Cancer Prev. 2020, 21, 1453–1458. [Google Scholar] [CrossRef]
- Barollo, S.; Bertazza, L.; Watutantrige-Fernando, S.; Censi, S.; Cavedon, E.; Galuppini, F.; Pennelli, G.; Fassina, A.; Citton, M.; Rubin, B.; et al. Overexpression of L-Type Amino Acid Transporter 1 (LAT1) and 2 (LAT2): Novel Markers of Neuroendocrine Tumors. PLoS ONE 2016, 11, e0156044. [Google Scholar] [CrossRef]
- Yoshida, G.J. Beyond the Warburg Effect: N-Myc Contributes to Metabolic Reprogramming in Cancer Cells. Front. Oncol. 2020, 10, 791. [Google Scholar] [CrossRef]
- Nakamura, E.; Sato, M.; Yang, H.; Miyagawa, F.; Harasaki, M.; Tomita, K.; Matsuoka, S.; Noma, A.; Iwai, K.; Minato, N. 4F2 (CD98) Heavy Chain Is Associated Covalently with an Amino Acid Transporter and Controls Intracellular Trafficking and Membrane Topology of 4F2 Heterodimer. J. Biol. Chem. 1999, 274, 3009–3016. [Google Scholar] [CrossRef]
- Kanai, Y.; Segawa, H.; Miyamoto, K.-I.; Uchino, H.; Takeda, E.; Endou, H. Expression Cloning and Characterization of a Transporter for Large Neutral Amino Acids Activated by the Heavy Chain of 4F2 Antigen (CD98). J. Biol. Chem. 1998, 273, 23629–23632. [Google Scholar] [CrossRef]
- Mastroberardino, L.; Spindler, B.; Pfeiffer, R.; Skelly, P.J.; Loffing, J.; Shoemaker, C.B.; Verrey, F. Amino-acid transport by heterodimers of 4F2hc/CD98 and members of a permease family. Nat. Cell Biol. 1998, 395, 288–291. [Google Scholar] [CrossRef]
- Meier, C.; Ristic, Z.; Klauser, S.; Verrey, F. Activation of system L heterodimeric amino acid exchangers by intracellular substrates. EMBO J. 2002, 21, 580–589. [Google Scholar] [CrossRef] [PubMed]
- Yanagida, O.; Kanai, Y.; Chairoungdua, A.; Kim, D.K.; Segawa, H.; Nii, T.; Cha, S.H.; Matsuo, H.; Fukushima, J.-I.; Fukasawa, Y.; et al. Human L-type amino acid transporter 1 (LAT1): Characterization of function and expression in tumor cell lines. Biochim. Biophys. Acta (BBA) Biomembr. 2001, 1514, 291–302. [Google Scholar] [CrossRef]
- Salisbury, T.B.; Arthur, S. The Regulation and Function of the L-Type Amino Acid Transporter 1 (LAT1) in Cancer. Int. J. Mol. Sci. 2018, 19, 2373. [Google Scholar] [CrossRef] [PubMed]
- Laplante, M.; Sabatini, D.M. mTOR signaling at a glance. J Cell Sci 2009, 122, 3589–3594. [Google Scholar] [CrossRef]
- Ben-Sahra, I.; Manning, B.D. mTORC1 signaling and the metabolic control of cell growth. Curr. Opin. Cell Biol. 2017, 45, 72–82. [Google Scholar] [CrossRef]
- Yoshida, G.J. Therapeutic strategies of drug repositioning targeting autophagy to induce cancer cell death: From pathophysiology to treatment. J. Hematol. Oncol. 2017, 10, 1–14. [Google Scholar] [CrossRef]
- Kato, H.; Nakajima, S.; Saito, Y.; Takahashi, S.; Katoh, R.; Kitamura, M. mTORC1 serves ER stress-triggered apoptosis via selective activation of the IRE1–JNK pathway. Cell Death Differ. 2011, 19, 310–320. [Google Scholar] [CrossRef]
- Saxton, R.A.; Sabatini, D.M. mTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 168, 960–976. [Google Scholar] [CrossRef]
