Metabolic Signatures of Tumor Responses to Doxorubicin Elucidated by Metabolic Profiling in Ovo

 ,
,  ,
, 
 and
and
Abstract
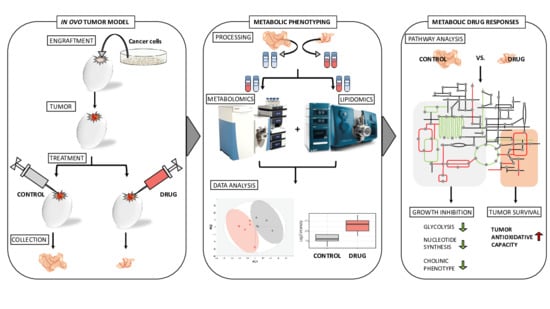
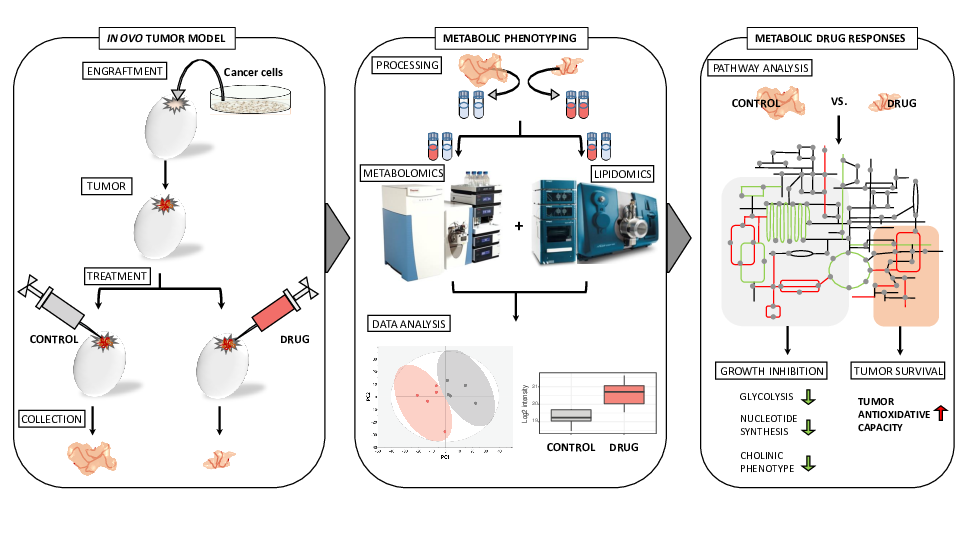
1. Introduction
2. Results
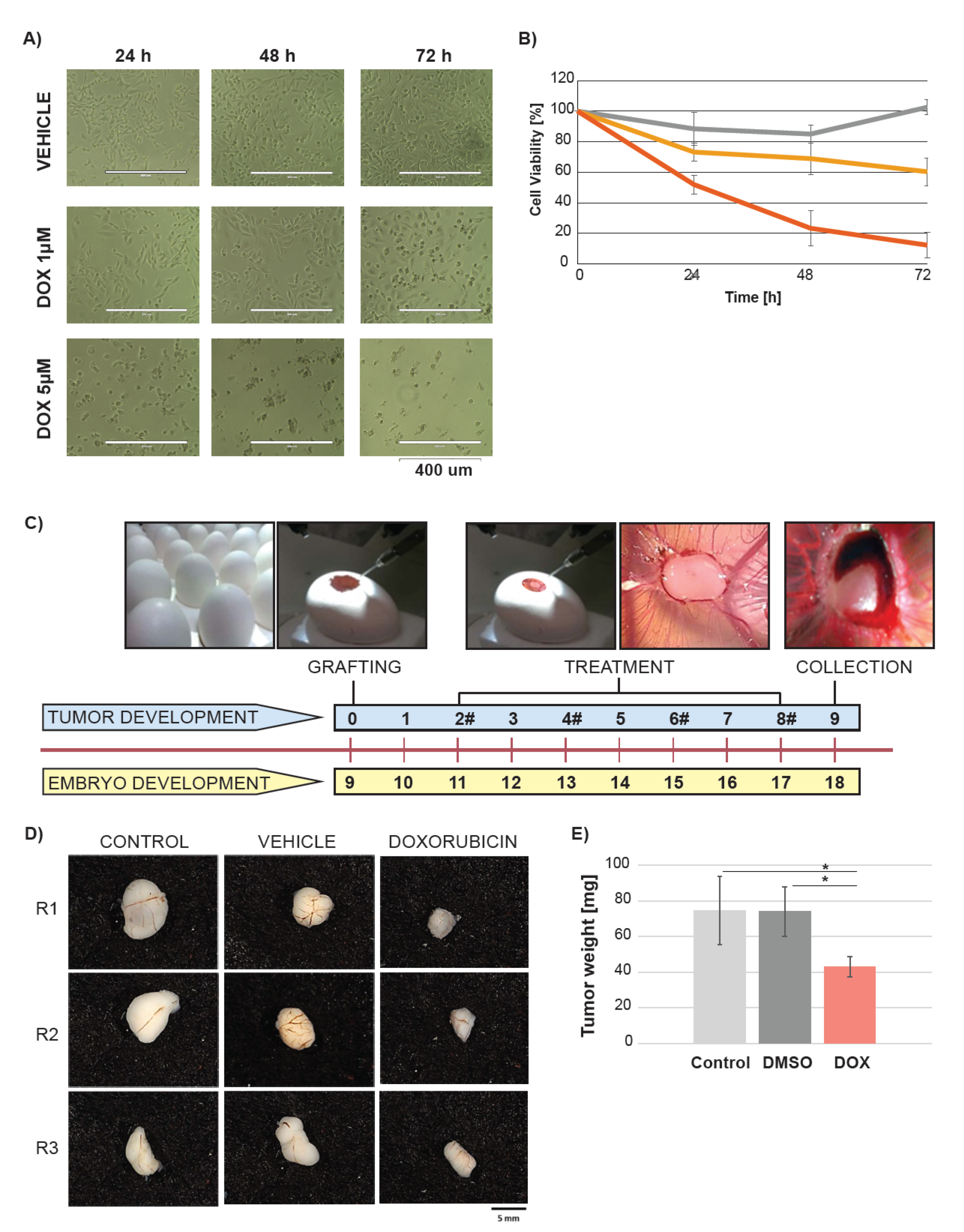
2.1. Doxorubicin Treatment Suppresses Tumor Growth in Ovo
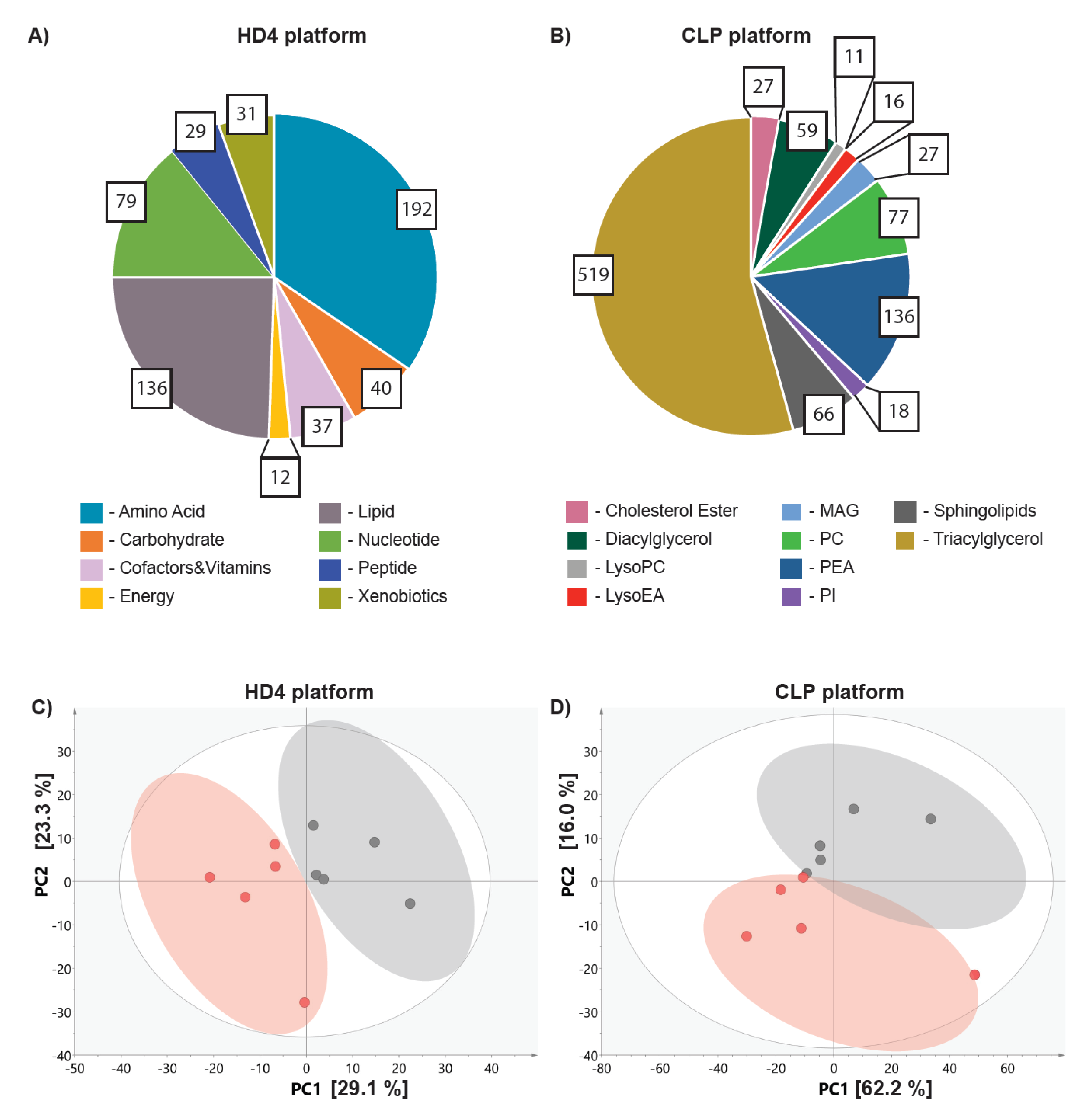
2.2. Metabolic Profiling in Ovo Reveals Alterations in Tumor Metabolism Triggered by Doxorubicin Treatment
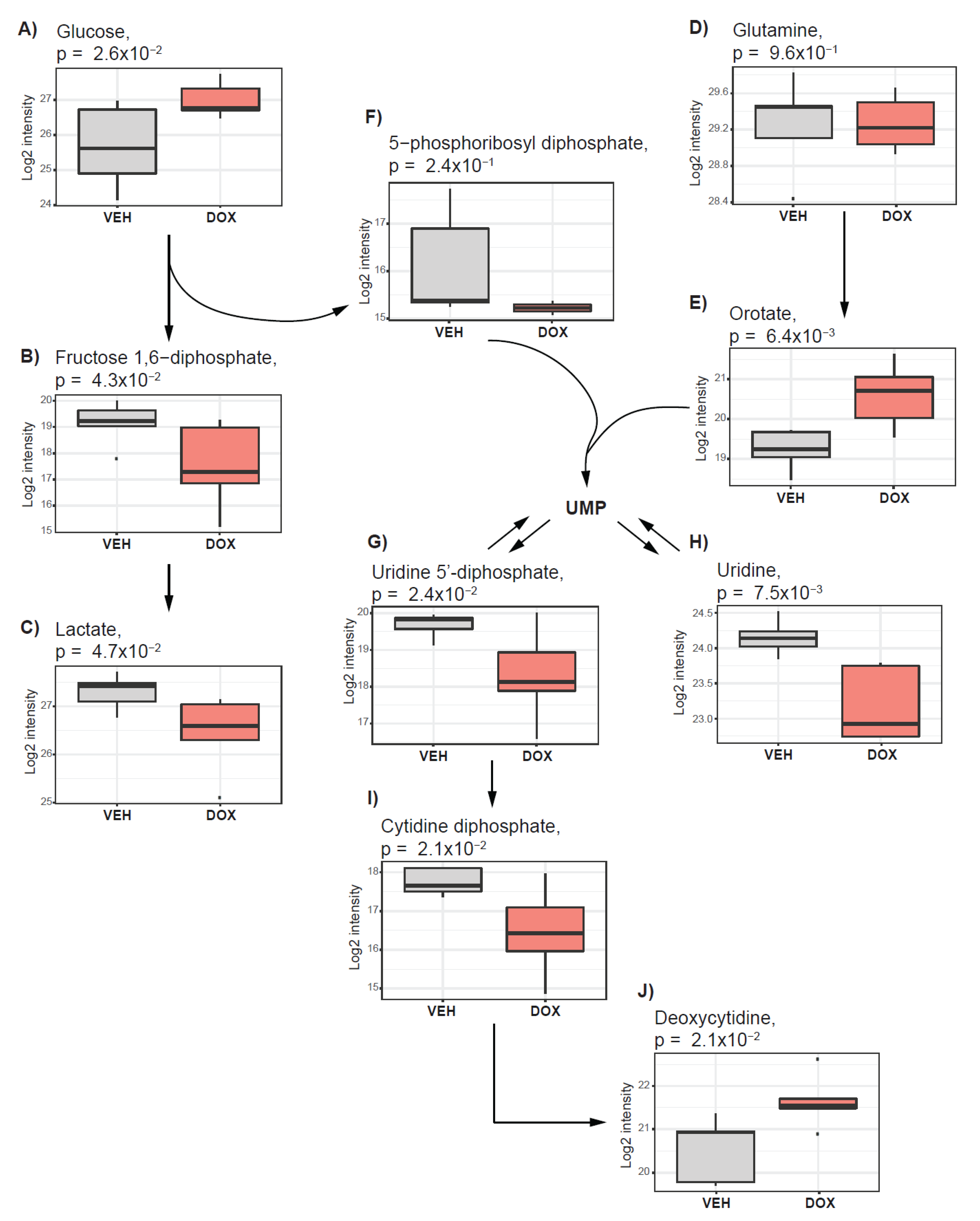
2.3. Doxorubicin Treatment Suppresses Glycolysis and Nucleotide Synthesis
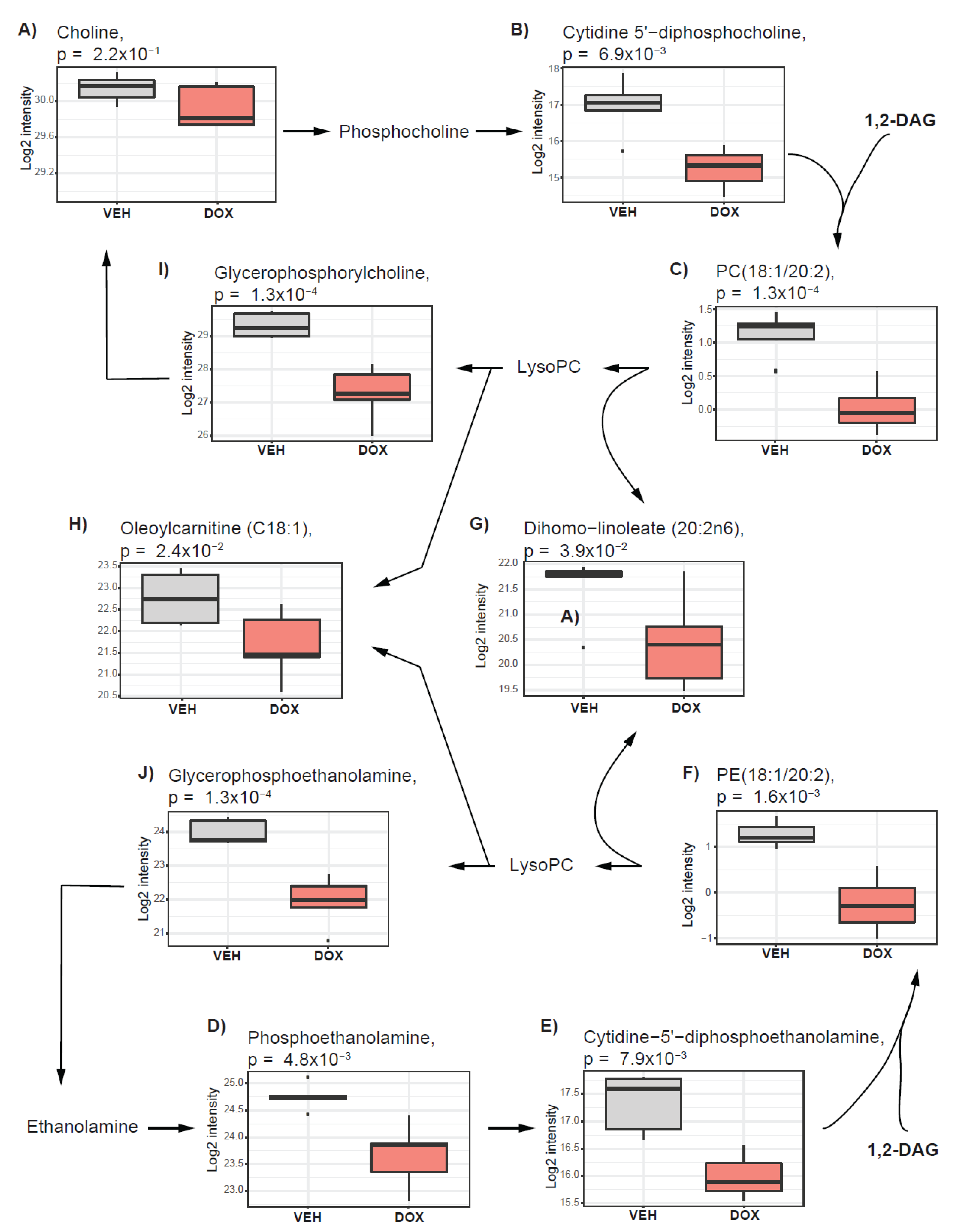
2.4. The Cholinic Phenotype and Fatty Acid Metabolism Are Suppressed by Doxorubicin Treatment
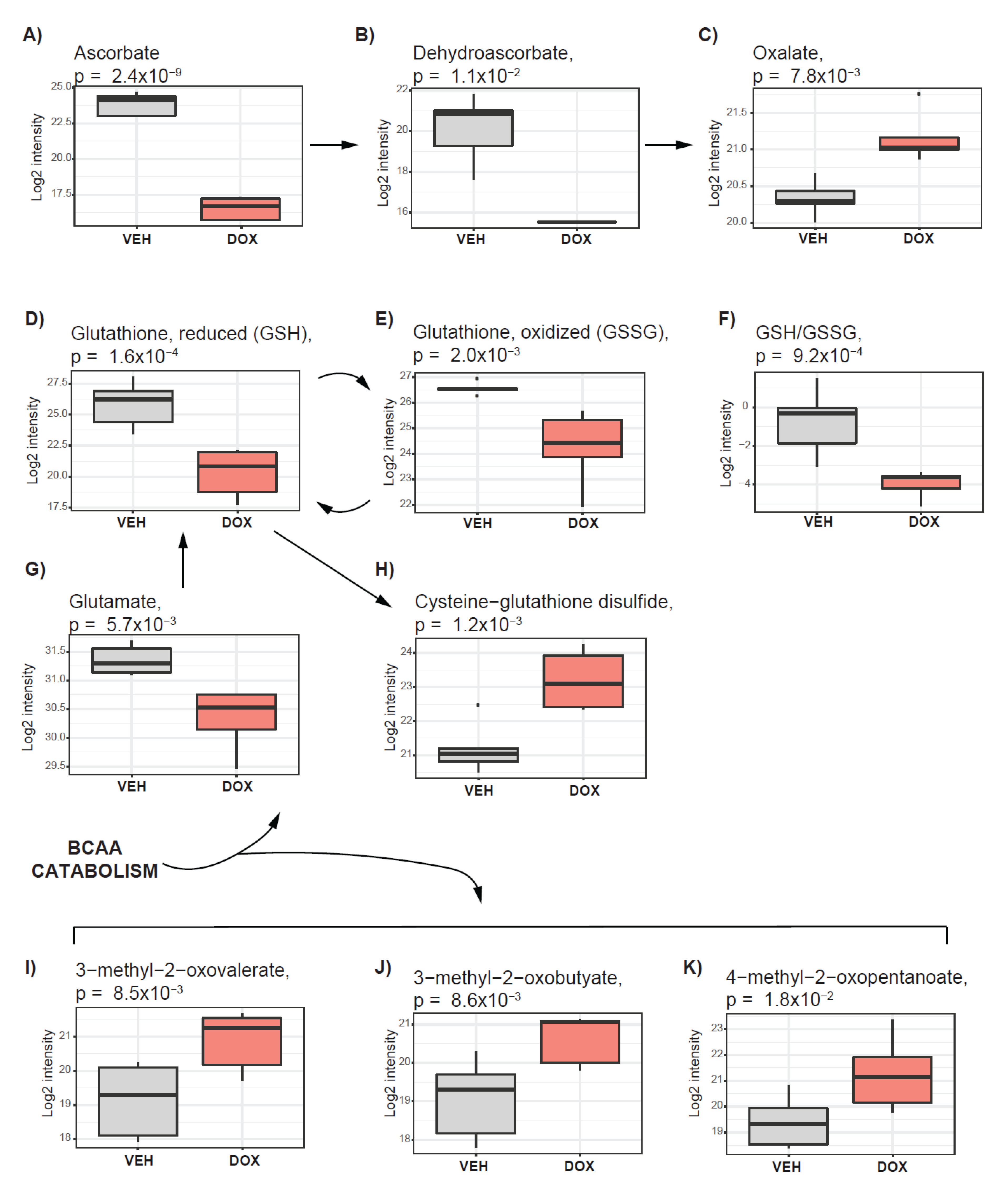
2.5. Pathways Supporting Tumor Antioxidative Capacity Are Activated in Response to Doxorubicin Treatment
3. Discussion
4. Materials and Methods
4.1. In Ovo Experiments
4.1.1. Chick Embryo Tumor Grafting and Treatment
4.1.2. Tumor Growth Analysis
4.1.3. Fresh Tumor Collection for Metabolic Analysis
4.2. Metabolic Profiling
4.2.1. Non-Targeted Metabolic Profiling (HD4 Platform)
4.2.2. Lipidomic Profiling (CLP Platform)
4.3. Viability Assay
4.4. Statistical Data Analysis
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Sorlie, T.; Perou, C.M.; Tibshirani, R.; Aas, T.; Geisler, S.; Johnsen, H.; Hastie, T.; Eisen, M.B.; van de Rijn, M.; Jeffrey, S.S.; et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc. Natl. Acad. Sci. USA 2001, 98, 10869–10874. [Google Scholar] [CrossRef] [PubMed]
- Brenton, J.D.; Carey, L.A.; Ahmed, A.; Caldas, C. Molecular classification and molecular forecasting of breast cancer: Ready for clinical application? J. Clin. Oncol. 2005, 23, 7350–7360. [Google Scholar] [CrossRef] [PubMed]
- Malorni, L.; Shetty, P.B.; De Angelis, C.; Hilsenbeck, S.; Rimawi, M.F.; Elledge, R.; Osborne, C.K.; De Placido, S.; Arpino, G. Clinical and biologic features of triple−negative breast cancers in a large cohort of patients with long−term follow−up. Breast Cancer Res. Treat. 2012, 136, 795–804. [Google Scholar] [CrossRef]
