Atom Identifiers Generated by a Neighborhood-Specific Graph Coloring Method Enable Compound Harmonization across Metabolic Databases



Abstract
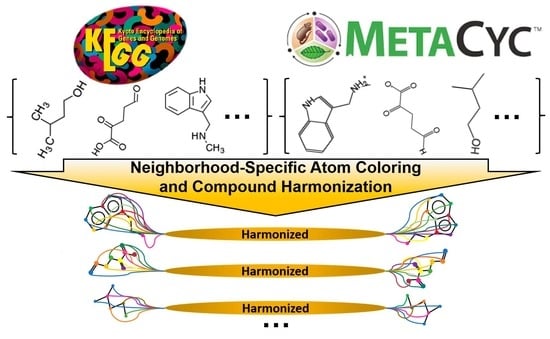
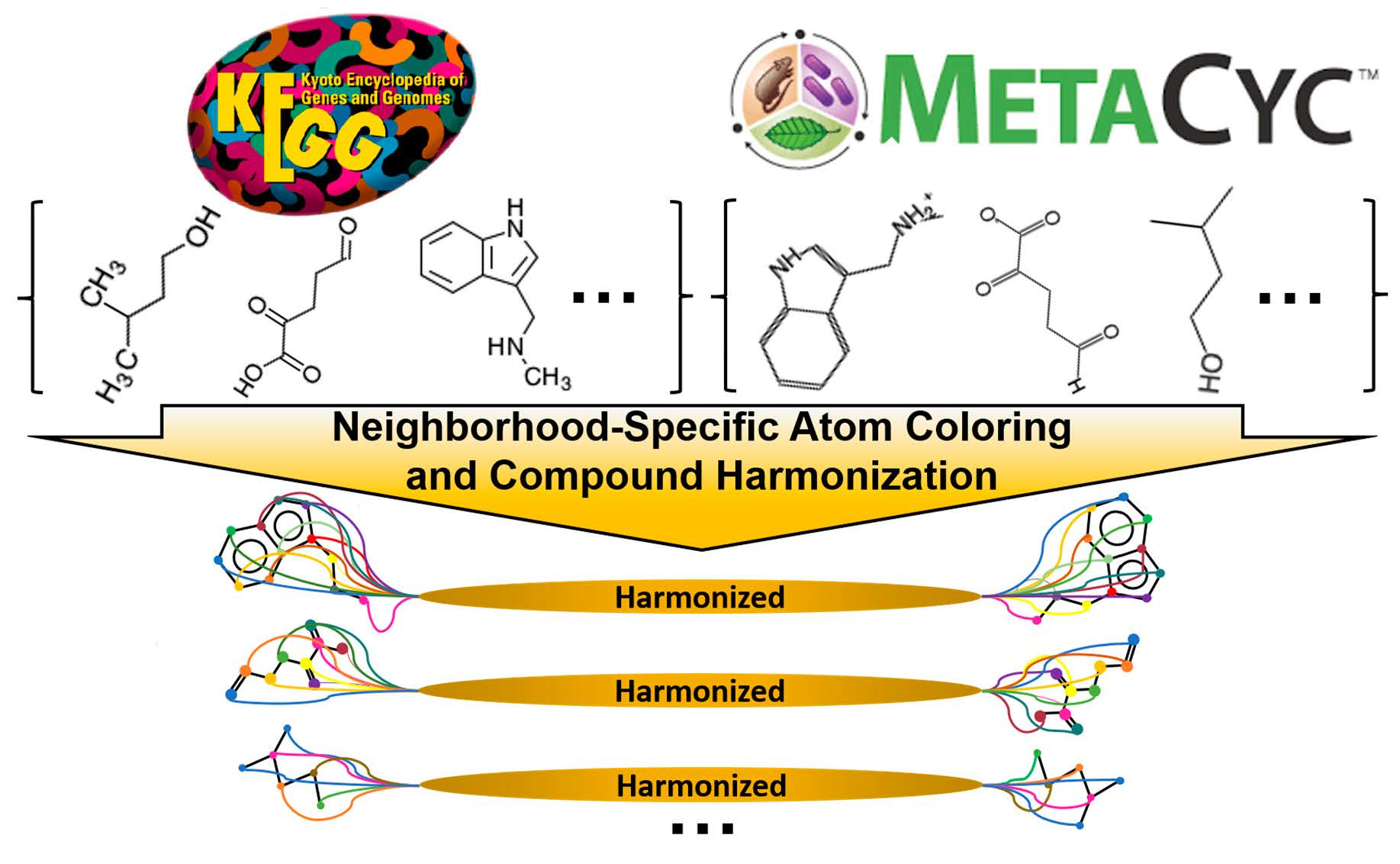
1. Introduction
2. Results
2.1. Overview of KEGG and MetaCyc Databases
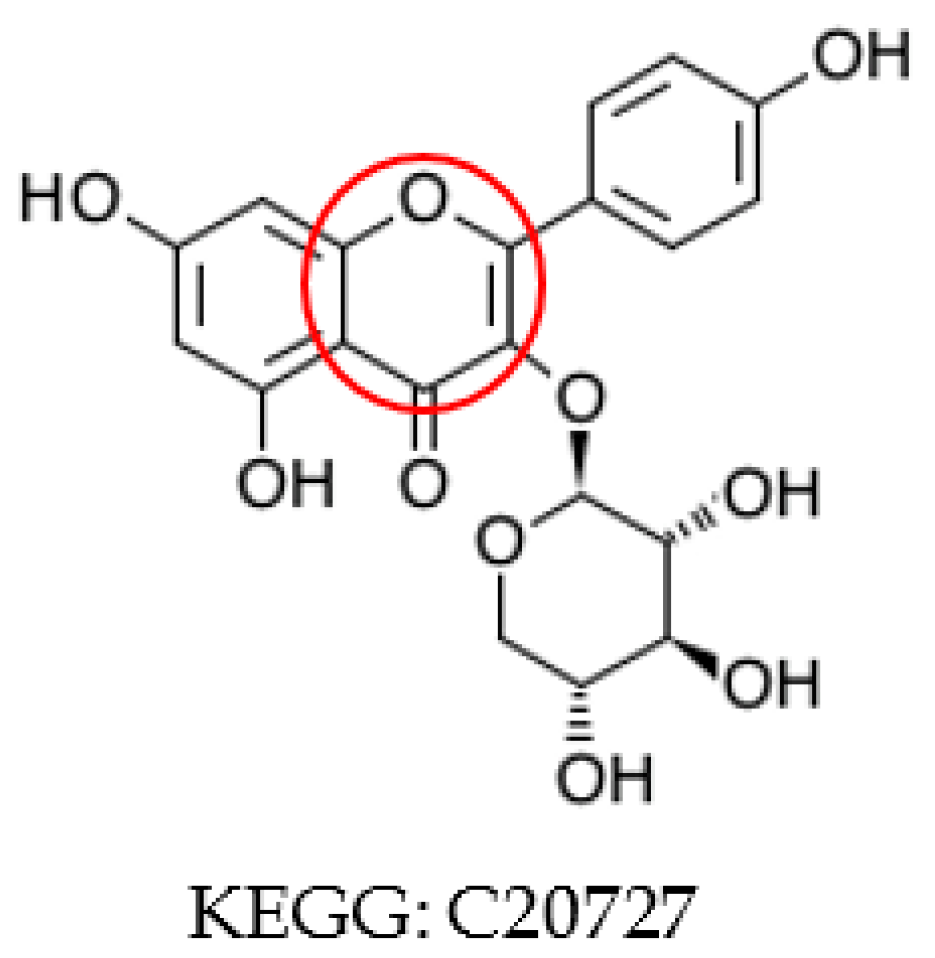
2.2. Aromatic Substructure Detection
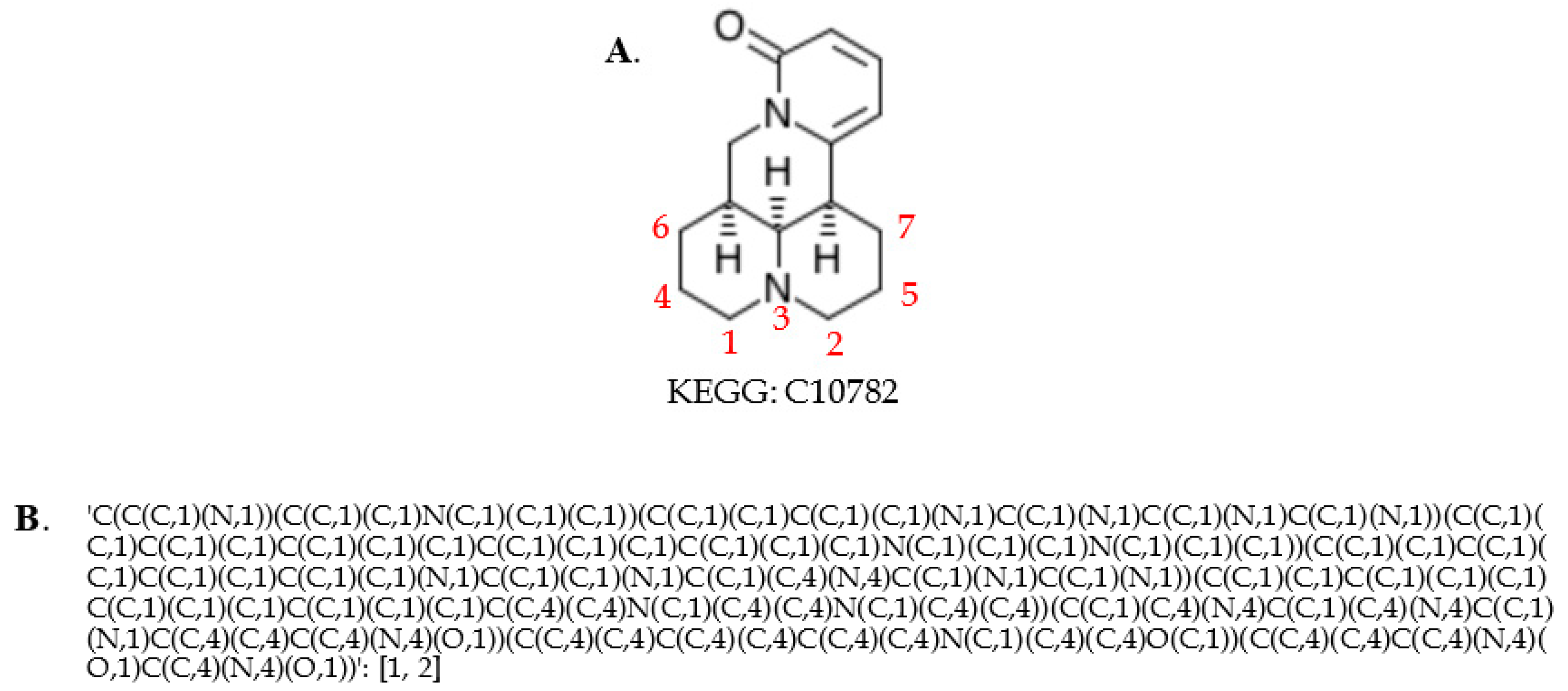
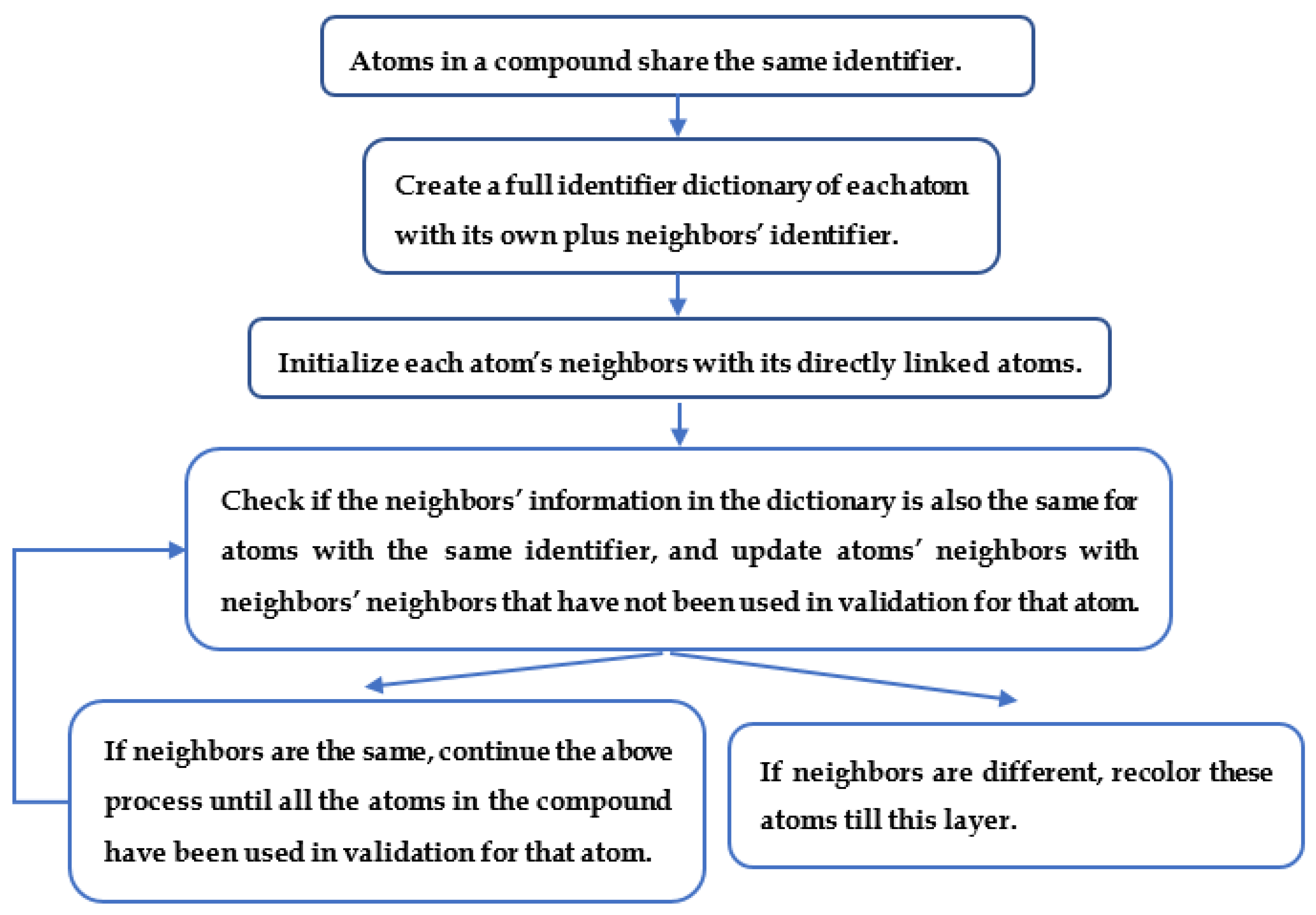
2.3. Generating Identifiers for Atoms Using a Graph Coloring Method
2.4. Detection of Correspondences Between KEGG and MetaCyc Compounds via Coloring Identifiers
2.5. Compound Representation Errors and Issues Detected in the KEGG and MetaCyc Databases
2.5.1. Incomplete KEGG Aromatic Atom Types
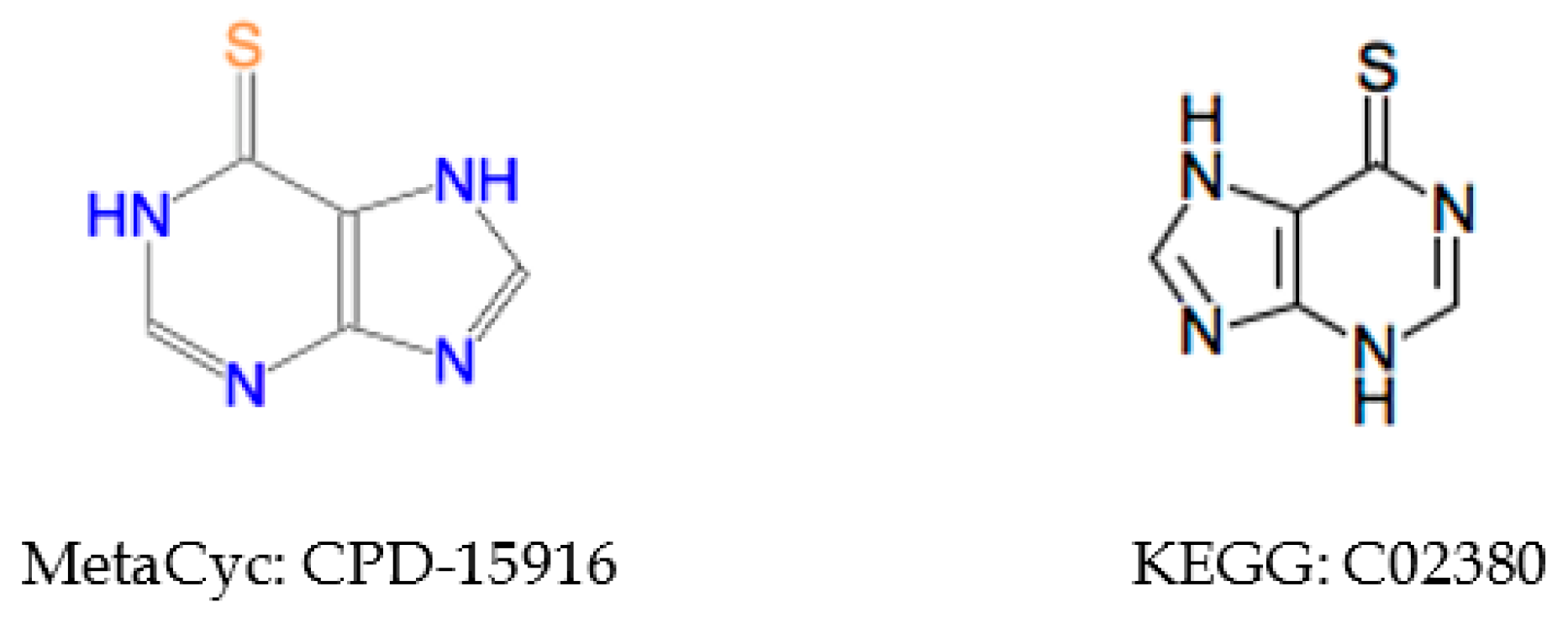
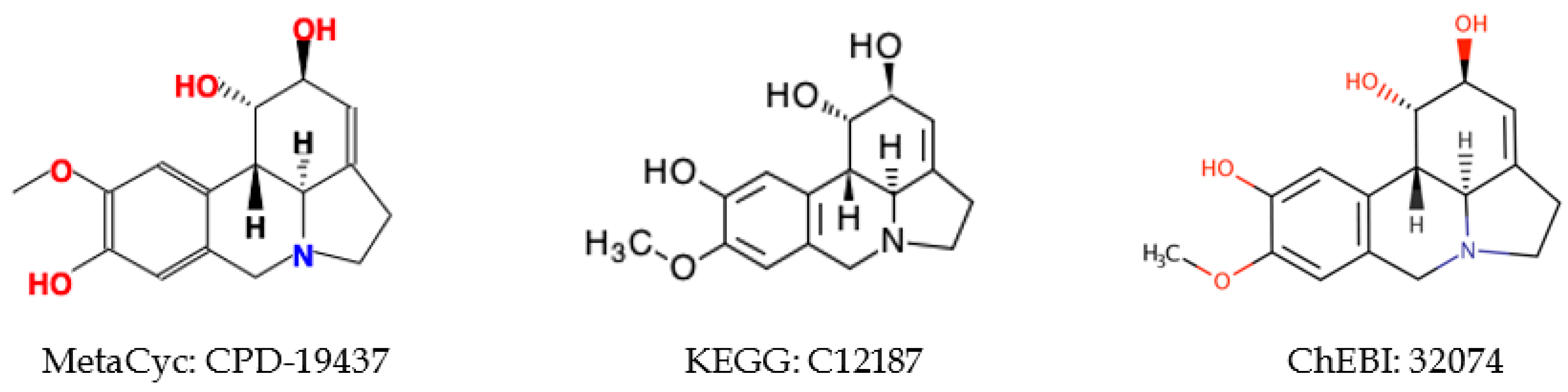
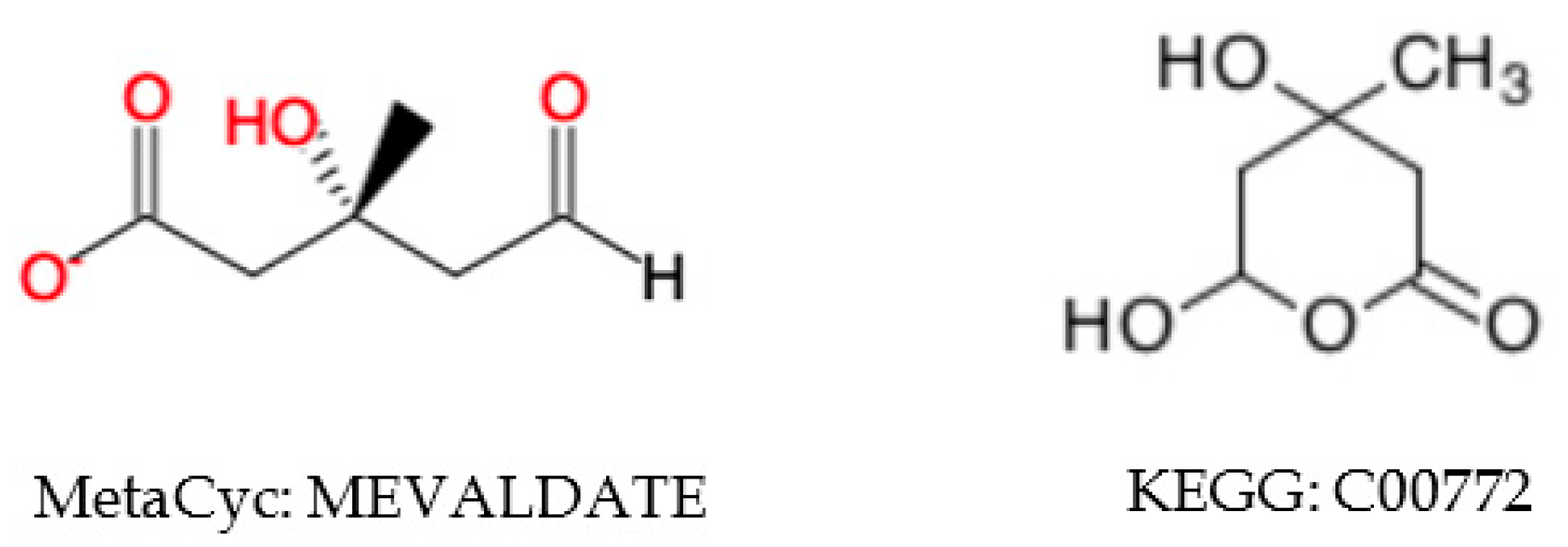
2.5.2. Inconsistent Compound Representations
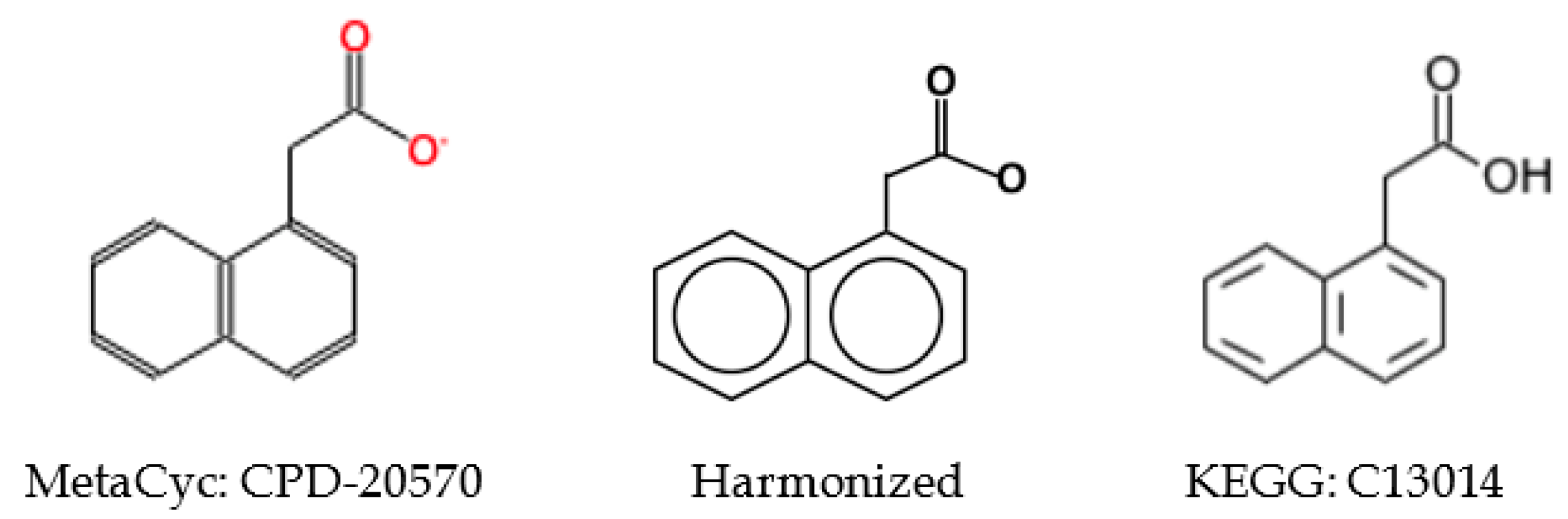
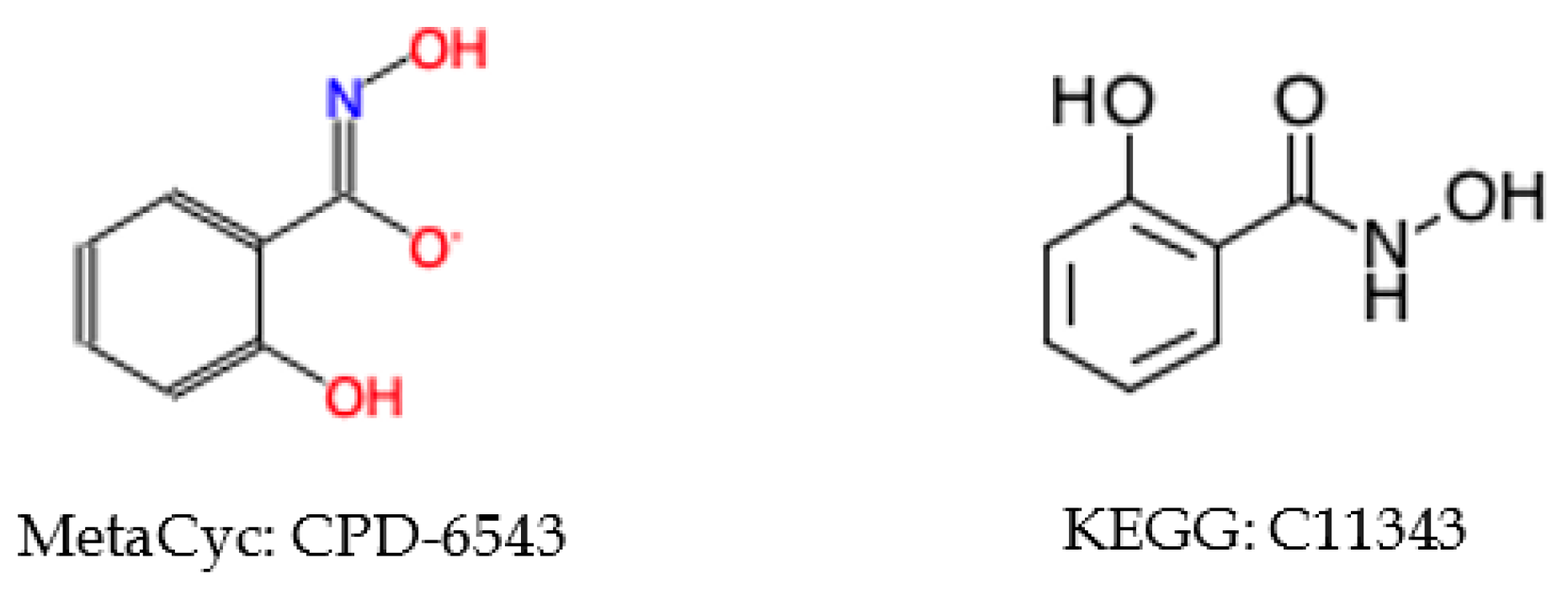
2.5.3. Incorrect Cross-Referencing
2.6. Estimating the Error Rate of the Graph Coloring Method
2.6.1. Ambiguous Coloring Identifiers
2.6.2. Pseudosymmetric Atoms
2.6.3. Changeable Graph Representation
3. Discussion
4. Materials and Methods
4.1. Compound and Metabolic Reaction Data