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.-L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 2011, 13, 132–141. [Google Scholar] [CrossRef]
- Chen, R.; Zou, Y.; Mao, D.; Sun, D.; Gao, G.; Shi, J.; Liu, X.; Zhu, C.; Yang, M.; Ye, W.; et al. The general amino acid control pathway regulates mTOR and autophagy during serum/glutamine starvation. J. Cell Biol. 2014, 206, 173–182. [Google Scholar] [CrossRef]
- Park, Y.; Reyna-Neyra, A.; Philippe, L.; Thoreen, C. mTORC1 Balances Cellular Amino Acid Supply with Demand for Protein Synthesis through Post-transcriptional Control of ATF4. Cell Rep. 2017, 19, 1083–1090. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, K.; Anzai, N. Novel therapeutic approaches targeting L-type amino acid transporters for cancer treatment. World J. Gastrointest. Oncol. 2017, 9, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Wullschleger, S.; Loewith, R.J.; Hall, M.N. TOR Signaling in Growth and Metabolism. Cell 2006, 124, 471–484. [Google Scholar] [CrossRef] [PubMed]
- Tokunaga, C.; Yoshino, K.-I.; Yonezawa, K. mTOR integrates amino acid- and energy-sensing pathways. Biochem. Biophys. Res. Commun. 2004, 313, 443–446. [Google Scholar] [CrossRef]
- Beaumatin, F.; O’Prey, J.; Barthet, V.J.; Zunino, B.; Parvy, J.-P.; Bachmann, A.M.; O’Prey, M.; Kania, E.; Gonzalez, P.S.; MacIntosh, R.; et al. mTORC1 Activation Requires DRAM-1 by Facilitating Lysosomal Amino Acid Efflux. Mol. Cell 2019, 76, 163–176. [Google Scholar] [CrossRef]
- Yoshida, G.J.; Saya, H. EpCAM expression in the prostate cancer makes the difference in the response to growth factors. Biochem. Biophys. Res. Commun. 2014, 443, 239–245. [Google Scholar] [CrossRef]
- Xu, D.; Hemler, M.E. Metabolic Activation-related CD147-CD98 Complex. Mol. Cell. Proteom. 2005, 4, 1061–1071. [Google Scholar] [CrossRef]
- Tayama, S.; Motohara, T.; Narantuya, D.; Li, C.; Fujimoto, K.; Sakaguchi, I.; Tashiro, H.; Saya, H.; Nagano, O.; Katabuchi, H. The impact of EpCAM expression on response to chemotherapy and clinical outcomes in patients with epithelial ovarian cancer. Oncotarget 2017, 8, 44312–44325. [Google Scholar] [CrossRef]
- Betsunoh, H.; Fukuda, T.; Anzai, N.; Nishihara, D.; Mizuno, T.; Yuki, H.; Masuda, A.; Yamaguchi, Y.; Abe, H.; Yashi, M.; et al. Increased expression of system large amino acid transporter (LAT)-1 mRNA is associated with invasive potential and unfavorable prognosis of human clear cell renal cell carcinoma. BMC Cancer 2013, 13, 509. [Google Scholar] [CrossRef]
- Yanagisawa, N.; Ichinoe, M.; Mikami, T.; Nakada, N.; Hana, K.; Koizumi, W.; Endou, H.; Okayasu, I. High expression of L-type amino acid transporter 1 (LAT1) predicts poor prognosis in pancreatic ductal adenocarcinomas. J. Clin. Pathol. 2012, 65, 1019–1023. [Google Scholar] [CrossRef]
- Kaira, K.; Nakamura, K.; Hirakawa, T.; Imai, H.; Tominaga, H.; Oriuchi, N.; Nagamori, S.; Kanai, Y.; Tsukamoto, N.; Oyama, T.; et al. Prognostic significance of L-type amino acid transporter 1 (LAT1) expression in patients with ovarian tumors. Am. J. Transl. Res. 2015, 7, 1161–1171. [Google Scholar] [PubMed]