- Hwang, S.Y.; Park, S.; Kwon, Y. Recent therapeutic trends and promising targets in triple negative breast cancer. Pharmacol. Ther. 2019, 199, 30–57. [Google Scholar] [CrossRef] [PubMed]
- Bianchini, G.; Balko, J.M.; Mayer, I.A.; Sanders, M.E.; Gianni, L. Triple−negative breast cancer: Challenges and opportunities of a heterogeneous disease. Nat. Rev. Clin. Oncol. 2016, 13, 674–690. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Warburg, O. The metabolism of carcinoma Cells. J. Cancer Res. 1925, 9, 148–163. [Google Scholar] [CrossRef]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytoto × icity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Lanning, N.J.; Castle, J.P.; Singh, S.J.; Leon, A.N.; Tovar, E.A.; Sanghera, A.; MacKeigan, J.P.; Filipp, F.V.; Graveel, C.R. Metabolic profiling of triple−negative breast cancer cells reveals metabolic vulnerabilities. Cancer Metab. 2017, 5, 6. [Google Scholar] [CrossRef]
- Cao, M.D.; Lamichhane, S.; Lundgren, S.; Bofin, A.; Fjøsne, H.; Giskeødegård, G.F.; Bathen, T.F. Metabolic characterization of triple negative breast cancer. BMC Cancer 2014, 14, 941. [Google Scholar] [CrossRef]
- Tayyari, F.; Gowda, G.A.N.; Olopade, O.F.; Berg, R.; Yang, H.H.; Lee, M.P.; Ngwa, W.F.; Mittal, S.K.; Raftery, D.; Mohammed, S.I. Metabolic profiles of triple−negative and luminal A breast cancer subtypes in African−American identify key metabolic differences. Oncotarget 2018, 9, 11677–11690. [Google Scholar] [CrossRef]
- Lehninger, A.L. Lehninger Principles of Biochemistry, Second Edition, 5th ed.; W.H. Freeman: New York, NY, USA, 1993; ISBN 071677108x. [Google Scholar]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [PubMed]
- Lunt, S.Y.; Vander Heiden, M.G. Aerobic glycolysis: Meeting the metabolic requirements of cell proliferation. Annu. Rev. Cell Dev. Biol. 2011, 27, 441–464. [Google Scholar] [CrossRef]