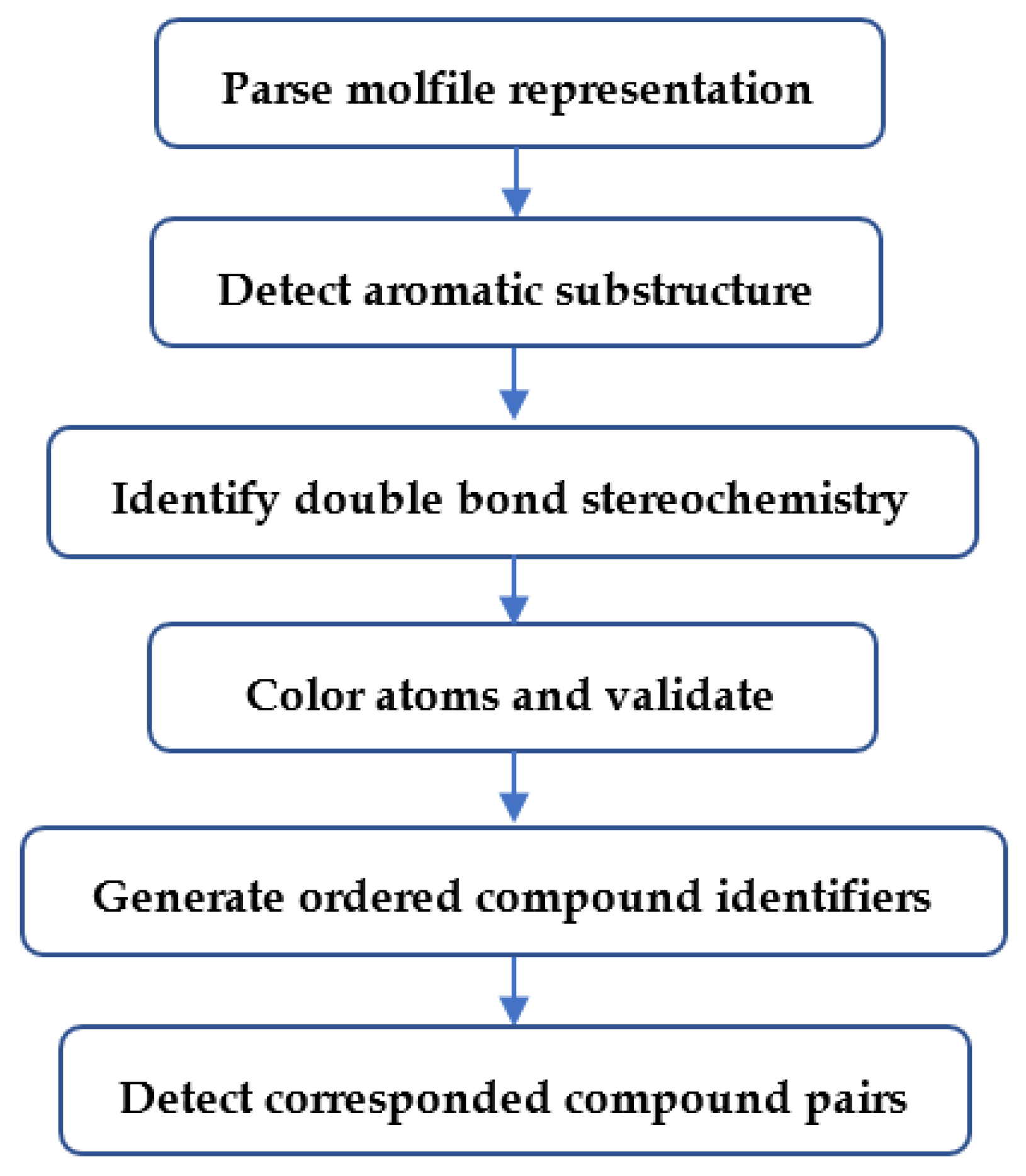
4.2. Overview of Major Analysis Steps
4.3. Molfile Parser
4.4. Aromatic Substructure Detection
4.5. Identification of Double Bond Stereochemistry
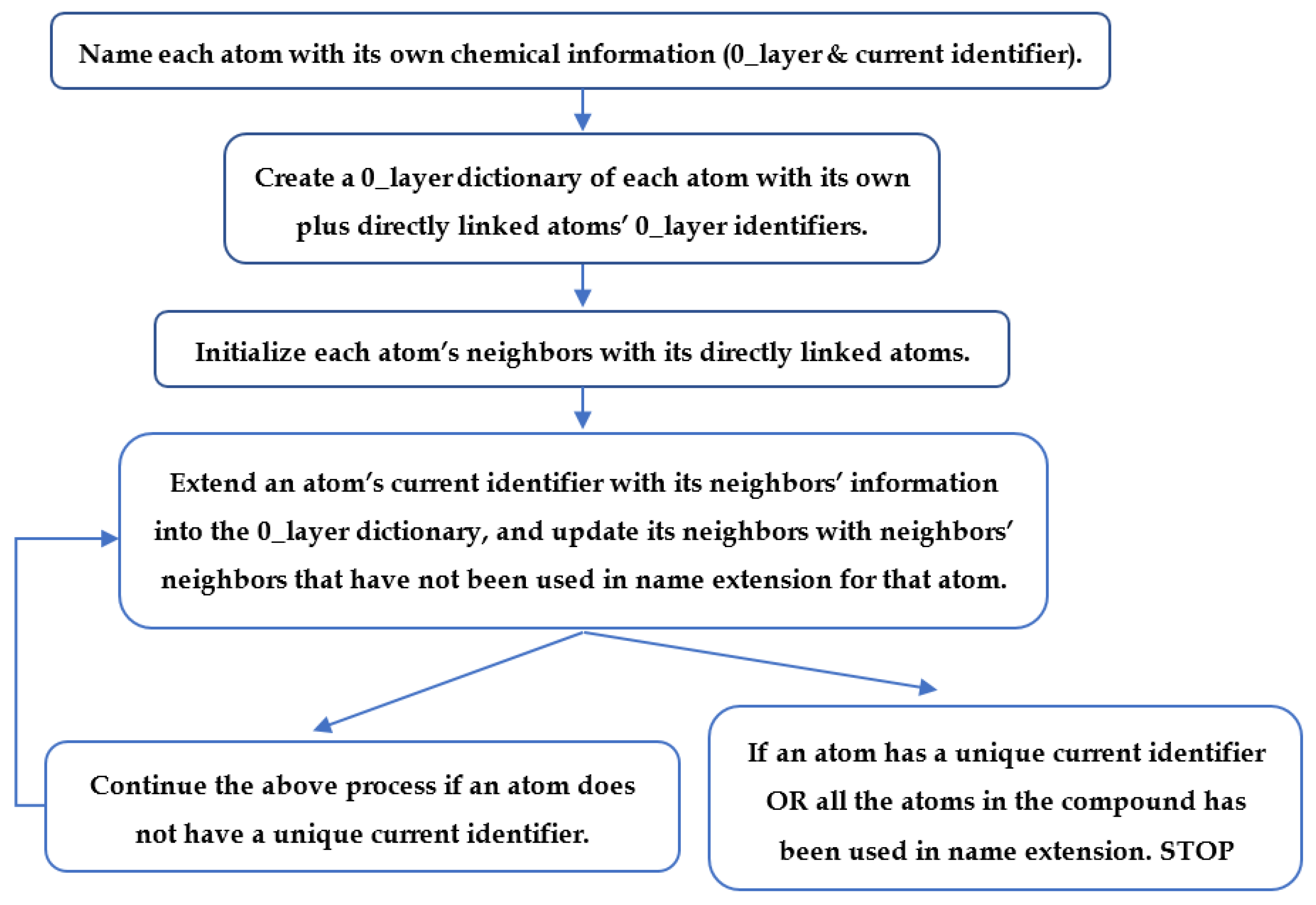
4.6. Neighborhood-Specific Graph Coloring Method
4.7. Atom Coloring Validation and Recolor
4.8. Creation of Compound Coloring Identifiers Based on Atom Coloring Identifiers
4.9. Prediction of Possible Compound Correspondence via Metabolic Reactions
Supplementary Materials
Data Availability
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Young, J.D. INCA: A computational platform for isotopically non-stationary metabolic flux analysis. Bioinformatics 2014, 30, 1333–1335. [Google Scholar] [CrossRef] [PubMed]
- Antoniewicz, M.R. Methods and advances in metabolic flux analysis: A mini-review. J. Ind. Microbiol. Biotechnol. 2015, 42, 317–325. [Google Scholar] [CrossRef]
- Moseley, H.N.; Lane, A.N.; Belshoff, A.C.; Higashi, R.M.; Fan, T.W. A novel deconvolution method for modeling UDP-N-acetyl-D-glucosamine biosynthetic pathways based on 13C mass isotopologue profiles under non-steady-state conditions. BMC Biol. 2011, 9, 37. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.; Moseley, H.N.B. Moiety modeling framework for deriving moiety abundances from mass spectrometry measured isotopologues. BMC Bioinform. 