Metabolic Dynamics of In Vitro CD8+ T Cell Activation


Abstract
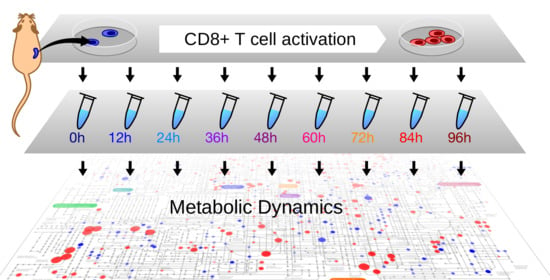
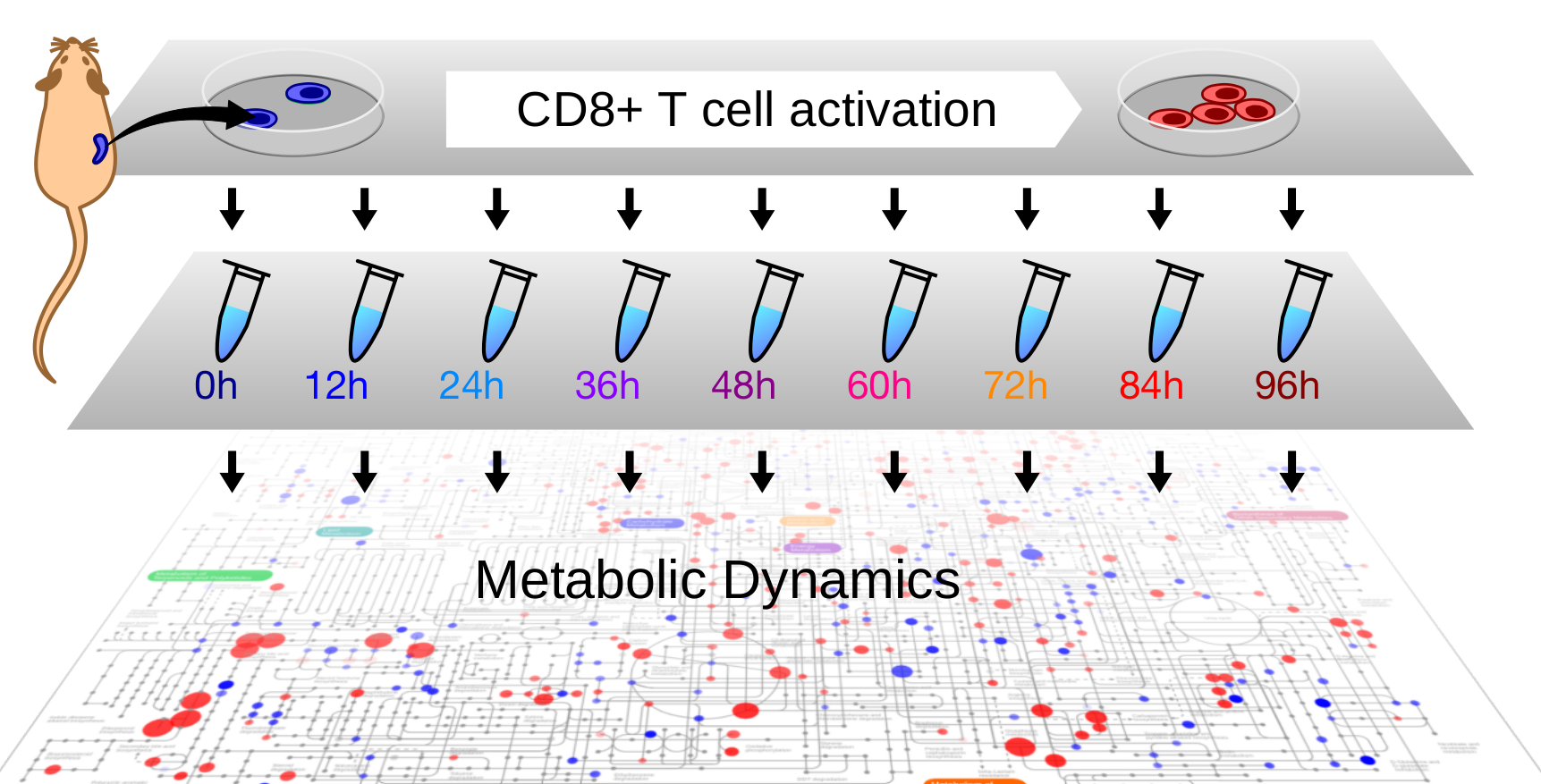
1. Introduction
2. Results
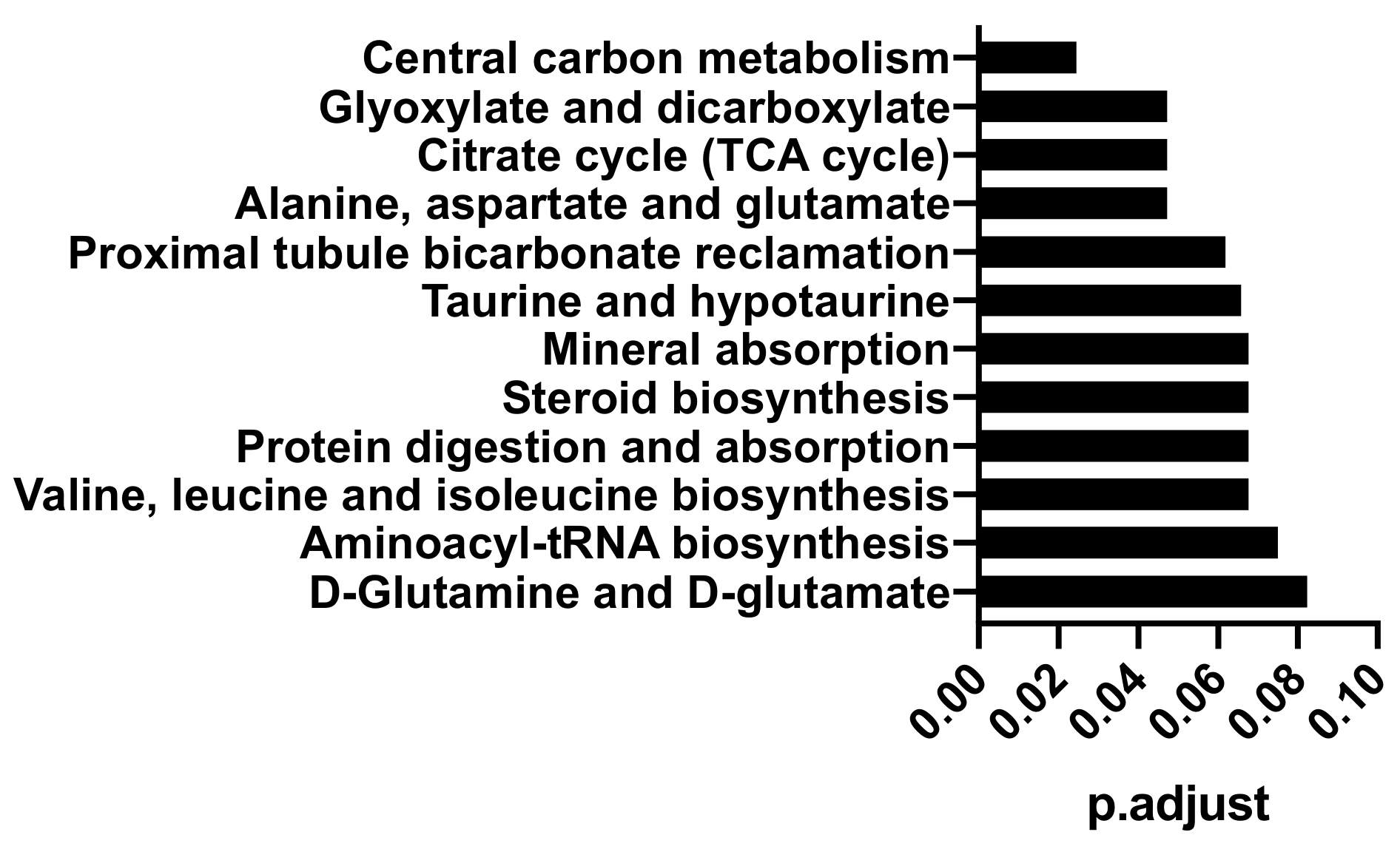
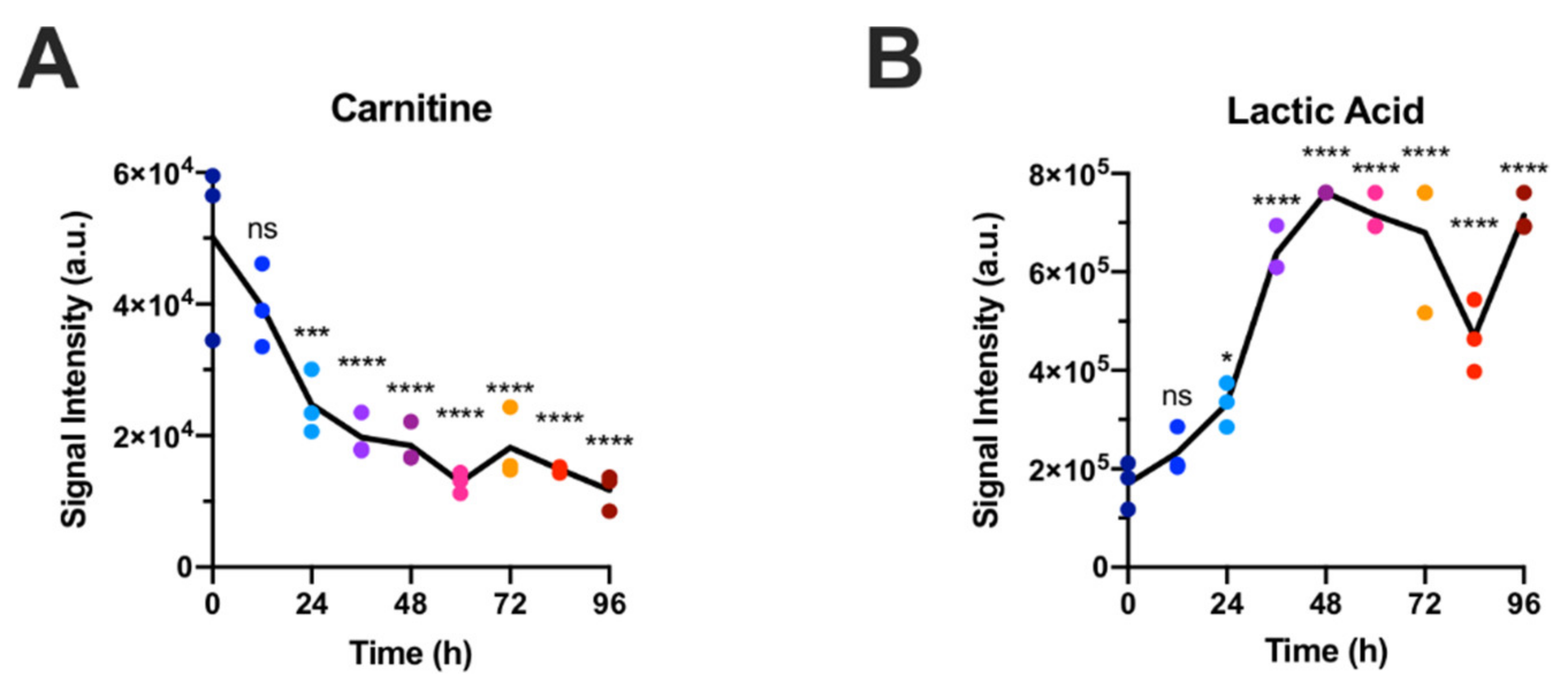
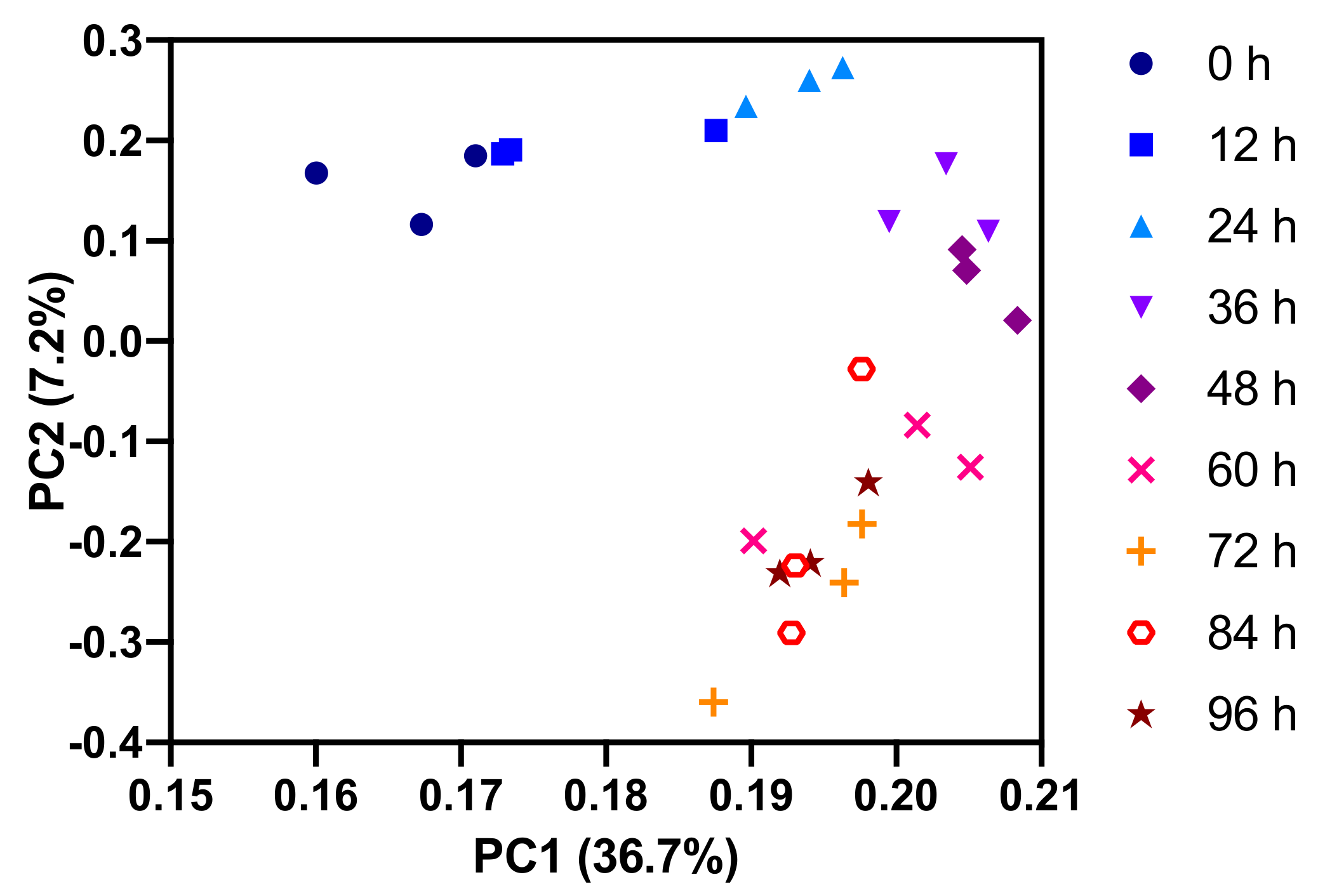
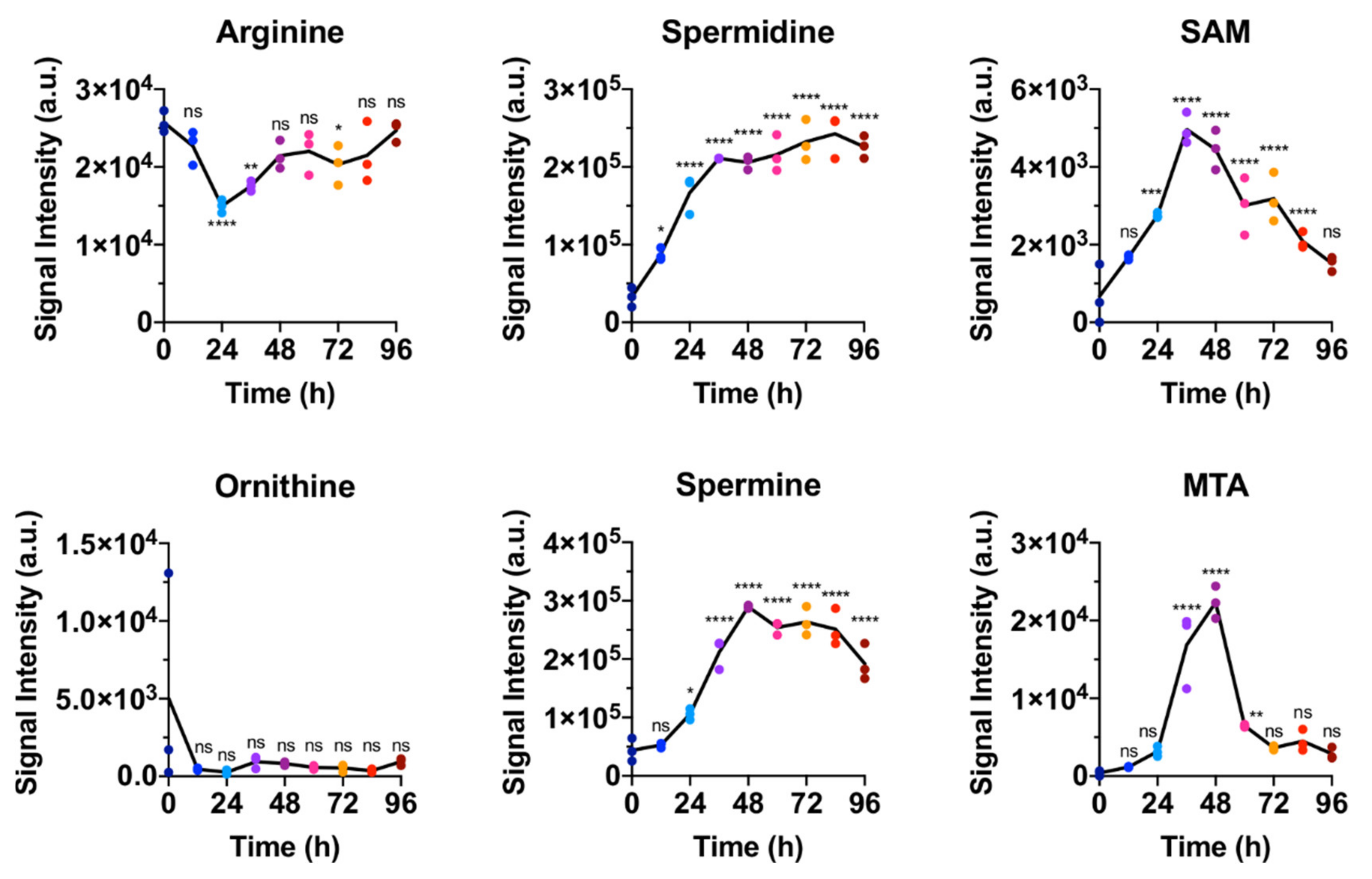
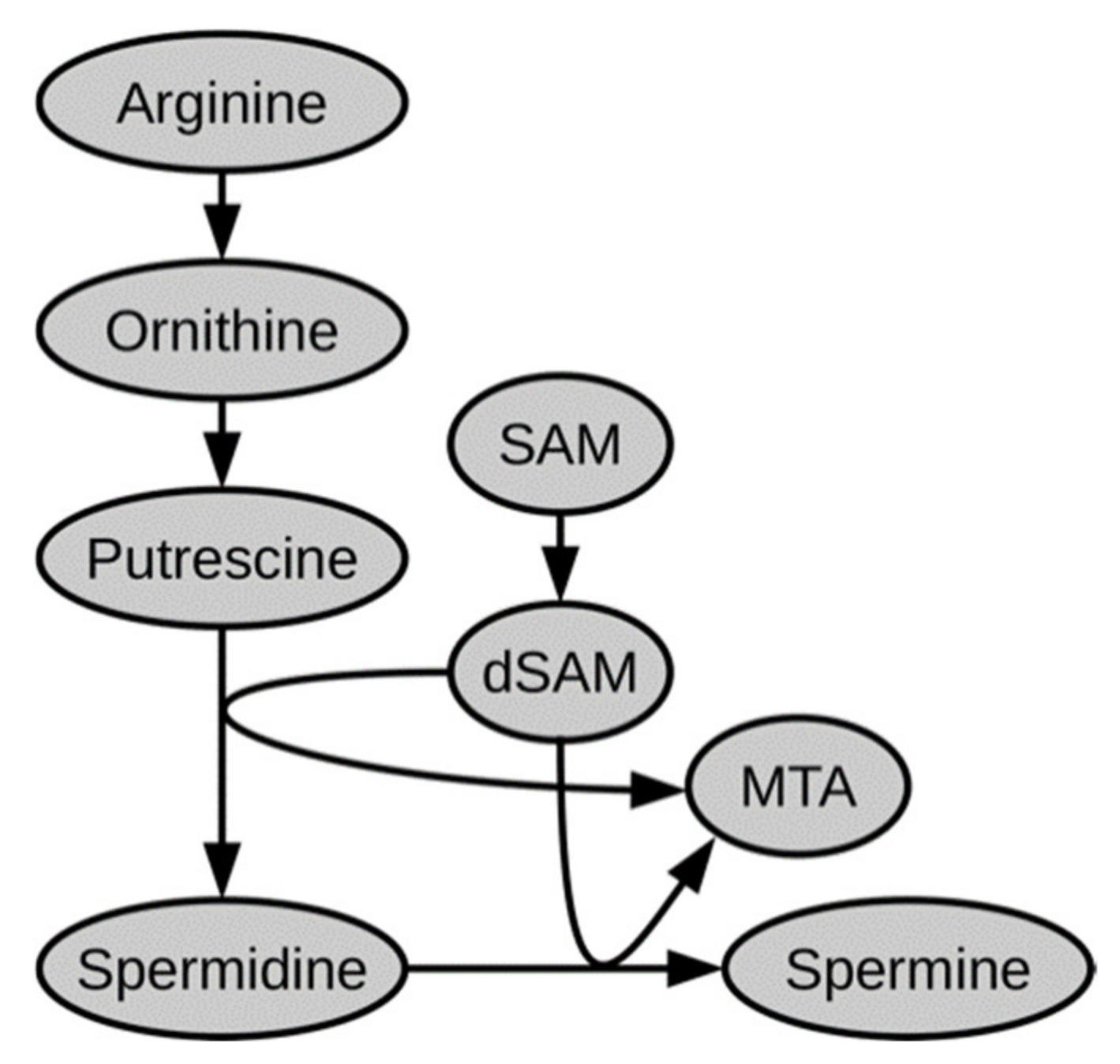
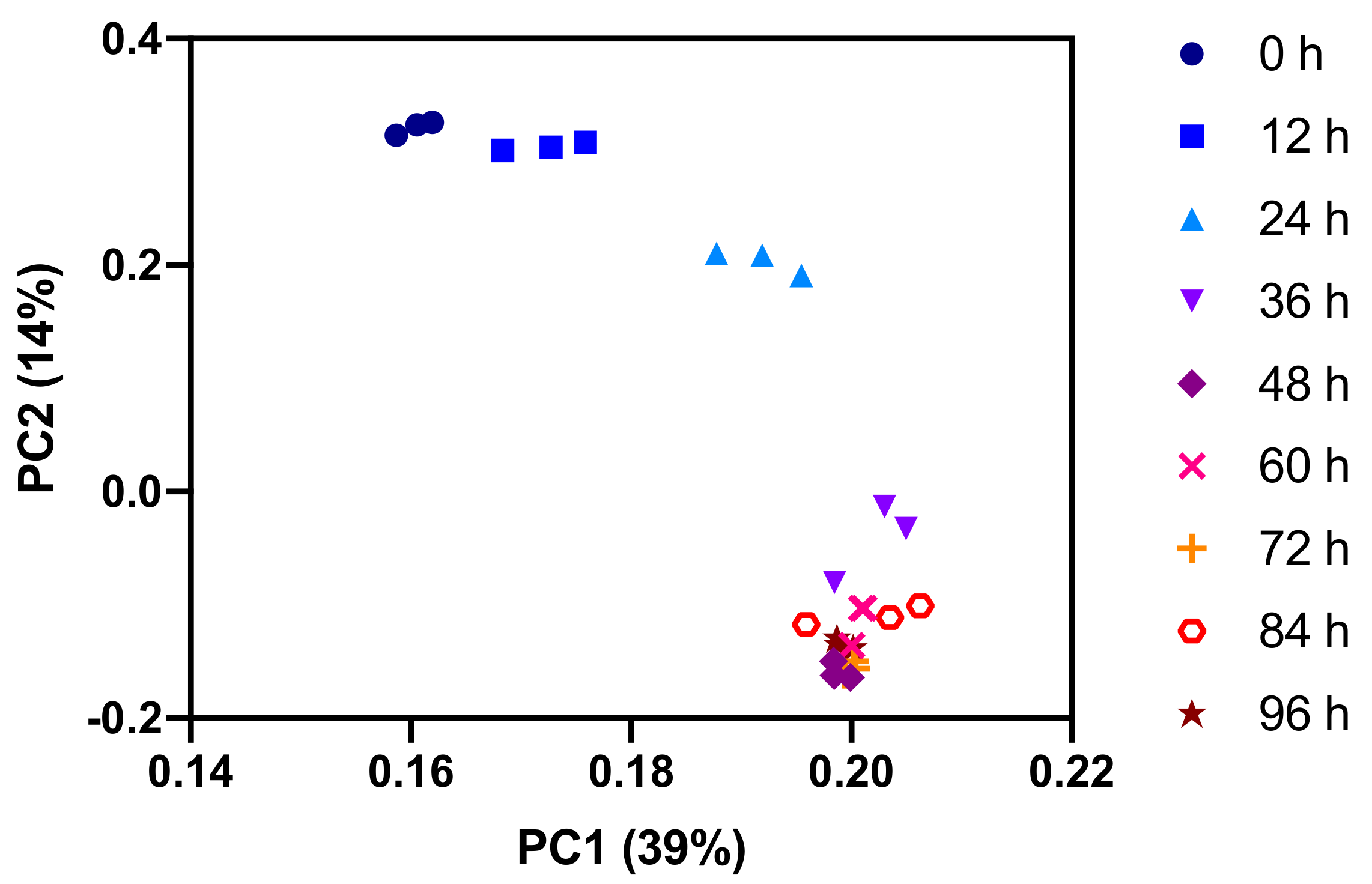
2.1. Polar Metabolites
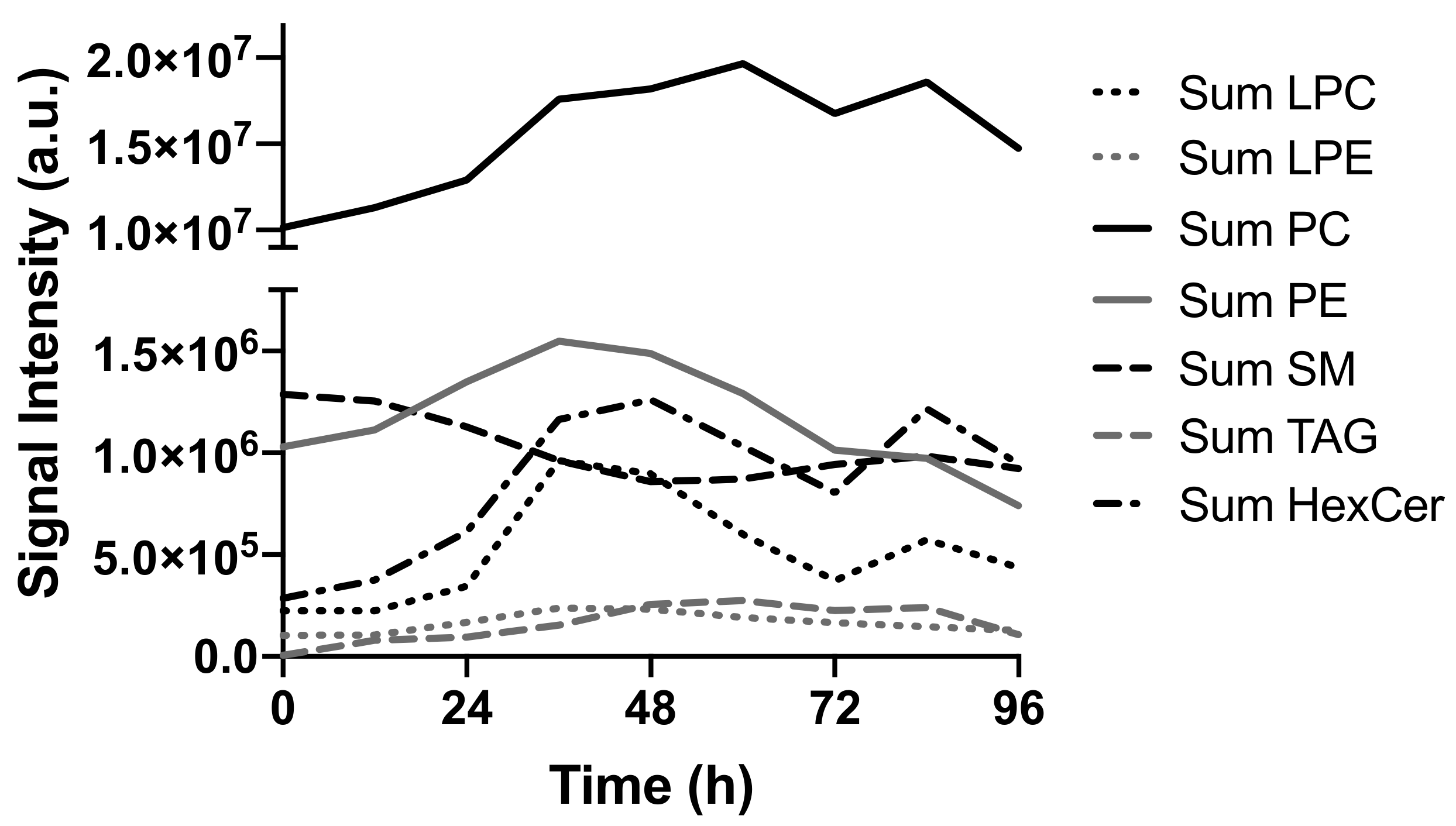
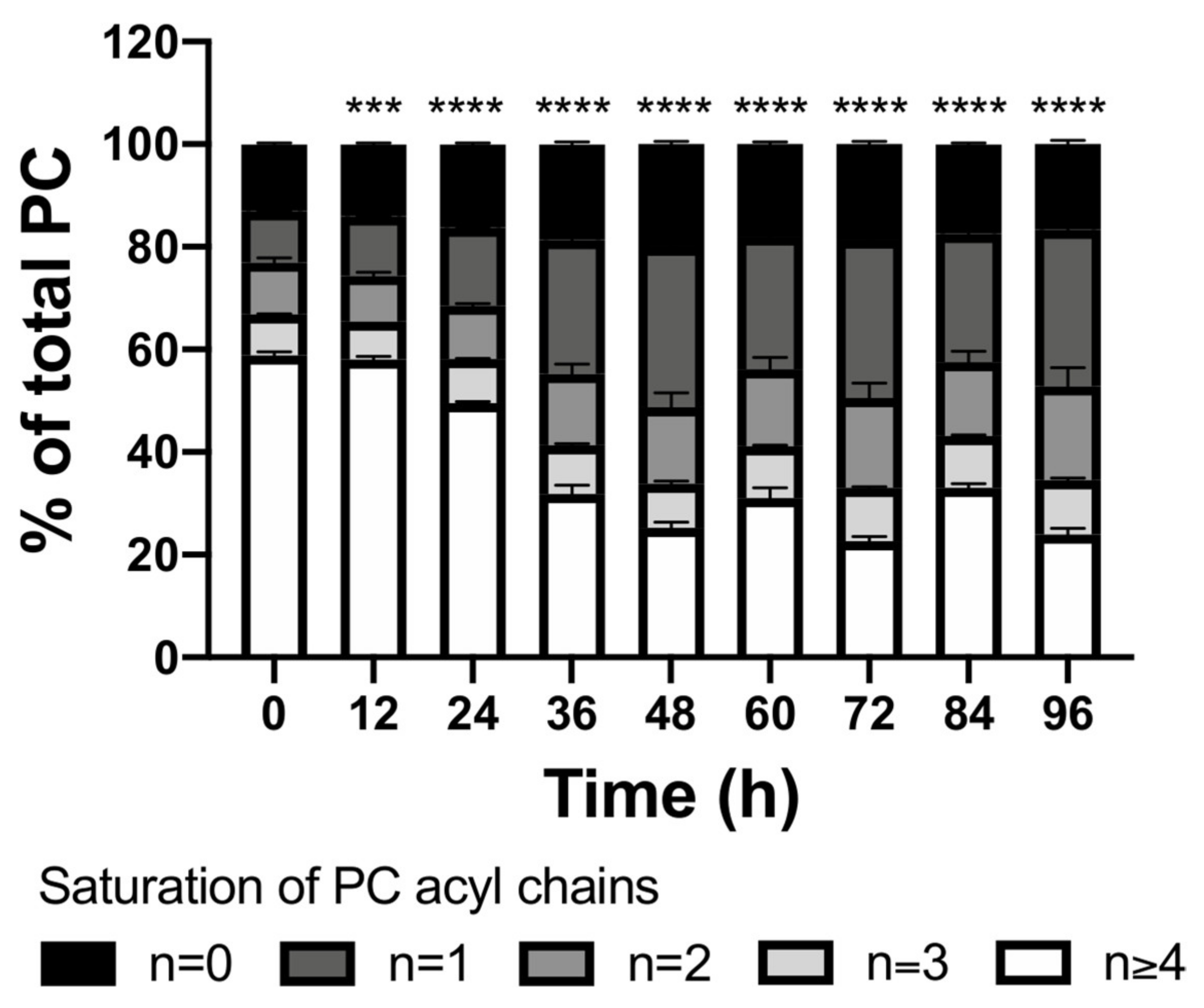
2.2. Lipids
3. Discussion
4. Materials and Methods
4.1. Mouse Lines
4.2. Primary T Cell Cultures
4.3. Sample Prep
4.4. FIA
4.5. LC-QTOF-HILIC
4.6. LC-QTOF-polarRP
4.7. LC-QTOF-Lipid
4.8. LC-MS/MS Data Processing
4.9. Statistical Analysis
4.10. Flow Cytometry
4.11. Graphical Abstract
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhang, N.; Bevan, M.J. CD8(+) T cells: Foot soldiers of the immune system. Immunity 2011, 35, 161–168. [Google Scholar] [CrossRef]
- Shyer, J.A.; Flavell, R.A.; Bailis, W. Metabolic signaling in T cells. Cell Res. 2020, 30, 649–659. [Google Scholar] [CrossRef]
- Chang, C.-H.; Qiu, J.; O’Sullivan, D.; Buck, M.D.; Noguchi, T.; Curtis, J.D.; Chen, Q.; Gindin, M.; Gubin, M.M.; van der Windt, G.J.W.; et al. Metabolic Competition in the Tumor Microenvironment Is a Driver of Cancer Progression. Cell 2015, 162, 1229–1241. [Google Scholar] [CrossRef]