- Kaira, K.; Arakawa, K.; Shimizu, K.; Oriuchi, N.; Nagamori, S.; Kanai, Y.; Oyama, T.; Takeyoshi, I. Relationship between CD147 and expression of amino acid transporters (LAT1 and ASCT2) in patients with pancreatic cancer. Am. J. Transl. Res. 2015, 7, 356–363. [Google Scholar] [PubMed]
- Yazawa, T.; Shimizu, K.; Kaira, K.; Nagashima, T.; Ohtaki, Y.; Atsumi, J.; Obayashi, K.; Nagamori, S.; Kanai, Y.; Oyama, T.; et al. Clinical significance of coexpression of L-type amino acid transporter 1 (LAT1) and ASC amino acid transporter 2 (ASCT2) in lung adenocarcinoma. Am. J. Transl. Res. 2015, 7, 1126–1139. [Google Scholar] [PubMed]
- Kaira, K.; Oriuchi, N.; Imai, H.; Shimizu, K.; Yanagitani, N.; Sunaga, N.; Hisada, T.; Kawashima, O.; Iijima, H.; Ishizuka, T.; et al. Expression of L-type amino acid transporter 1 (LAT1) in neuroendocrine tumors of the lung. Pathol. Res. Pr. 2008, 204, 553–561. [Google Scholar] [CrossRef] [PubMed]
- Kaira, K.; Oriuchi, N.; Imai, H.; Shimizu, K.; Yanagitani, N.; Sunaga, N.; Hisada, T.; Tanaka, S.; Ishizuka, T.; Kanai, Y.; et al. Prognostic significance of L-type amino acid transporter 1 expression in resectable stage I–III nonsmall cell lung cancer. Br. J. Cancer 2008, 98, 742–748. [Google Scholar] [CrossRef] [PubMed]
- Ichinoe, M.; Yanagisawa, N.; Mikami, T.; Hana, K.; Nakada, N.; Endou, H.; Okayasu, I.; Murakumo, Y. L-type amino acid transporter 1 (LAT1) expression in lymph node metastasis of gastric carcinoma: Its correlation with size of metastatic lesion and Ki-67 labeling. Pathol. Res. Pr. 2015, 211, 533–538. [Google Scholar] [CrossRef]
- Shi, L.; Luo, W.; Huang, W.; Huang, S.; Huang, G. Downregulation of L-type amino acid transporter 1 expression inhibits the growth, migration and invasion of gastric cancer cells. Oncol. Lett. 2013, 6, 106–112. [Google Scholar] [CrossRef]
- Janpipatkul, K.; Suksen, K.; Borwornpinyo, S.; Jearawiriyapaisarn, N.; Hongeng, S.; Piyachaturawat, P.; Chairoungdua, A. Downregulation of LAT1 expression suppresses cholangiocarcinoma cell invasion and migration. Cell. Signal. 2014, 26, 1668–1679. [Google Scholar] [CrossRef]
- Kaji, M.; Kabir-Salmani, M.; Anzai, N.; Jin, C.J.; Akimoto, Y.; Horita, A.; Sakamoto, A.; Kanai, Y.; Sakurai, H.; Iwashita, M. Properties of L-Type Amino Acid Transporter 1 in Epidermal Ovarian Cancer. Int. J. Gynecol. Cancer 2010, 20, 329–336. [Google Scholar] [CrossRef]
- Miranda-Gonçalves, V.; Honavar, M.; Pinheiro, C.; Martinho, O.; Pires, M.M.; Pinheiro, C.; Cordeiro, M.; Bebiano, G.; Costa, P.; Palmeirim, I.; et al. Monocarboxylate transporters (MCTs) in gliomas: Expression and exploitation as therapeutic targets. Neuro-Oncology 2012, 15, 172–188. [Google Scholar] [CrossRef]