2019, 20, 524. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.; Moseley, H.N.B. Robust Moiety Model Selection Using Mass Spectrometry Measured Isotopologues. Metabolites 2020, 10, 118. [Google Scholar] [CrossRef]
- Rathahao-Paris, E.; Alves, S.; Junot, C.; Tabet, J.-C. High resolution mass spectrometry for structural identification of metabolites in metabolomics. Metabolomics 2016, 12, 10. [Google Scholar] [CrossRef]
- Fan, T.W.-M.; Lorkiewicz, P.K.; Sellers, K.; Moseley, H.N.B.; Higashi, R.M.; Lane, A.N. Stable isotope-resolved metabolomics and applications for drug development. Pharmacol. Ther. 2012, 133, 366–391. [Google Scholar] [CrossRef]
- Creek, D.J.; Chokkathukalam, A.; Jankevics, A.; Burgess, K.E.V.; Breitling, R.; Barrett, M.P. Stable Isotope-Assisted Metabolomics for Network-Wide Metabolic Pathway Elucidation. Anal. Chem. 2012, 84, 8442–8447. [Google Scholar] [CrossRef]
- Aittokallio, T. Graph-based methods for analysing networks in cell biology. Brief Bioinform. 2006, 7, 243–255. [Google Scholar] [CrossRef]
- Arita, M. In Silico Atomic Tracing by Substrate-Product Relationships in Escherichia coli Intermediary Metabolism. Genome Resarch 2003, 13, 2455–2466. [Google Scholar] [CrossRef]