- Ma, X.; Bi, E.; Lu, Y.; Su, P.; Huang, C.; Liu, L.; Wang, Q.; Yang, M.; Kalady, M.F.; Qian, J.; et al. Cholesterol Induces CD8+ T Cell Exhaustion in the Tumor Microenvironment. Cell Metab. 2019, 30, 143–156.e5. [Google Scholar] [CrossRef] [PubMed]
- O’Sullivan, D.; Sanin, D.E.; Pearce, E.J.; Pearce, E.L. Metabolic interventions in the immune response to cancer. Nat. Rev. Immunol. 2019, 19, 324–335. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Shi, X.; Xu, C. Regulation of T cell signalling by membrane lipids. Nat. Rev. Immunol. 2016, 16, 690–701. [Google Scholar] [CrossRef]
- Bennett, B.D.; Kimball, E.H.; Gao, M.; Osterhout, R.; Van Dien, S.J.; Rabinowitz, J.D. Absolute metabolite concentrations and implied enzyme active site occupancy in Escherichia coli. Nat. Chem. Biol. 2009, 5, 593–599. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.W.; Freinkman, E.; Wang, T.; Birsoy, K.; Sabatini, D.M. Absolute Quantification of Matrix Metabolites Reveals the Dynamics of Mitochondrial Metabolism. Cell 2016, 166, 1324–1337.e11. [Google Scholar] [CrossRef] [PubMed]
- Lu, W.; Su, X.; Klein, M.S.; Lewis, I.A.; Fieh, O.; Rabinowitz, J.D. Metabolite Measurement: Pitfalls to Avoid and Practices to Follow | Annual Review of Biochemistry. Annu. Rev. Biochem. 2017, 86, 277–304. [Google Scholar] [CrossRef]
