Metabolomics and Multi-Omics Integration: A Survey of Computational Methods and Resources

 ,
, 

Abstract
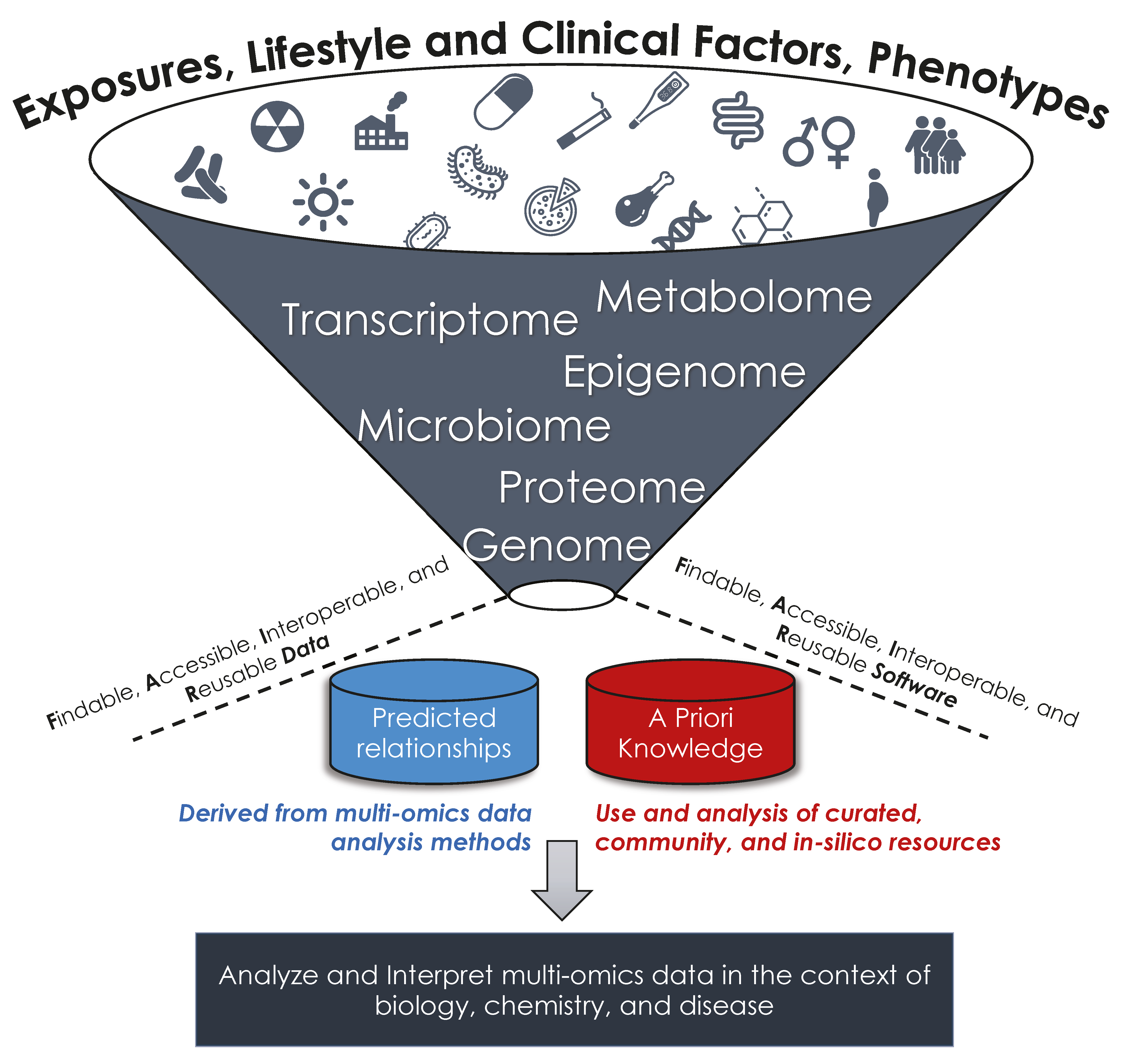
1. Introduction
2. Open-Source Tool Development and Data Guidelines
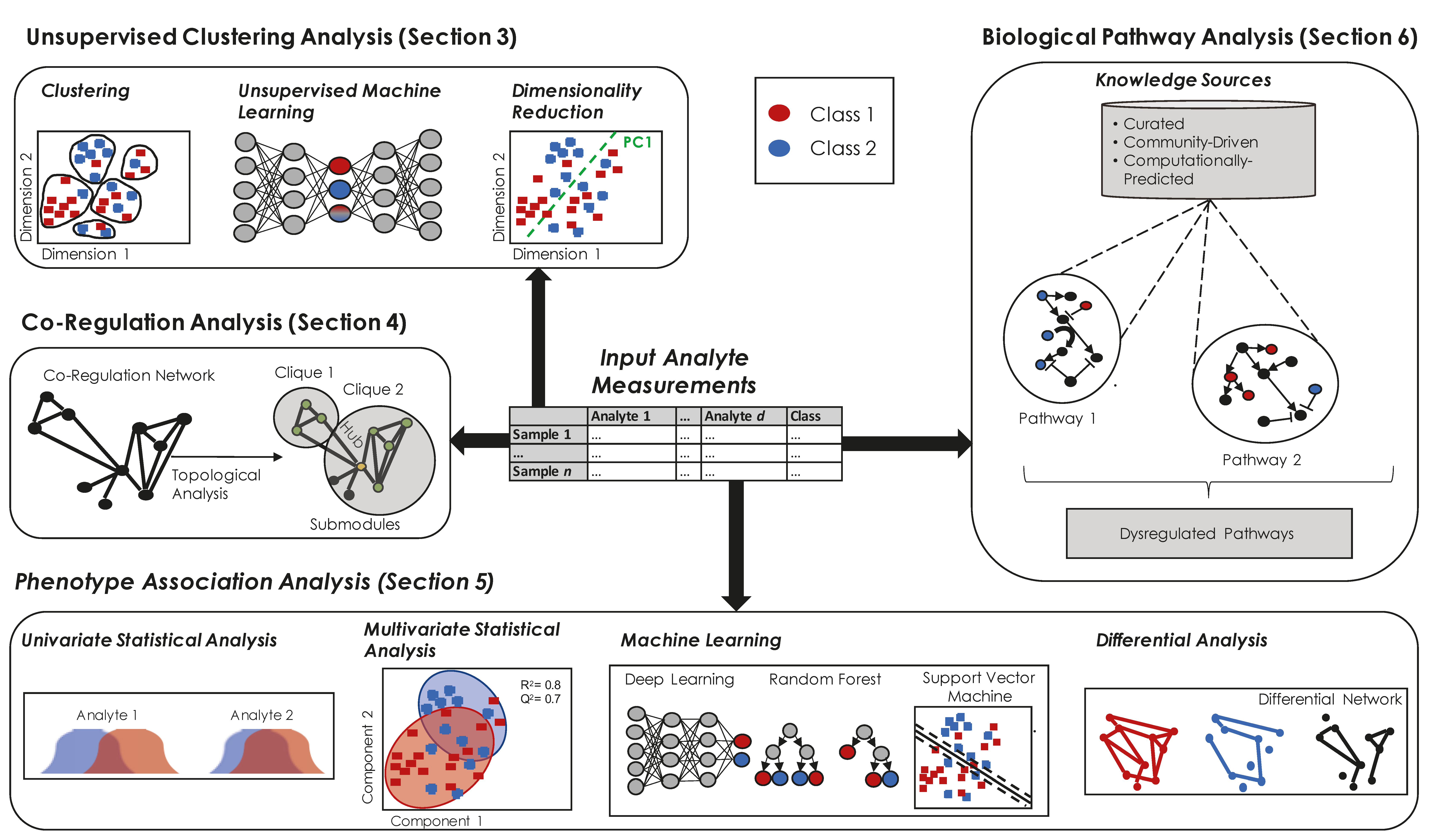
3. Unsupervised Clustering of Samples to Assess Data Quality or Separation by Sample Type
3.1. Dimensionality Reduction
3.2. Clustering
3.3. Other Machine Learning Methods
3.4. Time-Series Data
4. Identifying Groups of Multi-Omics Analytes that are Co-Regulated
4.1. Associative Networks
4.2. Topological Analysis of Networks
5. Identifying Multi-Omics Analytes Associated with Phenotype
5.1. Identifying Differentially Expressed Analytes (Univariate Statistical Methods)
5.2. Multivariate Statistical Methods
5.3. Identifying Analyte Relationships that Differ by Phenotype
5.4. Causative (Flux-Balance) Networks
5.5. Machine Learning Methods for Predicting Phenotype
6. Interpreting a List of Phenotype-Related Analytes in the Context of Biology, Diseases, or Chemistry
6.1. Pathway Enrichment Analysis
6.2. Visualization of Biological Pathways and Networks
6.3. Sources of A Priori Knowledge
6.3.1. Curated and Community Resources
6.3.2. Computationally Predicted Resources
6.3.3. Metrics Used to Define Confidence in Annotations
7. Discussion
8. Conclusions
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Pinu, F.R.; Beale, D.J.; Paten, A.M.; Kouremenos, K.; Swarup, S.; Schirra, H.J.; Wishart, D. Systems biology and multi-omics integration: Viewpoints from the metabolomics research community. Metabolites 2019, 9, 76. [Google Scholar] [CrossRef] [PubMed]
- Chong, J.; Soufan, O.; Li, C.; Caraus, I.; Li, S.; Bourque, G.; Wishart, D.S.; Xia, J. MetaboAnalyst 4.0: Towards more transparent and integrative metabolomics analysis. Nucleic Acids Res. 2018, 46, W486–W494. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.A.; Want, E.J.; O’Maille, G.; Abagyan, R.; Siuzdak, G. XCMS: Processing mass spectrometry data for metabolite profiling using nonlinear peak alignment, matching, and identification. Anal. Chem. 2006, 78, 779–787. [Google Scholar] [CrossRef] [PubMed]
- Rohart, F.; Gautier, B.; Singh, A.; Lê Cao, K.A. mixOmics: An R package for ‘omics feature selection and multiple data integration. PLoS Comput. Biol. 2017, 13, e1005752. [Google Scholar] [CrossRef] [PubMed]
- Ulfenborg, B. Vertical and horizontal integration of multi-omics data with miodin. BMC Bioinform. 2019, 20, 649. [Google Scholar] [CrossRef]
- Kumar, N.; Hoque, M.A.; Sugimoto, M. Robust volcano plot: Identification of differential metabolites in the presence of outliers. BMC Bioinform. 2018, 19, 128. [Google Scholar] [CrossRef]
- Greco, L.; Luta, G.; Krzywinski, M.; Altman, N. Analyzing outliers: Robust methods to the rescue. Nat. Methods 2019, 16, 275–276. [Google Scholar] [CrossRef]
- Taylor, S.L.; Ruhaak, L.R.; Kelly, K.; Weiss, R.H.; Kim, K. Effects of imputation on correlation: Implications for analysis of mass spectrometry data from multiple biological matrices. Brief. Bioinform. 2017, 18, 312–320. [Google Scholar] [CrossRef][Green Version]
- Hughes, R.A.; Heron, J.; Sterne, J.A.C.; Tilling, K. Accounting for missing data in statistical analyses: Multiple imputation is not always the answer. Int. J. Epidemiol. 2019, 48, 1294–1304. [Google Scholar] [CrossRef]
- Lin, D.; Zhang, J.; Li, J.; Xu, C.; Deng, H.-W.; Wang, Y.-P. An integrative imputation method based on multi-omics datasets. BMC Bioinform. 2016, 17, 247. [Google Scholar] [CrossRef]
- Zhu, H.; Li, G.; Lock, E.F. Generalized integrative principal component analysis for multi-type data with block-wise missing structure. Biostatistics 2020, 21, 302–318. [Google Scholar] [CrossRef] [PubMed]