- Arita, M.; Fujiwara, Y.; Nakanishi, Y. Map Editor for the Atomic Reconstruction of Metabolism (ARM). In Biotechnology in Agriculture and Forestry; Saito, K., Dixon, R.A., Willmitzer, L., Eds.; Springer: Berlin/Heidelberg, Germany, 2006; Volume 57, pp. 129–139. [Google Scholar]
- Chokkathukalam, A.; Kim, D.; Barrett, M.P.; Breitling, R.; Creek, D.J. Stable isotope-labeling studies in metabolomics: New insights into structure and dynamics of metabolic networks. Bioanalysis 2014, 6, 511–524. [Google Scholar] [CrossRef] [PubMed]
- Hadadi, N.; Hafner, J.; Soh, K.; Hatzimanikatis, V. Reconstruction of biological pathways and metabolic networks from in silico labeled metabolites. Biotech. J. 2017, 12, 1600464. [Google Scholar] [CrossRef] [PubMed]
- Dalby, A.; Nourse, J.G.; Hounshell, W.D.; Gushurst, A.K.; Grier, D.L.; Leland, B.A.; Laufer, J. Description of several chemical structure file formats used by computer programs developed at Molecular Design Limited. J. Chem. Inf. Comput. Sci. 1992, 32, 244–255. [Google Scholar] [CrossRef]
- Kotera, M.; Hattori, M.; Oh, M.A.; Yamamoto, R.; Komeno, T.; Yabuzaki, J.; Tonomura, K.; Goto, S.; Kanehisa, M. RPAIR: A reactant-pair database representing chemical changes in enzymatic reactions. Genome Inform. 2004, 15, P062. [Google Scholar]