- Kamburov, A.; Stelzl, U.; Lehrach, H.; Herwig, R. The ConsensusPathDB interaction database: 2013 update. Nucleic Acids Res. 2013, 41, D793–D800. [Google Scholar] [CrossRef]
- Ma, E.H.; Verway, M.J.; Johnson, R.M.; Roy, D.G.; Steadman, M.; Hayes, S.; Williams, K.S.; Sheldon, R.D.; Samborska, B.; Kosinski, P.A.; et al. Metabolic Profiling Using Stable Isotope Tracing Reveals Distinct Patterns of Glucose Utilization by Physiologically Activated CD8+ T Cells. Immunity 2019, 51, 856–870.e5. [Google Scholar] [CrossRef]
- Gerriets, V.A.; Rathmell, J.C. Metabolic pathways in T cell fate and function. Trends Immunol. 2012, 33, 168–173. [Google Scholar] [CrossRef]
- O’Brien, K.L.; Assmann, N.; O’Connor, E.; Keane, C.; Walls, J.; Choi, C.; Oefner, P.J.; Gardiner, C.M.; Dettmer, K.; Finlay, D.K. De novo polyamine synthesis supports metabolic and functional responses in activated murine Natural Killer cells. Eur. J. Immunol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Puleston, D.J.; Baixauli, F.; Sanin, D.E.; Villa, M.; Kabat, A.; Kaminski, M.M.; Weiss, H.; Grzes, K.; Flachsmann, L.J.; Field, C.S.; et al. Polyamine Metabolism Regulates the T Cell Epigenome Through Hypusination. bioRxiv Immunol. 2020. [Google Scholar] [CrossRef]
- Puleston, D.J.; Buck, M.D.; Klein Geltink, R.I.; Kyle, R.L.; Caputa, G.; O’Sullivan, D.; Cameron, A.M.; Castoldi, A.; Musa, Y.; Kabat, A.M.; et al. Polyamines and eIF5A Hypusination Modulate Mitochondrial Respiration and Macrophage Activation. Cell Metab. 2019, 30, 352–363. [Google Scholar] [CrossRef] [PubMed]