- Gross, M.I.; Demo, S.D.; Dennison, J.B.; Chen, L.; Chernov−Rogan, T.; Goyal, B.; Janes, J.R.; Laidig, G.J.; Lewis, E.R.; Li, J.; et al. Antitumor activity of the glutaminase inhibitor CB−839 in triple−negative breast cancer. Mol. Cancer Ther. 2014, 13, 890–901. [Google Scholar] [CrossRef] [PubMed]
- Glunde, K.; Bhujwalla, Z.M.; Ronen, S.M. Choline metabolism in malignant transformation. Nat. Rev. Cancer 2011, 11, 835–848. [Google Scholar] [CrossRef]
- Uchida, Y.; Itoh, M.; Taguchi, Y.; Yamaoka, S.; Umehara, H.; Ichikawa, S.-I.; Hirabayashi, Y.; Holleran, W.M.; Okazaki, T. Ceramide reduction and transcriptional up−regulation of glucosylceramide synthase through do xorubicin−activated Sp1 in drug−resistant HL−60/ADR cells. Cancer Res. 2004, 64, 6271–6279. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.-Y.; Yu, J.Y.; Yin, D.; Patwardhan, G.A.; Gupta, V.; Hirabayashi, Y.; Holleran, W.M.; Giuliano, A.E.; Jazwinski, S.M.; Gouaze−Andersson, V.; et al. A role for ceramide in driving cancer cell resistance to do xorubicin. FASEB J. 2008, 22, 2541–2551. [Google Scholar] [CrossRef]
- Xu, M.; Chen, S.; Yang, W.; Cheng, X.; Ye, Y.; Mao, J.; Wu, X.; Huang, L.; Ji, J. fgfr4 links glucose metabolism and chemotherapy resistance in breast cancer. Cell. Physiol. Biochem. 2018, 47, 151–160. [Google Scholar] [CrossRef]
- Clohessy, J.G.; Pandolfi, P.P. Mouse hospital and co−clinical trial project−from bench to bedside. Nat. Rev. Clin. Oncol. 2015, 12, 491–498. [Google Scholar] [CrossRef]
- Nagle, P.W.; Plukker, J.T.M.; Muijs, C.T.; van Luijk, P.; Coppes, R.P. Patient−derived tumor organoids for prediction of cancer treatment response. Semin. Cancer Biol. 2018, 53, 258–264. [Google Scholar] [CrossRef]
- Pauli, C.; Hopkins, B.D.; Prandi, D.; Shaw, R.; Fedrizzi, T.; Sboner, A.; Sailer, V.; Augello, M.; Puca, L.; Rosati, R.; et al. Personalized in vitro and in vivo cancer models to guide precision medicine. Cancer Discov. 2017, 7, 462–477. [Google Scholar] [CrossRef] [PubMed]