- Latendresse, M.; Malerich, J.P.; Travers, M.; Karp, P.D. Accurate Atom-Mapping Computation for Biochemical Reactions. J. Chem. Inf. Model. 2012, 52, 2970–2982. [Google Scholar] [CrossRef]
- Pitkänen, E.; Jouhten, P.; Rousu, J. Inferring branching pathways in genome-scale metabolic networks. BMC Syst. Biol. 2009, 3, 103. [Google Scholar] [CrossRef]
- Heath, A.P.; Bennett, G.N.; Kavraki, L.E. Finding metabolic pathways using atom tracking. Bioinformatics 2010, 26, 1548–1555. [Google Scholar] [CrossRef]
- Latendresse, M.; Krummenacker, M.; Karp, P.D. Optimal metabolic route search based on atom mappings. Bioinformatics 2014, 30, 2043–2050. [Google Scholar] [CrossRef]
- Altman, T.; Travers, M.; Kothari, A.; Caspi, R.; Karp, P.D. A systematic comparison of the MetaCyc and KEGG pathway databases. BMC Bioinform. 2013, 14, 112. [Google Scholar] [CrossRef]
- Dashti, H.; Westler, W.M.; Markley, J.L.; Eghbalnia, H.R. Unique identifiers for small molecules enable rigorous labeling of their atoms. Sci. Data 2017, 4, 170073. [Google Scholar] [CrossRef]
- Willighagen, E.; Mayfield, J. The Chemistry Development Kit (CDK) v2.0: Atom typing, depiction, molecular formulas, and substructure searching. J. Cheminform. 2017, 9, 33. [Google Scholar] [CrossRef] [PubMed]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An open chemical toolbox. J. Cheminform. 2011, 3, 33. [Google Scholar] [CrossRef] [PubMed]