- Harayama, T.; Riezman, H. Understanding the diversity of membrane lipid composition. Nat. Rev. Mol. Cell Biol. 2018, 19, 281–296. [Google Scholar] [CrossRef] [PubMed]
- Bi, J.; Ichu, T.-A.; Zanca, C.; Yang, H.; Zhang, W.; Gu, Y.; Chowdhry, S.; Reed, A.; Ikegami, S.; Turner, K.M.; et al. Oncogene Amplification in Growth Factor Signaling Pathways Renders Cancers Dependent on Membrane Lipid Remodeling. Cell Metab. 2019, 30, 525–538.e8. [Google Scholar] [CrossRef] [PubMed]
- Grüner, B.M.; Fendt, S.-M. Cancer cells stock up in lymph vessels to survive. Nature 2020, 585, 36–37. [Google Scholar] [CrossRef] [PubMed]
- Ubellacker, J.M.; Tasdogan, A.; Ramesh, V.; Shen, B.; Mitchell, E.C.; Martin-Sandoval, M.S.; Gu, Z.; McCormick, M.L.; Durham, A.B.; Spitz, D.R.; et al. Lymph protects metastasizing melanoma cells from ferroptosis. Nature 2020, 585, 113–118. [Google Scholar] [CrossRef] [PubMed]
- Rudd-Schmidt, J.A.; Hodel, A.W.; Noori, T.; Lopez, J.A.; Cho, H.-J.; Verschoor, S.; Ciccone, A.; Trapani, J.A.; Hoogenboom, B.W.; Voskoboinik, I. Lipid order and charge protect killer T cells from accidental death. Nat. Commun. 2019, 10, 5396. [Google Scholar] [CrossRef] [PubMed]