- Zabielska−Koczywąs, K.; Michalak, K.; Wojtalewicz, A.; Winiarczyk, M.; Adaszek, Ł.; Winiarczyk, S.; Lechowski, R. Proteomic differences in feline fibrosarcomas grown using doxorubicin−sensitive and −resistant cell lines in the chick embryo model. Int. J. Mol. Sci. 2018, 19, 576. [Google Scholar] [CrossRef] [PubMed]
- DeBord, L.C.; Pathak, R.R.; Villaneuva, M.; Liu, H.-C.; Harrington, D.A.; Yu, W.; Lewis, M.T.; Sikora, A.G. The chick chorioallantoic membrane (CAM) as a versatile patient−derived xenograft (PDX) platform for precision medicine and preclinical research. Am. J. Cancer Res. 2018, 8, 1642–1660. [Google Scholar] [PubMed]
- Stehelin, D.; Varmus, H.E.; Bishop, J.M.; Vogt, P.K. DNA related to the transforming gene(s) of avian sarcoma viruses is present in normal avian DNA. Nature 1976, 260, 170–173. [Google Scholar] [CrossRef] [PubMed]
- Al Dhaheri, Y.; Attoub, S.; Arafat, K.; AbuQamar, S.; Viallet, J.; Saleh, A.; Al Agha, H.; Eid, A.; Iratni, R. Anti−Metastatic and Anti−Tumor growth effects of origanum majorana on highly metastatic human breast cancer cells: Inhibition of NFκB signaling and reduction of nitric oxide production. PLoS ONE 2013, 8, e68808. [Google Scholar] [CrossRef] [PubMed]
- Kue, C.S.; Tan, K.Y.; Lam, M.L.; Lee, H.B. Chick embryo chorioallantoic membrane (CAM): An alternative predictive model in acute to xicological studies for anti−cancer drugs. Exp. Anim. 2014, 64, 129–138. [Google Scholar] [CrossRef] [PubMed]
- Chick Chorioallantoic Membrane (CAM) Assay as an in vivo Model to Study the Effect of Newly Identified Molecules on Ovarian Cancer Invasion and Met. Available online: https://www.ncbi.nlm.nih.gov/pubmed/?term=Chick+Chorioallantoic+Membrane+(CAM)+Assay+as+an+in+Vivo+Model+to+Study+the+Effect+of+Newly+Identified+Molecules+on+Ovarian+Cancer+Invasion+and+Metastasis (accessed on 5 May 2020).