- Heller, S.; McNaught, A.; Stein, S.; Tchekhovskoi, D.; Pletnev, I. InChI - the worldwide chemical structure identifier standard. J. Cheminform. 2013, 5, 7. [Google Scholar] [CrossRef]
- Dashti, H.; Wedell, J.R.; Westler, W.M.; Markley, J.L.; Eghbalnia, H.R. Automated evaluation of consistency within the PubChem Compound database. Sci. Data 2019, 6, 190023. [Google Scholar] [CrossRef]
- Mitchell, J.M.; Fan, T.W.-M.; Lane, A.N.; Moseley, H.N.B. Development and in silico evaluation of large-scale metabolite identification methods using functional group detection for metabolomics. Front. Genet. 2014, 5. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ramar, R.; Venkatasubramanian, S. Neighborhood Distinguishing Coloring in Graphs. Innov. Incid. Geometry Algebraic Topol. Comb. 2013, 13, 135. [Google Scholar] [CrossRef]
- Li, Y.; Razborov, A.; Rossman, B. On the Complexity of Subgraph Isomorphism. SIAM J. Comput. 2017, 46, 936–971. [Google Scholar] [CrossRef]
- Indigo Toolkit. Available online: https://lifescience.opensource.epam.com/indigo/index.html (accessed on 30 April 2020).