- Fang, J.; Quinones, Q.J.; Holman, T.L.; Morowitz, M.J.; Wang, Q.; Zhao, H.; Sivo, F.; Maris, J.M.; Wahl, M.L. The H+-Linked Monocarboxylate Transporter (MCT1/SLC16A1): A Potential Therapeutic Target for High-Risk Neuroblastoma. Mol. Pharmacol. 2006, 70, 2108–2115. [Google Scholar] [CrossRef] [PubMed]
- Pinheiro, C.; Longatto-Filho, A.; Ferreira, L.; Pereira, S.M.M.; Etlinger, D.; Moreira, M.A.R.; Jubé, L.F.; Queiroz, G.S.; Schmitt, F.; Baltazar, F. Increasing Expression of Monocarboxylate Transporters 1 and 4 Along Progression to Invasive Cervical Carcinoma. Int. J. Gynecol. Pathol. 2008, 27, 568–574. [Google Scholar] [CrossRef] [PubMed]
- De Oliveira, A.T.T.; Pinheiro, C.; Longatto-Filho, A.; Brito, M.J.; Martinho, O.; Matos, D.; Carvalho, A.L.; Vazquez, V.D.L.; Silva, T.B.; Neto, C.S.; et al. Co-expression of monocarboxylate transporter 1 (MCT1) and its chaperone (CD147) is associated with low survival in patients with gastrointestinal stromal tumors (GISTs). J. Bioenerg. Biomembr. 2012, 44, 171–178. [Google Scholar] [CrossRef] [PubMed]
- Pinheiro, C.; Longatto-Filho, A.; Scapulatempo, C.; Ferreira, L.; Martins, S.; Pellerin, L.; Rodrigues, M.; Alves, V.A.F.; Schmitt, F.; Baltazar, F. Increased expression of monocarboxylate transporters 1, 2, and 4 in colorectal carcinomas. Virchows Arch. 2008, 452, 139–146. [Google Scholar] [CrossRef]
- Pinheiro, C.; Albergaria, A.; Paredes, J.; Sousa, B.; Dufloth, R.; Vieira, D.; Schmitt, F.; Baltazar, F. Monocarboxylate transporter 1 is up-regulated in basal-like breast carcinoma. Histopathology 2010, 56, 860–867. [Google Scholar] [CrossRef]
- Pértega-Gomes, N.; Vizcaíno, J.R.; Miranda-Gonçalves, V.; Pinheiro, C.; Silva, J.; Pereira, H.; Monteiro, P.; Henrique, R.; Reis, R.M.; Lopes, C.; et al. Monocarboxylate transporter 4 (MCT4) and CD147 overexpression is associated with poor prognosis in prostate cancer. BMC Cancer 2011, 11, 312. [Google Scholar] [CrossRef]
- Gerlinger, M.; Santos, C.R.; Spencer-Dene, B.; Martinez, P.; Endesfelder, D.; Burrell, R.A.; Vetter, M.; Jiang, M.; Saunders, R.E.; Kelly, G.; et al. Genome-wide RNA interference analysis of renal carcinoma survival regulators identifies MCT4 as a Warburg effect metabolic target. J. Pathol. 2012, 227, 146–156. [Google Scholar] [CrossRef]
- Doherty, J.R.; Cleveland, J.L. Targeting lactate metabolism for cancer therapeutics. J. Clin. Investig. 2013, 123, 3685–3692. [Google Scholar] [CrossRef]
- Payen, V.L.; Mina, E.; Van Hée, V.F.; Porporato, P.E.; Sonveaux, P. Monocarboxylate transporters in cancer. Mol. Metab. 2020, 33, 48–66. [Google Scholar] [CrossRef]
- De Saedeleer, C.; Porporato, P.; Copetti, T.; Escuredo, J.P.; Payen, V.; Brisson, L.; Feron, O.; Sonveaux, P. Glucose deprivation increases monocarboxylate transporter 1 (MCT1) expression and MCT1-dependent tumor cell migration. Oncogene 2014, 33, 4060–4068. [Google Scholar] [CrossRef]
- Fan, Q.; Yang, L.; Zhang, X.; Ma, Y.; Li, Y.; Dong, L.; Zong, Z.; Hua, X.; Su, D.; Li, H.; et al. Autophagy promotes metastasis and glycolysis by upregulating MCT1 expression and Wnt/beta-catenin signaling pathway activation in hepatocellular carcinoma cells. J. Exp. Clin. Cancer Res. 2018, 37, 9. [Google Scholar] [CrossRef] [PubMed]