- Chu, S.H.; Huang, M.; Kelly, R.S.; Benedetti, E.; Siddiqui, J.K.; Zeleznik, O.A.; Pereira, A.; Herrington, D.; Wheelock, C.E.; Krumsiek, J.; et al. Integration of Metabolomic and Other Omics Data in Population-Based Study Designs: An Epidemiological Perspective. Metabolites 2019, 9, 117. [Google Scholar] [CrossRef] [PubMed]
- Tarazona, S.; Balzano-Nogueira, L.; Conesa, A. Multiomics Data Integration in Time Series Experiments. In Comprehensive Analytical Chemistry; Elsevier B.V.: Amsterdam, The Netherlands, 2018; Volume 82, pp. 505–532. ISBN 9780444640444. [Google Scholar]
- Ritchie, M.D.; Holzinger, E.R.; Li, R.; Pendergrass, S.A.; Kim, D. Methods of integrating data to uncover genotype-phenotype interactions. Nat. Rev. Genet. 2015, 16, 85–97. [Google Scholar] [CrossRef] [PubMed]
- Misra, B.B.; Langefeld, C.; Olivier, M.; Cox, L.A. Integrated omics: Tools, advances and future approaches. J. Mol. Endocrinol. 2019, 62, R21–R45. [Google Scholar] [CrossRef]
- Cavill, R.; Jennen, D.; Kleinjans, J.; Briedé, J.J. Transcriptomic and metabolomic data integration. Brief. Bioinform. 2016, 17, 891–901. [Google Scholar] [CrossRef] [PubMed]
- Stanstrup, J.; Broeckling, C.D.; Helmus, R.; Hoffmann, N.; Mathé, E.; Naake, T.; Nicolotti, L.; Peters, K.; Rainer, J.; Salek, R.M.; et al. The metaRbolomics Toolbox in Bioconductor and beyond. Metabolites 2019, 9, 200. [Google Scholar] [CrossRef]
- Liu, Z.; Ma, A.; Mathé, E.; Merling, M.; Ma, Q.; Liu, B. Network analyses in microbiome based on high-throughput multi-omics data. Brief. Bioinform. 2020. [Google Scholar] [CrossRef]
- Wilkinson, M.D.; Dumontier, M.; Aalbersberg, I.J.; Appleton, G.; Axton, M.; Baak, A.; Blomberg, N.; Boiten, J.W.; da Silva Santos, L.B.; Bourne, P.E.; et al. The FAIR Guiding Principles for scientific data management and stewardship. Sci. Data 2016, 3, 1–9. [Google Scholar] [CrossRef]
- Lamprecht, A.-L.; Garcia, L.; Kuzak, M.; Martinez, C.; Arcila, R.; Martin Del Pico, E.; Dominguez Del Angel, V.; van de Sandt, S.; Ison, J.; Martinez, P.A.; et al. Towards FAIR principles for research software. Data Sci. 2019, 1–23. [Google Scholar] [CrossRef]
- Silva, L.B.; Jimenez, R.C.; Blomberg, N.; Luis Oliveira, J. General guidelines for biomedical software development. F1000Research 2017, 6, 273. [Google Scholar] [CrossRef]
- Jiménez, R.C.; Kuzak, M.; Alhamdoosh, M.; Barker, M.; Batut, B.; Borg, M.; Capella-Gutierrez, S.; Chue Hong, N.; Cook, M.; Corpas, M.; et al. Four simple recommendations to encourage best practices in research software. F1000Research 2017, 6, 876. [Google Scholar] [CrossRef] [PubMed]
- Russell, P.H.; Johnson, R.L.; Ananthan, S.; Harnke, B.; Carlson, N.E. A large-scale analysis of bioinformatics code on GitHub. PLoS ONE 2018, 13, e0205898. [Google Scholar] [CrossRef] [PubMed]
- Begley, C.G.; Ellis, L.M. Drug development: Raise standards for preclinical cancer research. Nature 2012, 483, 531–533. [Google Scholar] [CrossRef] [PubMed]
- Brazma, A.; Hingamp, P.; Quackenbush, J.; Sherlock, G.; Spellman, P.; Stoeckert, C.; Aach, J.; Ansorge, W.; Ball, C.A.; Causton, H.C.; et al. Minimum information about a microarray experiment (MIAME) - Toward standards for microarray data. Nat. Genet. 2001, 29, 365–371. [Google Scholar] [CrossRef]
- Sumner, L.W.; Amberg, A.; Barrett, D.; Beale, M.H.; Beger, R.; Daykin, C.A.; Fan, T.W.-M.; Fiehn, O.; Goodacre, R.; Griffin, J.L.; et al. Proposed minimum reporting standards for chemical analysis Chemical Analysis Working Group (CAWG) Metabolomics Standards Initiative (MSI). Metabolomics 2007, 3, 211–221. [Google Scholar] [CrossRef]