- Evans, A.M.; Bridgewater, B.R.; Liu, Q.; Mitchell, M.W.; Robinson, R.J.; Dai, H.; Stewart, S.J.; DeHaven, C.D.; Miller, L.A.D. High resolution mass spectrometry improves data quantity and quality as compared to unit mass resolution mass spectrometry in high−throughput profiling metabolomics. Metabolomics 2014, 4, 1. [Google Scholar]
- Ubhi, B.K. Direct infusion−tandem mass spectrometry (DI−MS/MS) analysis of complex lipids in human plasma and serum using the lipidyzerTM platform. In Methods in Molecular Biology; Humana Press Inc.: New York, NY, USA; Springer: New York, NY, USA, 2018; Volume 1730, pp. 227–236. [Google Scholar]
- de Lima Junior, E.A.; Yamashita, A.S.; Pimentel, G.D.; De Sousa, L.G.O.; Santos, R.V.T.; Gonçalves, C.L.; Streck, E.L.; de Lira, F.S.; Rosa Neto, J.C. Do xorubicin caused severe hyperglycaemia and insulin resistance, mediated by inhibition in AMPk signalling in skeletal muscle. J. Cachexia Sarcopenia Muscle 2016, 7, 615–625. [Google Scholar] [CrossRef]
- Hove-Jensen, B.; Andersen, K.R.; Kilstrup, M.; Martinussen, J.; Switzer, R.L.; Willemoës, M. Phosphoribosyl Diphosphate (PRPP): Biosynthesis, enzymology, utilization, and metabolic significance. Microbiol. Mol. Biol. Rev. 2017, 81, e00040-16. [Google Scholar]
- Cheng, M.; Rizwan, A.; Jiang, L.; Bhujwalla, Z.M.; Glunde, K. Molecular Effects of Do xorubicin on choline metabolism in breast cancer. Neoplasia (U.S.) 2017, 19, 617–627. [Google Scholar] [CrossRef]
- Greenwood, H.E.; McCormick, P.N.; Gendron, T.; Glaser, M.; Pereira, R.; Maddocks, O.D.K.; Sander, K.; Zhang, T.; Koglin, N.; Lythgoe, M.F.; et al. Measurement of tumor antioxidant capacity and prediction of chemotherapy resistance in preclinical models of ovarian cancer by positron emission tomography. Clin. Cancer Res. 2019, 25, 2471–2482. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, A.; Moss, D.; Sée, V. The chorioallantoic membrane of the chick embryo to assess tumor formation and metastasis. In Methods in Molecular Biology; Humana Press Inc.: New York, NY, USA; Springer: New York, NY, USA, 2016; Volume 1464, pp. 97–105. [Google Scholar]
- Ribatti, D. The chick embryo chorioallantoic membrane (CAM). A multifaceted e × perimental model. Mech. Dev. 2016, 141, 70–77. [Google Scholar] [CrossRef] [PubMed]
- Beger, R.D.; Dunn, W.; Schmidt, M.A.; Gross, S.S.; Kirwan, J.A.; Cascante, M.; Brennan, L.; Wishart, D.S.; Oresic, M.; Hankemeier, T.; et al. Metabolomics enables precision medicine: “A White Paper, Community Perspective”. Metabolomics 2016, 12, 149. [Google Scholar] [CrossRef] [PubMed]
- Hirschey, M.D.; DeBerardinis, R.J.; Diehl, A.M.E.; Drew, J.E.; Frezza, C.; Green, M.F.; Jones, L.W.; Ko, Y.H.; Le, A.; Lea, M.A.; et al. Dysregulated metabolism contributes to oncogenesis. Semin. Cancer Biol. 2015, 35, S129–S150. [Google Scholar] [CrossRef] [PubMed]
- Halama, A.; Kulinski, M.; Dib, S.S.; Zaghlool, S.B.; Siveen, K.S.; Iskandarani, A.; Zierer, J.; Prabhu, K.S.; Satheesh, N.J.; Bhagwat, A.M.; et al. Accelerated lipid catabolism and autophagy are cancer survival mechanisms under inhibited glutaminolysis. Cancer Lett. 2018, 430, 133–147. [Google Scholar] [CrossRef]
- Pera, B.; Krumsiek, J.; Assouline, S.E.; Marullo, R.; Patel, J.; Phillip, J.M.; Román, L.; Mann, K.K.; Cerchietti, L. Metabolomic profiling reveals cellular reprogramming of B−Cell lymphoma by a lysine deacetylase inhibitor through the choline pathway. EBioMedicine 2018, 28, 80–89. [Google Scholar] [CrossRef]
- Alves de Lima, E.; Oliveira de Souza, C.; Abílio de Souza Teixeira, A.; Batatinha, H.A.; de Santos Lira, F.; Neto, J.C.R. Do xorubicin leads to impaired insulin signaling in skeletal muscle. Cancer Metab. 2014, 2, P2. [Google Scholar] [CrossRef]
- Yun, J.; Mullarky, E.; Lu, C.; Bosch, K.N.; Kavalier, A.; Rivera, K.; Roper, J.; Chio, I.I.C.; Giannopoulou, E.G.; Rago, C.; et al. Vitamin C selectively kills KRAS and BRAF mutant colorectal cancer cells by targeting GAPDH. Science (80) 2015, 350, 1391–1396. [Google Scholar] [CrossRef]
- Myers, M.B.; Banda, M.; McKim, K.L.; Wang, Y.; Powell, M.J.; Parsons, B.L. Breast cancer heterogeneity examined by High−Sensitivity Quantification of PIK3CA, KRAS, HRAS, and BRAF mutations in normal breast and ductal carcinomas. Neoplasia (U.S.) 2016, 18, 253–263. [Google Scholar] [CrossRef]
- Griffith, O.W.; Meister, A. Potent and specific inhibition of glutathione synthesis by buthionine sulfo × imine (S−n−butyl homocysteine sulfoximine). J. Biol. Chem. 1979, 254, 7558–7560. [Google Scholar]