- Antia, R.; Schlegel, R.A.; Williamson, P. Binding of perforin to membranes is sensitive to lipid spacing and not headgroup. Immunol. Lett. 1992, 32, 153–157. [Google Scholar] [CrossRef]
- Lone, A.M.; Taskén, K. Proinflammatory and immunoregulatory roles of eicosanoids in T cells. Front. Immunol. 2013, 4, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Nicolaou, A.; Mauro, C.; Urquhart, P.; Marelli-Berg, F. Polyunsaturated fatty acid-derived lipid mediators and T cell function. Front. Immunol. 2014, 5, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Fonteh, A.N.; Bass, D.A.; Marshall, L.A.; Seeds, M.; Samet, J.M.; Chilton, F.H. Evidence that secretory phospholipase A2 plays a role in arachidonic acid release and eicosanoid biosynthesis by mast cells. J. Immunol. 1994, 152, 5438–5446. [Google Scholar] [PubMed]
- Abraham, R.T.; McKinney, M.M.; Forray, C.; Shipley, G.D.; Handwerger, B.S. Stimulation of Nachidonic Acid Release and Eicosanoid Biosynthesis in an Interleukin 2-Dependent T Cell Line. Immunopharmacol. Immunotoxicol. 1986, 8, 165–204. [Google Scholar] [CrossRef]
- Bligh, E.G.; Dyer, W.J. Canadian Journal of Biochemistry and Physiology. Can. J. Biochem. Physiol. 1959, 37, 911–917. [Google Scholar] [CrossRef]
- Fuhrer, T.; Heer, D.; Begemann, B.; Zamboni, N. High-throughput, accurate mass metabolome profiling of cellular extracts by flow injection-time-of-flight mass spectrometry. Anal. Chem. 2011, 83, 7074–7080. [Google Scholar] [CrossRef]