- Castle, A.L.; Fiehn, O.; Kaddurah-Daouk, R.; Lindon, J.C. Metabolomics Standards Workshop and the development of international standards for reporting metabolomics experimental results. Brief. Bioinform. 2006, 7, 159–165. [Google Scholar] [CrossRef]
- Taylor, C.F.; Paton, N.W.; Lilley, K.S.; Binz, P.A.; Julian, R.K.; Jones, A.R.; Zhu, W.; Apweiler, R.; Aebersold, R.; Deutsch, E.W.; et al. The minimum information about a proteomics experiment (MIAPE). Nat. Biotechnol. 2007, 25, 887–893. [Google Scholar] [CrossRef]
- Ochsner, S.A.; Steffen, D.L.; Stoeckert, C.J.; McKenna, N.J. Much room for improvement in deposition rates of expression microarray datasets. Nat. Methods 2008, 5, 991. [Google Scholar] [CrossRef]
- Spicer, R.A.; Salek, R.; Steinbeck, C. Comment: A decade after the metabolomics standards initiative it’s time for a revision. Sci. Data 2017, 4, 170138. [Google Scholar] [CrossRef]
- Tomczak, K.; Czerwińska, P.; Wiznerowicz, M. The Cancer Genome Atlas (TCGA): An immeasurable source of knowledge. Wspolczesna Onkol. 2015, 19, A68–A77. [Google Scholar] [CrossRef]
- Barretina, J.; Caponigro, G.; Stransky, N.; Venkatesan, K.; Margolin, A.A.; Kim, S.; Wilson, C.J.; Lehár, J.; Kryukov, G.V.; Sonkin, D.; et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 2012, 483, 603–607. [Google Scholar] [CrossRef] [PubMed]
- National Cancer Institute Office of Cancer Genomics TARGET: Therapeutically Applicable Research to Generate Effective Treatments. Available online: https://ocg.cancer.gov/programs/target (accessed on 1 May 2020).
- Edwards, N.J.; Oberti, M.; Thangudu, R.R.; Cai, S.; McGarvey, P.B.; Jacob, S.; Madhavan, S.; Ketchum, K.A. The CPTAC data portal: A resource for cancer proteomics research. J. Proteome Res. 2015, 14, 2707–2713. [Google Scholar] [CrossRef] [PubMed]
- Shoemaker, R.H. The NCI60 human tumour cell line anticancer drug screen. Nat. Rev. Cancer 2006, 6, 813–823. [Google Scholar] [CrossRef] [PubMed]
- Haug, K.; Cochrane, K.; Nainala, V.; Williams, M.; Chang, J.; Jayaseelan, K.; O’Donovan, C. MetaboLights: A resource evolving in response to the needs of its scientific community. - PubMed - NCBI. Nucleic Acids Res. 2020, 48, D440–D444. [Google Scholar]
- Sud, M.; Fahy, E.; Cotter, D.; Azam, K.; Vadivelu, I.; Burant, C.; Edison, A.; Fiehn, O.; Higashi, R.; Nair, K.S.; et al. Metabolomics Workbench: An international repository for metabolomics data and metadata, metabolite standards, protocols, tutorials and training, and analysis tools. Nucleic Acids Res. 2016, 44, D463–D470. [Google Scholar] [CrossRef]
- Vizcaíno, J.A.; Côté, R.; Reisinger, F.; Foster, J.M.; Mueller, M.; Rameseder, J.; Hermjakob, H.; Martens, L. A guide to the Proteomics Identifications Database proteomics data repository. Proteomics 2009, 9, 4276–4283. [Google Scholar] [CrossRef]
- Clough, E.; Barrett, T. The Gene Expression Omnibus database. Methods Mol. Biol. 2016, 1418, 93–110. [Google Scholar]
- Leinonen, R.; Sugawara, H.; Shumway, M. The Sequence Read Archive. Nucleic Acids Res. 2011, 39, D19–D21. [Google Scholar] [CrossRef]
- Feingold, E.A.; Good, P.J.; Guyer, M.S.; Kamholz, S.; Liefer, L.; Wetterstrand, K.; Collins, F.S.; Gingeras, T.R.; Kampa, D.; Sekinger, E.A.; et al. The ENCODE (ENCyclopedia of DNA Elements) Project. Science 2004, 306, 636–640. [Google Scholar] [CrossRef]
- Methé, B.A.; Nelson, K.E.; Pop, M.; Creasy, H.H.; Giglio, M.G.; Huttenhower, C.; Gevers, D.; Petrosino, J.F.; Abubucker, S.; Badger, J.H.; et al. A framework for human microbiome research. Nature 2012, 486, 215–221. [Google Scholar]