- Hao, J.; Chen, H.; Madigan, M.C.; Cozzi, P.J.; Beretov, J.; Xiao, W.; Delprado, W.J.; Russell, P.J.; Li, Y. Co-expression of CD147 (EMMPRIN), CD44v3-10, MDR1 and monocarboxylate transporters is associated with prostate cancer drug resistance and progression. Br. J. Cancer 2010, 103, 1008–1018. [Google Scholar] [CrossRef] [PubMed]
- Slomiany, M.G.; Grass, G.D.; Robertson, A.D.; Yang, X.Y.; Maria, B.L.; Beeson, C.; Toole, B.P. Hyaluronan, CD44, and Emmprin Regulate Lactate Efflux and Membrane Localization of Monocarboxylate Transporters in Human Breast Carcinoma Cells. Cancer Res. 2009, 69, 1293–1301. [Google Scholar] [CrossRef] [PubMed]
- Kirk, P.; Wilson, M.; Heddle, C.; Brown, M.; Barclay, A.; Halestrap, A.P. CD147 is tightly associated with lactate transporters MCT1 and MCT4 and facilitates their cell surface expression. EMBO J. 2000, 19, 3896–3904. [Google Scholar] [CrossRef] [PubMed]
- Gallagher, S.M.; Castorino, J.J.; Wang, D.; Philp, N.J. Monocarboxylate Transporter 4 Regulates Maturation and Trafficking of CD147 to the Plasma Membrane in the Metastatic Breast Cancer Cell Line MDA-MB-231. Cancer Res. 2007, 67, 4182–4189. [Google Scholar] [CrossRef] [PubMed]
- Ke, X.; Fei, F.; Chen, Y.; Xu, L.; Zhang, Z.; Huang, Q.; Zhang, H.; Yang, H.; Chen, Z.; Xing, J. Hypoxia upregulates CD147 through a combined effect of HIF-1α and Sp1 to promote glycolysis and tumor progression in epithelial solid tumors. Carcinogenesis 2012, 33, 1598–1607. [Google Scholar] [CrossRef]
- Sprowl-Tanio, S.; Habowski, A.N.; Pate, K.T.; McQuade, M.M.; Wang, K.; Edwards, R.A.; Grun, F.; Lyou, Y.; Waterman, M.L. Lactate/pyruvate transporter MCT-1 is a direct Wnt target that confers sensitivity to 3-bromopyruvate in colon cancer. Cancer Metab. 2016, 4, 1–18. [Google Scholar] [CrossRef]
- Li, K.K.W.; Pang, J.C.-S.; Ching, A.K.-K.; Wong, C.K.; Kong, X.; Wang, Y.; Zhou, L.; Chen, Z.; Ng, H. miR-124 is frequently down-regulated in medulloblastoma and is a negative regulator of SLC16A1. Hum. Pathol. 2009, 40, 1234–1243. [Google Scholar] [CrossRef]
- Romero-Cordoba, S.; Rodriguez-Cuevas, S.; Bautista-Pina, V.; Maffuz-Aziz, A.; D’Ippolito, E.; Cosentino, G.; Baroni, S.; Iorio, M.V.; Hidalgo-Miranda, A. Loss of function of miR-342-3p results in MCT1 over-expression and contributes to oncogenic metabolic reprogramming in triple negative breast cancer. Sci. Rep. 2018, 8, 1–16. [Google Scholar] [CrossRef]
- Xu, W.; Zhang, Z.; Zou, K.; Cheng, Y.; Yang, M.; Chen, H.; Wang, H.; Zhao, J.; Chen, P.; He, L.; et al. MiR-1 suppresses tumor cell proliferation in colorectal cancer by inhibition of Smad3-mediated tumor glycolysis. Cell Death Dis. 2017, 8, e2761. [Google Scholar] [CrossRef]