- Danchin, A. Enzyme nomenclature, recommendations (1992) of the nomenclature committee or the international union of biochemistry and molecular biology. Biochimie 1993, 75, 501. [Google Scholar] [CrossRef]
- McDonald, A.G.; Boyce, S.; Tipton, K.F. ExplorEnz: The primary source of the IUBMB enzyme list. Nucleic Acids Res. 2009, 37, 593–597. [Google Scholar] [CrossRef]
- Szatylowicz, H.; Krygowski, T.M.; Solà, M.; Palusiak, M.; Dominikowska, J.; Stasyuk, O.A.; Poater, J. Why 1,2-quinone derivatives are more stable than their 2,3-analogues? Theor. Chem. Acc. 2015, 134, 35. [Google Scholar] [CrossRef]
- Ivanov, J.; Schüürmann, G. Simple Algorithms for Determining the Molecular Symmetry. J. Chem. Inf. Comput. Sci. 1999, 39, 728–737. [Google Scholar] [CrossRef]
- Tinhofer, G.; Klin, M. Algebraic Combinatorics in Mathematical Chemistry. Methods and Algorithms. III, Graph Invariants and Stabilization Methods (Preliminary Version); Technical Report, TUM-M9902; Technische Universitat Munchen: Munchen, Germany, 1999. [Google Scholar]
- Schneider, N.; Sayle, R.; Landrum, G. Get Your Atoms in Order—An Open-Source Implementation of a Novel and Robust Molecular Canonicalization Algorithm. J. Chem. Inf. Model. 2015, 55, 2111–2120. [Google Scholar] [CrossRef] [PubMed]
- Andrey, S. Ctfile. Available online: https://github.com/MoseleyBioinformaticsLab/ctfile (accessed on 26 November 2019).