- Oliveira, F.S.; Brestelli, J.; Cade, S.; Zheng, J.; Iodice, J.; Fischer, S.; Aurrecoechea, C.; Kissinger, J.C.; Brunk, B.P.; Stoeckert, C.J.; et al. MicrobiomeDB: A systems biology platform for integrating, mining and analyzing microbiome experiments. Nucleic Acids Res. 2018, 46. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Yu, W.-H.; Izard, J.; Baranova, O.V.; Lakshmanan, A.; Dewhirst, F.E. The Human Oral Microbiome Database: A web accessible resource for investigating oral microbe taxonomic and genomic information. Database (Oxford) 2010, 2010, baq013. [Google Scholar] [CrossRef] [PubMed]
- Sarkans, U.; Gostev, M.; Athar, A.; Behrangi, E.; Melnichuk, O.; Ali, A.; Minguet, J.; Rada, J.; Snow, C.; Tikhonov, A.; et al. The BioStudies database-one stop shop for all data supporting a life sciences study. Nucleic Acids Res. 2018, 46, D1266–D1270. [Google Scholar] [CrossRef] [PubMed]
- Sreng, N.; Champion, S.; Martin, J.C.; Khelaifia, S.; Christensen, J.E.; Padmanabhan, R.; Azalbert, V.; Blasco-Baque, V.; Loubieres, P.; Pechere, L.; et al. Resveratrol-mediated glycemic regulation is blunted by curcumin and is associated to modulation of gut microbiota. J. Nutr. Biochem. 2019, 72, 108218. [Google Scholar] [CrossRef] [PubMed]
- Tkachev, A.; Stepanova, V.; Zhang, L.; Khrameeva, E.; Zubkov, D.; Giavalisco, P.; Khaitovich, P. Differences in lipidome and metabolome organization of prefrontal cortex among human populations. Sci. Rep. 2019, 9, 18348. [Google Scholar] [CrossRef]
- Chaisaingmongkol, J.; Budhu, A.; Dang, H.; Rabibhadana, S.; Pupacdi, B.; Kwon, S.M.; Forgues, M.; Pomyen, Y.; Bhudhisawasdi, V.; Lertprasertsuke, N.; et al. Common Molecular Subtypes Among Asian Hepatocellular Carcinoma and Cholangiocarcinoma. Cancer Cell 2017, 32, 57–70. [Google Scholar] [CrossRef]
- Terunuma, A.; Putluri, N.; Mishra, P.; Mathé, E.A.; Dorsey, T.H.; Yi, M.; Wallace, T.A.; Issaq, H.J.; Zhou, M.; Killian, J.K.; et al. MYC-driven accumulation of 2-hydroxyglutarate is associated with breast cancer prognosis. J. Clin. Invest. 2014, 124, 398–412. [Google Scholar] [CrossRef]
- Overmyer, K.A.; Rhoads, T.W.; Merrill, A.E.; Ye, Z.; Westphall, M.S.; Acharya, A.; Shukla, S.K.; Coon, J.J. Proteomics, lipidomics, metabolomics and 16S DNA sequencing of dental plaque from patients with diabetes and periodontal disease. bioRxiv 2020. [Google Scholar] [CrossRef]
- Battaglioli, E.J.; Hale, V.L.; Chen, J.; Jeraldo, P.; Ruiz-Mojica, C.; Schmidt, B.A.; Rekdal, V.M.; Till, L.M.; Huq, L.; Smits, S.A.; et al. Clostridioides difficile uses amino acids associated with gut microbial dysbiosis in a subset of patients with diarrhea. Sci. Transl. Med. 2018, 10, eaam7019. [Google Scholar] [CrossRef]
- Athreya, A.; Iyer, R.; Neavin, D.; Wang, L.; Weinshilboum, R.; Kaddurah-Daouk, R.; Rush, J.; Frye, M.; Bobo, W. Augmentation of physician assessments with multi-omics enhances predictability of drug response: A case study of major depressive disorder. IEEE Comput. Intell. Mag. 2018, 13, 20–31. [Google Scholar] [CrossRef]
- Schmaler, M.; Colone, A.; Spagnuolo, J.; Zimmermann, M.; Lepore, M.; Kalinichenko, A.; Bhatia, S.; Cottier, F.; Rutishauser, T.; Pavelka, N.; et al. Modulation of bacterial metabolism by the microenvironment controls MAIT cell stimulation. Mucosal Immunol. 2018, 11, 1060–1070. [Google Scholar] [CrossRef] [PubMed]
- Knudsen, E.S.; Balaji, U.; Freinkman, E.; McCue, P.; Witkiewicz, A.K. Unique metabolic features of pancreatic cancer stroma: Relevance to the tumor compartment, prognosis, and invasive potential. Oncotarget 2016, 7, 78396–78411. [Google Scholar] [CrossRef] [PubMed]
- Chung, N.C.; Mirza, B.; Choi, H.; Wang, J.; Wang, D.; Ping, P.; Wang, W. Unsupervised classification of multi-omics data during cardiac remodeling using deep learning. Methods 2019, 166, 66–73. [Google Scholar] [CrossRef] [PubMed]