- Chambers, M.C.; Maclean, B.; Burke, R.; Amodei, D.; Ruderman, D.L.; Neumann, S.; Gatto, L.; Fischer, B.; Pratt, B.; Egertson, J.; et al. A cross-platform toolkit for mass spectrometry and proteomics. Nat. Biotechnol. 2012, 30, 918–920. [Google Scholar] [CrossRef]
- Wishart, D.S.; Feunang, Y.D.; Marcu, A.; Guo, A.C.; Liang, K.; Vázquez-Fresno, R.; Sajed, T.; Johnson, D.; Li, C.; Karu, N.; et al. HMDB 4.0: The human metabolome database for 2018. Nucleic Acids Res. 2018, 46, D608–D617. [Google Scholar] [CrossRef]
- Bajad, S.U.; Lu, W.; Kimball, E.H.; Yuan, J.; Peterson, C.; Rabinowitz, J.D. Separation and quantitation of water soluble cellular metabolites by hydrophilic interaction chromatography-tandem mass spectrometry. J. Chromatogr. A 2006, 1125, 76–88. [Google Scholar] [CrossRef]
- Paglia, G.; Magnúsdóttir, M.; Thorlacius, S.; Sigurjónsson, Ó.E.; Guðmundsson, S.; Palsson, B.Ø.; Thiele, I. Intracellular metabolite profiling of platelets: Evaluation of extraction processes and chromatographic strategies. J. Chromatogr. B 2012, 898, 111–120. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Sedlak, M.; Regnier, F.E.; Mosier, N.; Ho, N.; Adamec, J. Simultaneous quantification of metabolites involved in central carbon and energy metabolism using reversed-phase liquid chromatography-mass spectrometry and in vitro 13C labeling. Anal. Chem. 2008, 80, 9508–9516. [Google Scholar] [CrossRef]