- Tagde, A.; Singh, H.; Kang, M.H.; Reynolds, C.P. The glutathione synthesis inhibitor buthionine sulfoximine synergistically enhanced melphalan activity against preclinical models of multiple myeloma. Blood Cancer J. 2014, 4, e229. [Google Scholar] [CrossRef] [PubMed]
- Octavia, Y.; Tocchetti, C.G.; Gabrielson, K.L.; Janssens, S.; Crijns, H.J.; Moens, A.L. Do × orubicin−induced cardiomyopathy: From molecular mechanisms to therapeutic strategies. J. Mol. Cell. Cardiol. 2012, 52, 1213–1225. [Google Scholar] [CrossRef] [PubMed]
- BLIGH, E.G.; DYER, W.J. A rapid method of total lipid extraction and purification. Can. J. Biochem. Physiol. 1959, 37, 911–917. [Google Scholar] [CrossRef] [PubMed]
- Löfgren, L.; Ståhlman, M.; Forsberg, G.-B.; Saarinen, S.; Nilsson, R.; Hansson, G.I. The BUME method: A novel automated chloroform−free 96−well total lipid extraction method for blood plasma. J. Lipid Res. 2012, 53, 1690–1700. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Metabolite | Pathway | Sub-Pathway Metabolism | Beta | p-Value |
|---|---|---|---|---|
| N-acetylaspartate (NAA) | Amino Acid | Alanine and Aspartate | −2.50 | 1.08 × 10−3 |
| N-acetylalanine | −1.25 | 1.49 × 10−3 | ||
| N-acetylglutamate | Glutamate | −3.10 | 1.67 × 10−4 | |
| N-acetyl-aspartyl-glutamate | −2.25 | 3.73 × 10−3 | ||
| Beta-citrylglutamate | −1.65 | 4.68 × 10−3 | ||
| Glutamate | −1.02 | 5.69 × 10−3 | ||
| Glutathione, reduced (GSH) | Glutathione | −5.53 | 1.63 × 10−4 | |
| Glutathione, oxidized (GSSG) | −2.31 | 2.05 × 10−3 | ||
| S-(1,2-dicarboxyethyl)glutathione | −2.01 | 2.76 × 10−3 | ||
| S-methylglutathione | −1.45 | 9.82 × 10−3 | ||
| Cysteine-glutathione disulfide | 2.00 | 1.16 × 10−3 | ||
| N-acetylserine | Glycine, Serine, and Threonine | −0.74 | 1.68 × 10−2 | |
| Glycine | −0.66 | 3.47 × 10−2 | ||
| N-acetylglycine | 1.02 | 1.15 × 10−2 | ||
| 1-methylhistamine | Histidine | −1.39 | 2.95 × 10−3 | |
| N-acetylhistamine | −1.26 | 3.00 × 10−2 | ||
| Imidazole lactate | 0.90 | 4.47 × 10−2 | ||
| 3-methyl-2-oxobutyrate | Leucine, Isoleucine, and Valine | 1.57 | 8.59 × 10−3 | |
| 3-methyl-2-oxovalerate | 1.75 | 8.47 × 10−3 | ||
| 4-methyl-2-oxopentanoate | 1.87 | 1.80 × 10−2 | ||
| N2-acetyllysine | Lysine | −0.98 | 3.23 × 10−3 | |
| Fructosyllysine | 0.80 | 4.47 × 10−2 | ||
| N-acetylmethionine | Methionine, Cysteine, SAM, and Taurine | −1.71 | 5.53 × 10−4 | |
| S-adenosylmethionine (SAM) | −1.20 | 6.85 × 10−3 | ||
| N-acetylmethionine sulfoxide | −0.71 | 3.39 × 10−2 | ||
| Hypotaurine | −0.85 | 3.77 × 10−2 | ||
| N(’1)-acetylspermidine | Polyamine | −1.96 | 2.82 × 10−3 | |
| Spermidine | −0.71 | 1.55 × 10−2 | ||
| N-acetylputrescine | −0.91 | 3.19 × 10−2 | ||
| N1,N12-diacetylspermine | 2.67 | 1.16 × 10−2 | ||
| Serotonin | Tryptophan | −1.29 | 1.14 × 10−2 | |
| N-formylanthranilic acid | 1.34 | 6.32 × 10−3 | ||
| O-methyltyrosine | Tyrosine | −1.24 | 9.36 × 10−3 | |
| 1-carboxyethyltyrosine | −1.13 | 1.54 × 10−2 | ||
| N-formylphenylalanine | 0.89 | 2.06 × 10−2 | ||
| N-monomethylarginine | Urea cycle | −1.54 | 3.10 × 10−2 | |
| N-acetylglucosaminylasparagine | Carbohydrate | Aminosugar | −0.95 | 3.57 × 10−3 |
| N-acetylglucosamine 6-P | −1.03 | 2.32 × 10−2 | ||
| N-acetylneuraminate | −0.73 | 3.75 × 10−2 | ||
| Glucose 1,6-diphosphate | Glycolysis, Gluconeogenesis, and Pyruvate | −1.62 | 4.27 × 10−2 | |
| Lactate | −0.86 | 4.71 × 10−2 | ||
| Glucose | 1.33 | 2.56 × 10−2 | ||
| UDP-glucose | Nucleotide Sugar | −1.50 | 2.35 × 10−3 | |
| UDP-glucuronate | −0.79 | 8.12 × 10−3 | ||
| UDP-galactose | −2.28 | 1.41 × 10−2 | ||
| UDP-N-acetylglucosamine | −0.97 | 2.46 × 10−2 | ||
| Ascorbate (Vitamin C) | Cof & Vit. | Ascorbate and Aldarate | −7.29 | 2.44 × 10−9 |
| Dehydroascorbate | −4.57 | 1.10 × 10−2 | ||
| Oxalate (ethanedioate) | 0.83 | 7.79 × 10−3 | ||
| Nicotinamide | Nicotinate and Nicotinamide | −1.42 | 1.76 × 10−3 | |
| Nicotinamide adenine dinucleotide reduced (NADH) | −1.43 | 9.38 × 10−3 | ||
| Nicotinamide adenine dinucleotide (NAD+) | −1.00 | 9.61 × 10−3 | ||
| Nicotinate | −0.80 | 9.71 × 10−3 | ||
| Adenosine 5’-diphosphoribose | −1.10 | 2.92 × 10−2 | ||
| Nicotinamide ribonucleotide | 1.22 | 1.71 × 10−2 | ||
| Pantothenate | Pantothenate and CoA | −1.14 | 7.54 × 10−4 | |
| Flavin adenine dinucleotide (FAD) | Riboflavin | −0.72 | 3.53 × 10−2 | |
| Thiamin (Vitamin B1) | Thiamine | −1.35 | 2.91 × 10−3 | |
| Alpha-tocopherol | Tocopherol | −2.30 | 2.76 × 10−2 | |
| Pyridoxal phosphate | Vitamin B6 | −1.18 | 2.50 × 10−3 | |
| Pyridoxamine phosphate | −1.83 | 3.79 × 10−3 | ||
| Pyridoxamine | −0.99 | 3.07 × 10−2 | ||
| Phosphate | Energy | Oxidative Phosphorylation | −0.61 | 2.24 × 10−2 |
| Succinate | TCA Cycle | −1.03 | 1.01 × 10−2 | |
| Malate | −0.86 | 1.63 × 10−2 | ||