- Argelaguet, R.; Velten, B.; Arnol, D.; Dietrich, S.; Zenz, T.; Marioni, J.C.; Buettner, F.; Huber, W.; Stegle, O. Multi-Omics Factor Analysis—A framework for unsupervised integration of multi-omics data sets. Mol. Syst. Boil. 2018, 14, e8124. [Google Scholar] [CrossRef] [PubMed]
- Meng, C.; Kuster, B.; Culhane, A.C.; Gholami, A.M. A multivariate approach to the integration of multi-omics datasets. BMC Bioinform. 2014, 15, 162. [Google Scholar] [CrossRef] [PubMed]
- Hout, M.C.; Papesh, M.H.; Goldinger, S.D. Multidimensional scaling. Wiley Interdiscip. Rev. Cogn. Sci. 2013, 4, 93–103. [Google Scholar] [CrossRef]
- Kuczynski, J.; Liu, Z.; Lozupone, C.; McDonald, D.; Fierer, N.; Knight, R. Microbial community resemblance methods differ in their ability to detect biologically relevant patterns. Nat. Methods 2010, 7, 813–819. [Google Scholar] [CrossRef]
- Van Der Maaten, L.; Hinton, G. Visualizing Data using t-SNE. J. March. Learn. Res. 2008, 9, 2579–2605. [Google Scholar]
- Wattenberg, M.; Viégas, F.; Johnson, I. How to Use t-SNE Effectively. Distill 2016, 1, e2. [Google Scholar] [CrossRef]
- Kimes, P.K.; Liu, Y.; Neil Hayes, D.; Marron, J.S. Statistical significance for hierarchical clustering. Biometrics 2017, 73, 811–821. [Google Scholar] [CrossRef]
- Macqueen, J.; Macqueen, J. Some methods for classification and analysis of multivariate observations. In Proceedings of the 5th Berkeley Symposium on Mathematical Statistics and Probability, Berkeley, CA, USA, 21 June–18 July 1965; pp. 281–297. [Google Scholar]
- Kaufman, L.; Rousseeuw, P. Finding Groups in Data: An Introduction to Cluster Analysis; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 1990. [Google Scholar]
- Rousseeuw, P.J. Silhouettes: A graphical aid to the interpretation and validation of cluster analysis. J. Comput. Appl. Math. 1987, 20, 53–65. [Google Scholar] [CrossRef]
- Tibshirani, R.; Walther, G.; Hastie, T. Estimating the number of clusters in a data set via the gap statistic. J. R. Stat. Soc. 2001, 63, 411–423. [Google Scholar] [CrossRef]
- Kohonen, T. The self-organizing map. Neurocomputing 1998, 21, 1–6. [Google Scholar] [CrossRef]
- Hamel, L.; Ott, B. A Population Based Convergence Criterion for Self-Organizing Maps. In Proceedings of the 2012 International Conference on Data Mining, Brussels, Belgium, 10–13 December 2012; pp. 98–104. [Google Scholar]
- Kiviluoto, K. Topology preservation in self-organizing maps. In Proceedings of the International Conference on Neural Networks (ICNN’96), Washington, DC, USA, 2–7 June 1996; pp. 294–299. [Google Scholar]
- Milone, D.H.; Stegmayer, G.S.; Kamenetzky, L.; López, M.; Lee, J.M.; Giovannoni, J.J.; Carrari, F. *omeSOM: A software for clustering and visualization of transcriptional and metabolite data mined from interspecific crosses of crop plants. BMC Bioinform. 2010, 11, 438. [Google Scholar] [CrossRef]
- Duda, R.O.; Hart, P.E.; Stork, D.G. Pattern Classification. In Pattern Classification and Scene Analysis; John Wiley & Sons: Hoboken, NJ, USA, 1995; pp. 6–22. [Google Scholar]
- Schwarz, G. Estimating the Dimension of a Model. Ann. Stat. 1978, 6, 461–464. [Google Scholar] [CrossRef]
- Akaike, H. Information theory and an extension of the maximum likelihood principle. In Proceedings of the 2nd International Symposium on Information Theory, Akadémiai Kiadó, Budapest, Hungary, 2–8 September 1971; pp. 267–281. [Google Scholar]
- Ester, M.; Kriegel, H.-P.; Sander, J.; Xu, X. A Density-Based Algorithm for Discovering Clusters in Large Spatial Databases with Noise. In Proceedings of the Second International Conference on Knowledge Discovery and Data Mining, Portland, OR, USA, 2–4 August 1996. [Google Scholar]