- Si-Hung, L.; Causon, T.J.; Hann, S. Comparison of fully-wettable RPLC stationary phases for LC-MS-based cellular metabolomics. Electrophoresis 2017, 38, 2287–2295. [Google Scholar] [CrossRef]
- Hermann, G.; Schwaiger, M.; Volejnik, P.; Koellensperger, G. 13C-labelled yeast as internal standard for LC–MS/MS and LC high resolution MS based amino acid quantification in human plasma. J. Pharm. Biomed. Anal. 2018, 155, 329–334. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.; Van Dommelen, J.; Van Der Heljden, R.; Spijksma, G.; Reijmers, T.H.; Wang, M.; Slee, E.; Lu, X.; Xu, G.; Van Der Greef, J.; et al. RPLC-lon-trap-FTMS method for lipid profiling of plasma: Method validation And application to p53 mutant mouse model. J. Proteome Res. 2008, 7, 4982–4991. [Google Scholar] [CrossRef] [PubMed]
- Bird, S.S.; Marur, V.R.; Sniatynski, M.J.; Greenberg, H.K.; Kristal, B.S. Lipidomics profiling by high-resolution LC-MS and high-energy collisional dissociation fragmentation: Focus on characterization of mitochondrial cardiolipins and monolysocardiolipins. Anal. Chem. 2011, 83, 940–949. [Google Scholar] [CrossRef] [PubMed]
- Bolstad, B.M. preprocessCore: A Collection of Pre-Processing Functions. R Package Version 1.50.0. 2020. Available online: https://bioconductor.org/packages/release/bioc/html/preprocessCore.html (accessed on 8 August 2020).