- Pavlides, S.; Tsirigos, A.; Vera, I.; Frank, P.; Casimiro, M.C.; Addya, S.; Sotgia, F.; Flomenberg, N.; Wang, C.; Fortina, P.; et al. Loss of stromal caveolin-1 leads to oxidative stress, mimics hypoxia and drives inflammation in the tumor microenvironment, conferring the “reverse Warburg effect”: A transcriptional informatics analysis with validation. Cell Cycle 2010, 9, 2201–2219. [Google Scholar] [CrossRef] [PubMed]
- Pértega-Gomes, N.; Vizcaíno, J.R.; Attig, J.; Jurmeister, S.; Lopes, C.; Baltazar, F. A lactate shuttle system between tumour and stromal cells is associated with poor prognosis in prostate cancer. BMC Cancer 2014, 14, 352. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Escuredo, J.; Dadhich, R.K.; Dhup, S.; Cacace, A.; Van Hée, V.F.; De Saedeleer, C.J.; Sboarina, M.; Rodriguez, F.; Fontenille, M.-J.; Brisson, L.; et al. Lactate promotes glutamine uptake and metabolism in oxidative cancer cells. Cell Cycle 2016, 15, 72–83. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. Tumor metabolism: Cancer cells give and take lactate. J. Clin. Investig. 2008, 118, 3835–3837. [Google Scholar] [CrossRef] [PubMed]
- Sonveaux, P.; Végran, F.; Schroeder, T.; Wergin, M.C.; Verrax, J.; Rabbani, Z.N.; De Saedeleer, C.J.; Kennedy, K.M.; Diepart, C.; Jordan, B.F.; et al. Targeting lactate-fueled respiration selectively kills hypoxic tumor cells in mice. J. Clin. Investig. 2008, 118, 3930–3942. [Google Scholar] [CrossRef] [PubMed]
- Rho, J.M. Inhibition of Lactate Dehydrogenase to Treat Epilepsy. N. Engl. J. Med. 2015, 373, 187–189. [Google Scholar] [CrossRef] [PubMed]
- Cahu, J.; Bustany, S.; Sola, B. Senescence-associated secretory phenotype favors the emergence of cancer stem-like cells. Cell Death Dis. 2012, 3, e446. [Google Scholar] [CrossRef]
- Ogasawara, M.; Zhang, H. Redox Regulation and Its Emerging Roles in Stem Cells and Stem-Like Cancer Cells. Antioxid. Redox Signal. 2009, 11, 1107–1122. [Google Scholar] [CrossRef]
- Yoshida, G.J.; Saya, H. Inversed relationship between CD44 variant and c-Myc due to oxidative stress-induced canonical Wnt activation. Biochem. Biophys. Res. Commun. 2014, 443, 622–627. [Google Scholar] [CrossRef]
- Rich, J.N. Cancer stem cells: Understanding tumor hierarchy and heterogeneity. Medicine (Baltimore) 2016, 95, S2–S7. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yoshida, G.J. The Harmonious Interplay of Amino Acid and Monocarboxylate Transporters Induces the Robustness of Cancer Cells. Metabolites 2021, 11, 27. https://doi.org/10.3390/metabo11010027
Yoshida GJ. The Harmonious Interplay of Amino Acid and Monocarboxylate Transporters Induces the Robustness of Cancer Cells. Metabolites. 2021; 11(1):27. https://doi.org/10.3390/metabo11010027
Chicago/Turabian StyleYoshida, Go J. 2021. "The Harmonious Interplay of Amino Acid and Monocarboxylate Transporters Induces the Robustness of Cancer Cells" Metabolites 11, no. 1: 27. https://doi.org/10.3390/metabo11010027
APA StyleYoshida, G. J. (2021). The Harmonious Interplay of Amino Acid and Monocarboxylate Transporters Induces the Robustness of Cancer Cells. Metabolites, 11(1), 27. https://doi.org/10.3390/metabo11010027