- Breiman, L. Random forests. Mach. Learn. 2001, 45, 5–32. [Google Scholar] [CrossRef]
- Boser, B.E.; Guyon, I.M.; Vapnik, V.N. Training algorithm for optimal margin classifiers. In Proceedings of the Fifth Annual ACM Workshop on Computational Learning Theory; Publ by ACM, Pittsburgh, PA, USA, 27–29 July 1992; pp. 144–152. [Google Scholar]
- Winters-Hilt, S.; Merat, S. SVM clustering. In Proceedings of the BMC Bioinformatics, BioMed Central, New Orleans, LA, USA, 1–3 Febuary 2007; p. S18. [Google Scholar]
- Ballard, D.H. Modular Learning in Neural Networks. In Proceedings of the Association for the Advancement of Artificial Intelligence Sixth National Conference on Artificial Intelligence, Seattle, WA, USA, 13–17 July 1987. [Google Scholar]
- Samek, W.; Wiegand, T.; Müller, K.-R. Explainable Artificial Intelligence: Understanding, Visualizing, and Interpreting Deep Learning Models. ITU J. ICT Discov. 2017. [Google Scholar]
- Karim, M.R.; Beyan, O.; Zappa, A.; Costa, I.G.; Rebholz-Schuhmann, D.; Cochez, M.; Decker, S. Deep learning-based clustering approaches for bioinformatics. Brief. Bioinform. 2020. [Google Scholar] [CrossRef]
- Bar-Joseph, Z.; Gerber, G.K.; Gifford, D.K.; Jaakkola, T.S.; Simon, I. Continuous Representations of Time-Series Gene Expression Data. J. Comput. Biol. 2003, 10, 341–356. [Google Scholar] [CrossRef]
- Déjean, S.; Martin, P.; Baccini, A.; Besse, P. Clustering Time-Series Gene Expression Data Using Smoothing Spline Derivatives. EURASIP J. Bioinform. Syst. Biol. 2007, 2007, 70561. [Google Scholar] [CrossRef]
- Corduas, M.; Piccolo, D. Time series clustering and classification by the autoregressive metric. Comput. Stat. Data Anal. 2008, 52, 1860–1872. [Google Scholar] [CrossRef]
- Kalpakis, K.; Gada, D.; Puttagunta, V. Distance measures for effective clustering of ARIMA time-series. In Proceedings of the IEEE International Conference on Data Mining, San Jose, CA, USA, 29 November–2 December 2001; pp. 273–280. [Google Scholar]
- Smyth, P. Clustering Sequences with Hidden Markov Models. Adv. Neural Inf. Process. Syst. 1997, 9, 648–654. [Google Scholar]
- Zeng, Y.; Garcia-Frias, J. A novel HMM-based clustering algorithm for the analysis of gene expression time-course data. Comput. Stat. Data Anal. 2006, 50, 2472–2494. [Google Scholar] [CrossRef]
- Jaskowiak, P.A.; Campello, R.J.G.B.; Costa, I.G. On the selection of appropriate distances for gene expression data clustering. BMC Bioinform. 2014, 15. [Google Scholar] [CrossRef]
- Giorgino, T. Computing and visualizing dynamic time warping alignments in R: The dtw package. J. Stat. Softw. 2009, 31, 1–24. [Google Scholar] [CrossRef]
- Chandereng, T.; Gitter, A. Lag penalized weighted correlation for time series clustering. BMC Bioinform. 2020, 21, 21. [Google Scholar] [CrossRef]
- Camacho, D.; de la Fuente, A.; Mendes, P. The origin of correlations in metabolomics data. Metabolomics 2005, 1, 53–63. [Google Scholar] [CrossRef]
- Do, K.T.; Kastenmüller, G.; Mook-Kanamori, D.O.; Yousri, N.A.; Theis, F.J.; Suhre, K.; Krumsiek, J. Network-based approach for analyzing intra- and interfluid metabolite associations in human blood, urine, and saliva. J. Proteome Res. 2015, 14, 1183–1194. [Google Scholar] [CrossRef]
- Wahl, S.; Vogt, S.; Stückler, F.; Krumsiek, J.; Bartel, J.; Kacprowski, T.; Schramm, K.; Carstensen, M.; Rathmann, W.; Roden, M.; et al. Multi-omic signature of body weight change: Results from a population-based cohort study. BMC Med. 2015, 13, 48. [Google Scholar] [CrossRef]