| Deoxycarnitine | Lipid | Carnitine | −1.09 | 2.94 × 10−3 |
| Docosahexaenoyl ethanolamide | Endocannabinoid | −0.74 | 1.51 × 10−2 | |
| Arachidoylcarnitine (C20) | Fatty Acid (Acyl Carnitine) | −1.19 | 3.11 × 10−2 | |
| Palmitoleoylcarnitine (C16:1) | −1.24 | 1.80 × 10−2 | ||
| Oleoylcarnitine (C18:1) | −1.09 | 2.40 × 10−2 | ||
| Arachidonoylcarnitine (C20:4) | −1.89 | 5.38 × 10−3 | ||
| Linoleoylcarnitine (C18:2) | −1.91 | 8.02 × 10−3 | ||
| Acetylcarnitine (C2) | −0.72 | 4.67 × 10−2 | ||
| Butyrylcarnitine (C4) | −0.83 | 2.34 × 10−2 | ||
| Glycerophosphoglycerol | Glycerolipid | −0.77 | 1.29 × 10−2 | |
| Glycerol | −0.68 | 2.10 × 10−2 | ||
| Myo-inositol | Inositol | −1.35 | 2.29 × 10−3 | |
| Docosapentaenoate (22:5n3) | Long Chain Polyunsaturated Fatty Acid (n3 and n6) | −1.70 | 8.61 × 10−3 | |
| Tetradecadienoate (14:2) | −0.61 | 2.75 × 10−2 | ||
| Dihomo-linoleate (20:2n6) | −1.10 | 3.85 × 10−2 | ||
| Docosapentaenoate (22:5n6) | −1.42 | 4.75 × 10−2 | ||
| Caproate (6:0) | Medium Chain Fatty Acid | 0.98 | 7.18 × 10−3 | |
| 3-hydroxy-3-methylglutarate | Mevalonate | −1.15 | 4.22 × 10−3 | |
| 1-palmitoyl-2-oleoyl-GPG (16:0/18:1) | Phosphatidylglycerol (PG) | −1.16 | 3.95 × 10−3 | |
| 1-stearoyl-2-oleoyl-GPS (18:0/18:1) | Phosphatidylserine (PS) | −0.84 | 9.50 × 10−3 | |
| Glycerophosphoethanolamine | Phospholipid | −2.05 | 1.30 × 10−4 | |
| Glycerophosphorylcholine (GPC) | −2.05 | 2.30 × 10−4 | ||
| Phosphoethanolamine | −1.09 | 4.80 × 10−3 | ||
| Cytidine 5’-diphosphocholine | −1.72 | 6.90 × 10−3 | ||
| Cytidine-5’-diphosphoethanolamine | −1.34 | 7.86 × 10−3 | ||
| Glycerophosphoinositol | −0.66 | 2.83 × 10−2 | ||
| Inosine | Nucleotide | Purine ((Hypo)Xanthine/Inosine) | −1.30 | 1.46 × 10−3 |
| Hypoxanthine | −0.82 | 1.22 × 10−2 | ||
| Allantoic acid | −0.86 | 3.68 × 10−2 | ||
| 2’-deoxyinosine | −0.73 | 4.88 × 10−2 | ||
| Adenylosuccinate | Purine (Adenine) | −1.66 | 3.99 × 10−3 | |
| Adenosine | −1.16 | 4.36 × 10−3 | ||
| 2’-deoxyadenosine | −1.07 | 4.83 × 10−2 | ||
| Guanosine | Purine (Guanine) | −1.40 | 1.30 × 10−3 | |
| Guanine | −1.87 | 1.55 × 10−3 | ||
| Guanosine 5’-diphosphate (GDP) | −1.56 | 2.36 × 10−2 | ||
| 2’-deoxyguanosine | −0.72 | 4.79 × 10−2 | ||
| Cytidine diphosphate | Pyrimidine (Cytidine) | −1.29 | 2.30 × 10−2 | |
| Cytidine 5’-monophosphate | −0.66 | 2.41 × 10−2 | ||
| 2’-deoxycytidine | 1.10 | 2.09 × 10−2 | ||
| 5-methylcytidine | 1.53 | 1.38 × 10−2 | ||
| Orotate | Pyrimidine (Orotate) | 1.36 | 6.40 × 10−3 | |
| 5,6-dihydrothymine | Pyrimidine (Thymine) | −0.76 | 2.21 × 10−2 | |
| Uridine 2’-monophosphate | Pyrimidine (Uracil) | −1.45 | 2.77 × 10−3 | |
| Uridine | −0.97 | 7.47 × 10−3 | ||
| Uracil | −1.00 | 8.67 × 10−3 | ||
| Uridine 5’-diphosphate (UDP) | −1.36 | 2.42 × 10−2 | ||
| 5-methyluridine (ribothymidine) | −0.83 | 4.42 × 10−2 | ||
| Phenylacetylglycine | Peptide | Acetylated Peptides | −1.95 | 2.44 × 10−4 |
| Glycylvaline | Dipeptide | −1.46 | 9.67 × 10−4 | |
| Glycylleucine | −1.45 | 1.39 × 10−3 | ||
| Leucylglycine | −1.37 | 5.91 × 10−3 | ||
| Phenylalanylglycine | −0.90 | 2.21 × 10−2 | ||
| Alanylleucine | −1.13 | 2.64 × 10−2 | ||
| Gamma-glutamylglutamate | Gamma-glutamyl Amino Acid | −1.48 | 2.01 × 10−2 | |
| Gamma-glutamylserine | 0.82 | 1.86 × 10−2 | ||
| Gamma-glutamylhistidine | 0.89 | 3.67 × 10−2 | ||
| Gamma-glutamyl-alpha-lysine | 0.99 | 2.54 × 10−2 | ||
| Gamma-glutamylthreonine | 1.06 | 2.03 × 10−2 | ||
| Gamma-glutamylalanine | 1.17 | 2.73 × 10−2 | ||
| Catechol sulfate | Xenobiotics | Benzoate | 1.15 | 2.76 × 10−2 |
| Ethyl glucuronide | Chemical | −1.84 | 1.02 × 10−2 |
| Metabolite | Pathway | Beta | p-Value |
|---|---|---|---|
| DAG(16:0/22:5) | Diacylglycerol Ester | −1.21 | 2.10 × 10−2 |
| DAG(16:0/22:6) | −1.30 | 1.69 × 10−2 | |
| DAG(16:1/22:6) | −1.08 | 3.34 × 10−2 | |
| DAG(18:2/22:5) | −1.04 | 3.54 × 10−2 | |
| DAG(18:2/22:6) | −1.11 | 3.51 × 10−2 | |
| LPE(22:5) | Lysophosphatidylethanolamine Ester | −0.85 | 2.78 × 10−2 |
| MAG(18:1) | Monoacylglycerol Ester | −0.87 | 1.01 × 10−2 |
| PC(14:0/18:1) | Phosphatidylcholine Ester | −0.85 | 3.31 × 10−2 |
| PC(16:0/14:0) | −1.48 | 2.27 × 10−3 | |
| PC(16:0/16:1) | −1.08 | 1.84 × 10−2 | |
| PC(16:0/18:0) | −0.79 | 3.42 × 10−2 | |
| PC(16:0/20:1) | −1.00 | 1.10 × 10−2 | |
| PC(16:0/20:2) | −1.07 | 7.59 × 10−3 | |
| PC(18:0/14:0) | −1.07 | 1.15 × 10−2 | |
| PC(18:0/16:1) | −1.09 | 3.49 × 10−2 | |
| PC(18:0/18:1) | −1.01 | 2.50 × 10−2 | |
| PC(18:1/16:1) | −1.04 | 1.12 × 10−2 | |
| PC(18:1/18:1) | −1.31 | 6.00 × 10−3 | |
| PC(18:1/18:2) | −0.76 | 3.81 × 10−2 | |
| PC(18:1/20:2) | −1.10 | 4.77 × 10−3 | |
| PC(18:1/20:3) | −0.83 | 3.02 × 10−2 | |