- Mevik, B.-H.; Wehrens, R.; Liland, K.H. pls: Partial Least Squares and Principal Component Regression. R Package Version 2.7-3. 2020. Available online: http://cran.r-project.org/package=pls (accessed on 8 August 2020).
- Zeileis, A.; Horthorn, T. Diagnostic Checking in Regression Relationships. R News 2002, 2, 7–10. [Google Scholar]
- Wood, S. Stable and efficient multiple smoothing parameter estimation for generalized additive models. J. Am. Stat. Assoc. 2004, 99, 673–686. [Google Scholar] [CrossRef]
- Darzi, Y.; Letunic, I.; Bork, P.; Yamada, T. iPath3.0: Interactive pathways explorer v3. Nucleic Acids Res. 2018, 46, W510–W513. [Google Scholar] [CrossRef]
- Haug, K.; Cochrane, K.; Nainala, V.C.; Williams, M.; Chang, J.; Jayaseelan, K.V.; O’Donovan, C. MetaboLights: A resource evolving in response to the needs of its scientific community. Nucleic Acids Res. 2020, 48, D440–D444. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Method Name | Chromatography/Stationary Phase | Coverage | Elution Order | Detection | Polarity |
|---|---|---|---|---|---|
| FIA | none | All compounds that ionize well | No retention | MS1 | + and − |
| LC-QTOF HILIC | HILIC/aminopropyl | Amino acids, nucleotides, sugar phosphates, soluble cofactors, organic acids | Hydrophilic moieties: increase retention, hydrophobic moieties: little influence | MS1 and MS2 | - |
| LC-QTOF polar RP | Reversed phase/C18 | Acyl carnitines, nucleosides, nucleobases, some cofactors, some amino acids | Hydrophilic moieties: reduce retention, hydrophobic moieties: increase retention | MS1 and MS2 | + |
| LC-QTOF Lipids | Reversed phase/C18 | Glycerolipids, glycerophospholipids, sterols, sphingolipids | Hydrophilic moieties: reduce retention, hydrophobic moieties: increase retention | MS1 and MS2 | + and − |
| Method Name | Polarity | # Features | % Features above Blank | % Features above Blank and with Time Trend |
|---|---|---|---|---|
| FIA | - | 1887 | 44.4 | 31.0 |
| FIA | + | 2416 | 52.7 | 28.4 |
| LC-QTOF HILIC | - | 1671 | 31.2 | 26.3 |
| LC-QTOF Polar RP | + | 1549 | 32.8 | 25.8 |
| LC-QTOF Lipids | - | 1745 | 57.9 | 53.4 |
| LC-QTOF Lipids | + | 1819 | 57.6 | 56.6 |
| Filter Parameters | |
|---|---|
| Minimum # Features for Extraction | 1 |
| Presence of features in minimum # of analyses | 3 |
| T-ReX 3D Processing Parameters | |
| Intensity threshold | 4000 (polar metabolites), 3000 (lipids neg), 6000 (lipids pos) |
| Minimum Peak Length | 12 |
| Enable Recursive Feature Extraction | true |
| Minimum Peak Length (recursive) | 7 |
| Perform MS/MS import | true |
| MS/MS import method | average |
| Ion Deconvolution Parameters | |
| EIC correlation | 0.8 |
| Primary ion (negative mode) | [M-H]- |
| Primary ion (positive mode) | [M+H]+ |
| Seed ions (negative mode) | [M+Cl]- |
| Seed ions (positive mode) | [M+Na]+, [M+K]+, [M+NH4]+ |
| Common ions (negative mode) | [M-H-H2O]-, [M+COOH]- |
| Common ions (positive mode) | [M-H-H2O]+ |
| Mass Calibration Parameters | |
| Lock Mass Calibration | false |
| Mass Recalibration | true, calibration segment 0.1-0.4 min |
| Expert settings | |
| FerraWorkflow.chargeMax | 1 (only for polar metabolites) |
| Smart Formula Parameters | |
|---|---|
| m/z tolerance | 1 mDa (narrow), 3 mDa (wide) |
| mSigma | 15 (narrow), 50 (wide) |
| Elements | CHNOPS |
| Upper formula | S1 |
| Element ratio filters | Common |
| Electron configuration | Both |
| Analyte List Parameters | |
| m/z tolerance | 1 mDa (narrow), 3 mDa (wide) |
| Retention time tolerance | 0.2 min (narrow), 0.4 min (wide) |
| mSigma | 15 (narrow), 50 (wide) |
| Spectral Library Parameters | |
| Libraries (polar metabolites) | In house library, Bruker MetaboBASE 3.0, GNPS export (downloaded July 2020) |
| Libraries (lipids) | In house library, Bruker MetaboBASE 3.0, MSDIAL LipidDB VS68 (neg and pos) |
| m/z tolerance | 1 mDa (narrow), 3 mDa (wide) |
| mSigma | 20 (narrow), 200 (wide) |
| MS/MS score | 900 (narrow), 700 (wide) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Edwards-Hicks, J.; Mitterer, M.; Pearce, E.L.; Buescher, J.M. Metabolic Dynamics of In Vitro CD8+ T Cell Activation. Metabolites 2021, 11, 12. https://doi.org/10.3390/metabo11010012
Edwards-Hicks J, Mitterer M, Pearce EL, Buescher JM. Metabolic Dynamics of In Vitro CD8+ T Cell Activation. Metabolites. 2021; 11(1):12. https://doi.org/10.3390/metabo11010012
Chicago/Turabian StyleEdwards-Hicks, Joy, Michael Mitterer, Erika L. Pearce, and Joerg M. Buescher. 2021. "Metabolic Dynamics of In Vitro CD8+ T Cell Activation" Metabolites 11, no. 1: 12. https://doi.org/10.3390/metabo11010012
APA StyleEdwards-Hicks, J., Mitterer, M., Pearce, E. L., & Buescher, J. M. (2021). Metabolic Dynamics of In Vitro CD8+ T Cell Activation. Metabolites, 11(1), 12. https://doi.org/10.3390/metabo11010012