- Lloyd-Price, J.; Arze, C.; Ananthakrishnan, A.N.; Schirmer, M.; Avila-Pacheco, J.; Poon, T.W.; Andrews, E.; Ajami, N.J.; Bonham, K.S.; Brislawn, C.J.; et al. Multi-omics of the gut microbial ecosystem in inflammatory bowel diseases. Nature 2019, 569, 655–662. [Google Scholar] [CrossRef]
- Li, S.; Sullivan, N.L.; Rouphael, N.; Yu, T.; Banton, S.; Maddur, M.S.; McCausland, M.; Chiu, C.; Canniff, J.; Dubey, S.; et al. Metabolic Phenotypes of Response to Vaccination in Humans. Cell 2017, 169, 862–877. [Google Scholar] [CrossRef] [PubMed]
- Aho, V.; Ollila, H.M.; Kronholm, E.; Bondia-Pons, I.; Soininen, P.; Kangas, A.J.; Hilvo, M.; Seppälä, I.; Kettunen, J.; Oikonen, M.; et al. Prolonged sleep restriction induces changes in pathways involved in cholesterol metabolism and inflammatory responses. Sci. Rep. 2016, 6, 24828. [Google Scholar] [CrossRef] [PubMed]
- Acharjee, A.; Ament, Z.; West, J.A.; Stanley, E.; Griffin, J.L. Integration of metabolomics, lipidomics and clinical data using a machine learning method. BMC Bioinform. 2016, 17, 37–49. [Google Scholar] [CrossRef] [PubMed]
- Schubert, K.O.; Stacey, D.; Arentz, G.; Clark, S.R.; Air, T.; Hoffmann, P.; Baune, B.T. Targeted proteomic analysis of cognitive dysfunction in remitted major depressive disorder: Opportunities of multi-omics approaches towards predictive, preventive, and personalized psychiatry. J. Proteomics 2018, 188, 63–70. [Google Scholar] [CrossRef] [PubMed]
- Kelly, R.S.; Chawes, B.L.; Blighe, K.; Virkud, Y.V.; Croteau-Chonka, D.C.; McGeachie, M.J.; Clish, C.B.; Bullock, K.; Celedón, J.C.; Weiss, S.T.; et al. An Integrative Transcriptomic and Metabolomic Study of Lung Function in Children With Asthma. Chest 2018, 154, 335–348. [Google Scholar] [CrossRef]
- Heiland, D.H.; Wörner, J.; Haaker, J.G.; Delev, D.; Pompe, N.; Mercas, B.; Franco, P.; Gäbelein, A.; Heynckes, S.; Pfeifer, D.; et al. The integrative metabolomic-transcriptomic landscape of glioblastome multiforme. Oncotarget 2017, 8, 49178–49190. [Google Scholar] [CrossRef][Green Version]
- Feng, J.; Zhang, Q.; Zhou, Y.; Yu, S.; Hong, L.; Zhao, S.; Yang, J.; Wan, H.; Xu, G.; Zhang, Y.; et al. Integration of Proteomics and Metabolomics Revealed Metabolite–Protein Networks in ACTH-Secreting Pituitary Adenoma. Front. Endocrinol. (Lausanne) 2018, 9, 678. [Google Scholar] [CrossRef]
- Price, N.D.; Magis, A.T.; Earls, J.C.; Glusman, G.; Levy, R.; Lausted, C.; McDonald, D.T.; Kusebauch, U.; Moss, C.L.; Zhou, Y.; et al. A wellness study of 108 individuals using personal, dense, dynamic data clouds. Nat. Biotechnol. 2017, 35, 747–756. [Google Scholar] [CrossRef]
- Butte, A.J.; Kohane, I.S. Relevance Networks: A First Step Toward Finding Genetic Regulatory Networks Within Microarray Data. In The Analysis of Gene Expression Data; Springer: New York, NY, USA, 2003; pp. 428–446. [Google Scholar]
- Kayano, M.; Imoto, S.; Yamaguchi, R.; Miyano, S. Multi-omics approach for estimating metabolic networks using low-order partial correlations. J. Comput. Biol. 2013, 20, 571–582. [Google Scholar] [CrossRef]
- Li, Z.; Zuo, Y.; Xu, C.; Varghese, R.S.; Ressom, H.W. INDEED: R package for network based differential expression analysis. In Proceedings of the 2018 IEEE International Conference on Bioinformatics and Biomedicine, Madrid, Spain, 3–6 December 2018; pp. 2709–2712. [Google Scholar]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef]
- Longabaugh, W.J.R. Combing the hairball with BioFabric: A new approach for visualization of large networks. BMC Bioinform. 2012, 13, 275. [Google Scholar] [CrossRef] [PubMed]
- Jiang, P.; Wang, H.; Li, W.; Zang, C.; Li, B.; Wong, Y.J.; Meyer, C.; Liu, J.S.; Aster, J.C.; Liu, X.S. Network analysis of gene essentiality in functional genomics experiments. Genome Biol. 2015, 16, 239. [Google Scholar] [CrossRef] [PubMed]