| PC(18:1/22:4) | −0.88 | 2.23 × 10−2 | |
| PC(18:1/22:6) | −0.88 | 1.69 × 10−2 | |
| PC(18:2/16:1) | −0.67 | 4.95 × 10−2 | |
| PC(20:0/18:1) | −0.84 | 3.37 × 10−2 | |
| PE(16:0/16:0) | Phosphatidylethanolamine Ester | −0.76 | 3.29 × 10−2 |
| PE(16:0/18:1) | −0.73 | 4.69 × 10−2 | |
| PE(16:0/20:1) | −0.81 | 1.38 × 10−2 | |
| PE(16:0/20:2) | −1.23 | 1.76 × 10−3 | |
| PE(16:0/22:4) | −0.82 | 3.72 × 10−2 | |
| PE(16:0/22:6) | −0.89 | 3.35 × 10−2 | |
| PE(18:0/16:0) | −0.86 | 4.24 × 10−2 | |
| PE(18:0/18:1) | −1.19 | 7.65 × 10−3 | |
| PE(18:0/20:1) | −1.26 | 7.23 × 10−3 | |
| PE(18:0/20:2) | −1.56 | 6.21 × 10−3 | |
| PE(18:0/22:4) | −0.96 | 2.53 × 10−2 | |
| PE(18:0/22:5) | −1.30 | 7.27 × 10−3 | |
| PE(18:0/22:6) | −1.31 | 7.55 × 10−3 | |
| PE(18:1/16:1) | −0.98 | 1.30 × 10−2 | |
| PE(18:1/18:1) | −1.46 | 3.23 × 10−3 | |
| PE(18:1/20:1) | −1.58 | 9.29 × 10−4 | |
| PE(18:1/20:2) | −1.52 | 1.59 × 10−3 | |
| PE(18:1/20:3) | −1.23 | 1.38 × 10−2 | |
| PE(18:1/20:4) | −0.80 | 3.89 × 10−2 | |
| PE(18:1/22:4) | −1.41 | 3.15 × 10−3 | |
| PE(18:1/22:5) | −1.48 | 1.83 × 10−3 | |
| PE(18:1/22:6) | −1.46 | 3.45 × 10−3 | |
| PE(18:2/22:4) | −0.54 | 4.71 × 10−2 | |
| PE(O-18:0/16:0) | −1.19 | 9.90 × 10−3 | |
| PE(P-16:0/20:5) | Phosphatidylethanolamine Plasmalogen | −0.95 | 3.47 × 10−2 |
| PE(P-18:0/16:0) | −1.24 | 6.36 × 10−3 | |
| PE(P-18:0/20:2) | −1.10 | 2.16 × 10−2 | |
| PE(P-18:0/20:5) | −0.94 | 4.43 × 10−2 | |
| PE(P-18:0/22:5) | −0.88 | 3.48 × 10−2 | |
| PE(P-18:0/22:6) | −0.83 | 4.23 × 10−2 | |
| PE(P-18:1/16:0) | −1.34 | 5.72 × 10−3 | |
| PE(P-18:1/18:1) | −0.90 | 2.94 × 10−2 | |
| PE(P-18:1/20:3) | −1.05 | 2.28 × 10−2 | |
| PE(P-18:1/20:4) | −1.01 | 2.45 × 10−2 | |
| PE(P-18:1/22:5) | −1.00 | 3.45 × 10−2 | |
| PE(P-18:1/22:6) | −1.12 | 1.10 × 10−2 | |
| PI(18:0/16:0) | Phosphatidylinositol Ester | −0.82 | 3.65 × 10−2 |
| SM(22:1) | Sphingomyelin | −0.81 | 3.13 × 10−2 |
| SM(24:0) | −1.09 | 1.68 × 10−2 | |
| SM(24:1) | −0.95 | 2.35 × 10−2 | |
| SM(26:0) | −0.98 | 2.57 × 10−2 | |
| SM(26:1) | −1.20 | 3.52 × 10−3 | |
| TAG56:3−FA20:2 | Triacylglycerol Ester | −1.15 | 4.07 × 10−2 |
| TAG56:4−FA22:4 | −1.38 | 3.19 × 10−2 | |
| TAG56:5−FA22:5 | −1.49 | 2.66 × 10−2 | |
| TAG56:6−FA22:6 | −1.45 | 3.16 × 10−2 | |
| TAG58:10−FA22:5 | −1.35 | 3.27 × 10−2 | |
| TAG58:10−FA22:6 | −1.31 | 4.34 × 10−2 | |
| TAG58:6−FA16:0 | −1.36 | 3.69 × 10−2 | |
| TAG58:6−FA18:0 | −1.25 | 4.19 × 10−2 | |
| TAG58:6−FA22:4 | −1.31 | 3.42 × 10−2 | |
| TAG58:6−FA22:5 | −1.43 | 2.79 × 10−2 | |
| TAG58:7−FA16:0 | −1.22 | 3.33 × 10−2 | |
| TAG58:7−FA18:0 | −1.30 | 3.26 × 10−2 | |
| TAG58:7−FA18:1 | −1.33 | 3.09 × 10−2 | |
| TAG58:7−FA18:2 | −1.23 | 3.79 × 10−2 | |
| TAG58:7−FA22:4 | −1.35 | 3.49 × 10−2 | |
| TAG58:7−FA22:5 | −1.47 | 2.35 × 10−2 | |
| TAG58:7−FA22:6 | −1.51 | 2.94 × 10−2 | |
| TAG58:8−FA18:1 | −1.24 | 4.39 × 10−2 | |
| TAG58:8−FA18:2 | −1.37 | 3.27 × 10−2 | |
| TAG58:8−FA22:5 | −1.42 | 2.63 × 10−2 | |
| TAG58:8−FA22:6 | −1.49 | 2.72 × 10−2 | |
| TAG58:9−FA18:2 | −1.35 | 3.24 × 10−2 | |
| TAG58:9−FA22:5 | −1.37 | 4.12 × 10−2 | |
| TAG58:9−FA22:6 | −1.53 | 2.74 × 10−2 | |
| TAG60:10−FA22:5 | −1.79 | 1.11 × 10−2 | |
| TAG60:10−FA22:6 | −1.70 | 1.33 × 10−2 | |
| TAG60:11−FA22:5 | −1.83 | 8.01 × 10−3 | |
| TAG60:11−FA22:6 | −1.78 | 1.14 × 10−2 | |
| TAG60:12−FA22:6 | −1.89 | 8.91 × 10−3 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Achkar, I.W.; Kader, S.; Dib, S.S.; Junejo, K.; Al-Bader, S.B.; Hayat, S.; Bhagwat, A.M.; Rousset, X.; Wang, Y.; Viallet, J.; et al. Metabolic Signatures of Tumor Responses to Doxorubicin Elucidated by Metabolic Profiling in Ovo. Metabolites 2020, 10, 268. https://doi.org/10.3390/metabo10070268
Achkar IW, Kader S, Dib SS, Junejo K, Al-Bader SB, Hayat S, Bhagwat AM, Rousset X, Wang Y, Viallet J, et al. Metabolic Signatures of Tumor Responses to Doxorubicin Elucidated by Metabolic Profiling in Ovo. Metabolites. 2020; 10(7):268. https://doi.org/10.3390/metabo10070268
Chicago/Turabian StyleAchkar, Iman W., Sara Kader, Shaima S. Dib, Kulsoom Junejo, Salha Bujassoum Al-Bader, Shahina Hayat, Aditya M. Bhagwat, Xavier Rousset, Yan Wang, Jean Viallet, and et al. 2020. "Metabolic Signatures of Tumor Responses to Doxorubicin Elucidated by Metabolic Profiling in Ovo" Metabolites 10, no. 7: 268. https://doi.org/10.3390/metabo10070268
APA StyleAchkar, I. W., Kader, S., Dib, S. S., Junejo, K., Al-Bader, S. B., Hayat, S., Bhagwat, A. M., Rousset, X., Wang, Y., Viallet, J., Suhre, K., & Halama, A. (2020). Metabolic Signatures of Tumor Responses to Doxorubicin Elucidated by Metabolic Profiling in Ovo. Metabolites, 10(7), 268. https://doi.org/10.3390/metabo10070268