- Bray, T. The JavaScript Object Notation (JSON) Data Interchange Format. Available online: https://www.rfc-editor.org/info/rfc7159 (accessed on 10 September 2020).
- Hattori, M.; Okuno, Y.; Goto, S.; Kanehisa, M. Development of a Chemical Structure Comparison Method for Integrated Analysis of Chemical and Genomic Information in the Metabolic Pathways. J. Am. Chem. Soc. 2003, 125, 11853–11865. [Google Scholar] [CrossRef]
- Teixeira, A.L.; Leal, J.P.; Falcao, A.O. Automated Identification and Classification of Stereochemistry: Chirality and Double Bond Stereoisomerism. Available online: https://arxiv.org/abs/1303.1724 (accessed on 7 March 2013).
- Cormen, T.H.; Leiserson, C.E.; Rivest, R.L.; Stein, C. Introduction to Algorithms, 2nd ed.; MIT Press: Cambridge, MA, USA, 2001. [Google Scholar]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
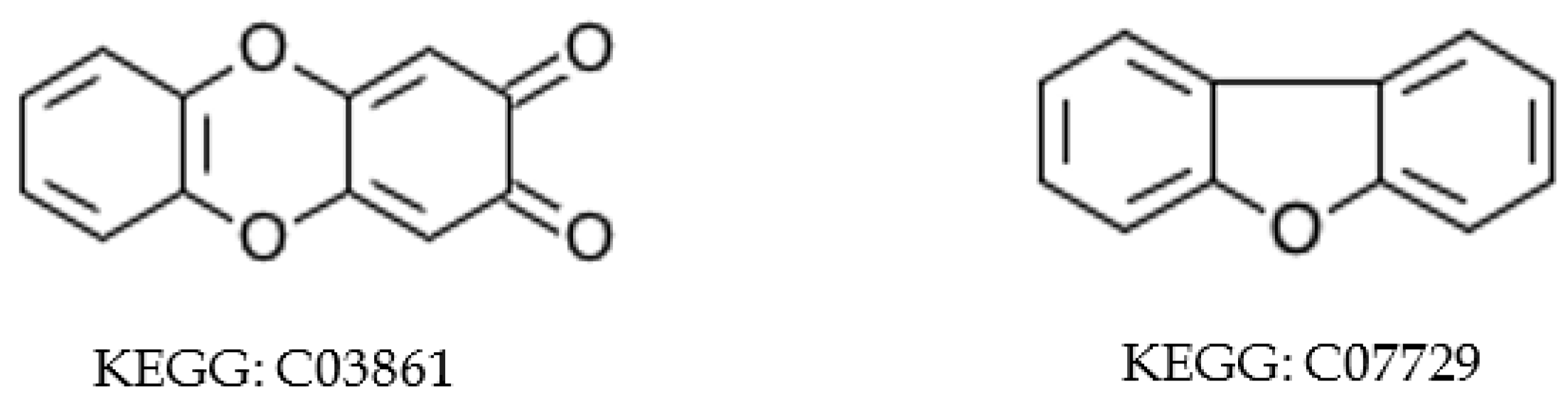{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Data Types | KEGG | MetaCyc | MetaCyc/KEGG a |
|---|---|---|---|
| Compounds | 18636 | 20264 | 1.09 |
| Reactions | 11427 | 17203 | 1.51 |
| Atom-resolved reactions | 10282 | 15909 | 1.53 |
| Identifiers | KEGG | MetaCyc | Correspondences |
|---|---|---|---|
| InChI | 13216 (70.9%) | 15076 (74.4%) | 2336 |
| ChEBI | 15353 (82.4%) | 8404 (41.5%) | 3106 |
| KEGG | 18636 (100%) | 5402 (26.7%) | 5402 |
| Either-ID | 18636 (100%) | 15216 (75.1%) | 5681 |
| Databases | BASS | Indigo |
|---|---|---|
| KEGG | 0 | ~1500 |
| MetaCyc | 30 | ~1700 |
| Databases | Count |
|---|---|
| KEGG | 9204 (49.4%) |
| MetaCyc | 8292 (40.9%) |
| Identifiers | Color Matched Pairs | ID Verified Pairs |
|---|---|---|
| Tight coloring identifier | 1763 | 1448 |
| Loose coloring identifier | 8865 | 5451 |
| EC Types | KEGG (Count/Percentage) | MetaCyc (Count/Percentage) |
|---|---|---|
| No EC | 1263/11.05% | 3427/19.92% |
| 1-leveled EC | 24/0.21% | 11/0.06% |
| 2-leveled EC | 126/1.10% | 67/0.39% |
| 3-leveled EC | 1081/9.46% | 2958/17.19% |
| 4-leveled EC | 8933/78.17% | 10740/62.43% |
| Conditions | ID-Confirmed Pairs | Possible Pairs |
|---|---|---|
| Pairs not in reaction | 1224 | 1122 |
| Pairs in reactions | 4227 | 2292 |
| Verified by 3-leveled EC | 3810 | 1848 |
| Verified by 4-leveled EC | 3580 | 1540 |
| Databases | Tight Coloring Identifier | Loose Coloring Identifier |
|---|---|---|
| KEGG | 99 (0.5%) | 968 (4.8%) |
| MetaCyc | 117 (0.6%) | 1144 (5.6%) |
| Databases | Atom Stereochemistry | Atom Charge | Bond Stereochemistry |
|---|---|---|---|
| KEGG | 232 | 38 | 169 |
| MetaCyc | 219 | 148 | 227 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jin, H.; Mitchell, J.M.; Moseley, H.N.B. Atom Identifiers Generated by a Neighborhood-Specific Graph Coloring Method Enable Compound Harmonization across Metabolic Databases. Metabolites 2020, 10, 368. https://doi.org/10.3390/metabo10090368
Jin H, Mitchell JM, Moseley HNB. Atom Identifiers Generated by a Neighborhood-Specific Graph Coloring Method Enable Compound Harmonization across Metabolic Databases. Metabolites. 2020; 10(9):368. https://doi.org/10.3390/metabo10090368
Chicago/Turabian StyleJin, Huan, Joshua M. Mitchell, and Hunter N. B. Moseley. 2020. "Atom Identifiers Generated by a Neighborhood-Specific Graph Coloring Method Enable Compound Harmonization across Metabolic Databases" Metabolites 10, no. 9: 368. https://doi.org/10.3390/metabo10090368
APA StyleJin, H., Mitchell, J. M., & Moseley, H. N. B. (2020). Atom Identifiers Generated by a Neighborhood-Specific Graph Coloring Method Enable Compound Harmonization across Metabolic Databases. Metabolites, 10(9), 368. https://doi.org/10.3390/metabo10090368