- Azevedo, H.; Moreira-Filho, C.A. Topological robustness analysis of protein interaction networks reveals key targets for overcoming chemotherapy resistance in glioma. Sci. Rep. 2015, 5, 16830. [Google Scholar] [CrossRef] [PubMed]
- Jalili, M. Functional Brain Networks: Does the Choice of Dependency Estimator and Binarization Method Matter? Sci. Rep. 2016, 6, 29780. [Google Scholar] [CrossRef] [PubMed]
- Waller, T.C.; Berg, J.A.; Lex, A.; Chapman, B.E.; Rutter, J. Compartment and hub definitions tune metabolic networks for metabolomic interpretations. Gigascience 2020, 9. [Google Scholar] [CrossRef] [PubMed]
- Wagner, A.; Fell, D.A. The small world inside large metabolic networks. Proc. R. Soc. B Biol. Sci. 2001, 268, 1803–1810. [Google Scholar] [CrossRef] [PubMed]
- Kitsak, M.; Sharma, A.; Menche, J.; Guney, E.; Ghiassian, S.D.; Loscalzo, J.; Barabási, A.-L. Tissue Specificity of Human Disease Module. Sci. Rep. 2016, 6, 35241. [Google Scholar] [CrossRef]
- Kim, S.; Thapa, I.; Zhang, L.; Ali, H. A novel graph theoretical approach for modeling microbiomes and inferring microbial ecological relationships. BMC Genomics 2019, 20, 1–13. [Google Scholar] [CrossRef]
- Celik, S.; Logsdon, B.; Lee, S. Efficient Dimensionality Reduction for High-Dimensional Network Estimation. In Proceedings of the 31st International Conference on Machine Learning, Beijing, China, 21–26 June 2014; pp. 1953–1961. [Google Scholar]
- Blondel, V.D.; Guillaume, J.-L.; Lambiotte, R.; Lefebvre, E. Fast unfolding of communities in large networks. J. Stat. Mech. Theory Exp. 2008, 2008. [Google Scholar] [CrossRef]
- Gaynor, S.M.; Lin, X.; Quackenbush, J. Spectral clustering in regression-based biological networks. bioRxiv 2019, 651950. [Google Scholar]
- Lu, Z.; Wahlström, J.; Nehorai, A. Community Detection in Complex Networks via Clique Conductance. Sci. Rep. 2018, 8, 5982. [Google Scholar] [CrossRef] [PubMed]
- Teran Hidalgo, S.J.; Ma, S. Clustering multilayer omics data using MuNCut. BMC Genomics 2018, 19, 198. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Li, C.-L.; Tu, B.-J.; Yang, K.; Mo, T.-T.; Zhang, R.-Y.; Cheng, S.-Q.; Chen, C.-Z.; Jiang, X.-J.; Han, T.-L.; et al. Integrated Epigenetics, Transcriptomics, and Metabolomics to Analyze the Mechanisms of Benzo[a]pyrene Neurotoxicity in the Hippocampus. Toxicol. Sci. 2018, 166, 65–81. [Google Scholar] [CrossRef] [PubMed]
- Yoon, H.; Yoon, D.; Yun, M.; Choi, J.S.; Park, V.Y.; Kim, E.K.; Jeong, J.; Koo, J.S.; Yoon, J.H.; Moon, H.J.; et al. Metabolomics of Breast Cancer Using High-Resolution Magic Angle Spinning Magnetic Resonance Spectroscopy: Correlations with 18F-FDG Positron Emission Tomography-Computed Tomography, Dynamic Contrast-Enhanced and Diffusion-Weighted Imaging MRI. PLoS ONE 2016, 11, e0159949. [Google Scholar] [CrossRef] [PubMed]
- Huan, T.; Troyer, D.A.; Li, L. Metabolite Analysis and Histology on the Exact Same Tissue: Comprehensive Metabolomic Profiling and Metabolic Classification of Prostate Cancer. Sci. Rep. 2016, 6, 1–13. [Google Scholar] [CrossRef]
- Clos-Garcia, M.; Andrés-Marin, N.; Fernández-Eulate, G.; Abecia, L.; Lavín, J.L.; van Liempd, S.; Cabrera, D.; Royo, F.; Valero, A.; Errazquin, N.; et al. Gut microbiome and serum metabolome analyses identify molecular biomarkers and altered glutamate metabolism in fibromyalgia. EBioMedicine 2019, 46, 499–511. [Google Scholar] [CrossRef]
- Lee, H.; Choi, J.M.; Cho, J.Y.; Kim, T.E.; Lee, H.J.; Jung, B.H. Regulation of endogenic metabolites by rosuvastatin in hyperlipidemia patients: An integration of metabolomics and lipidomics. Chem. Phys. Lipids 2018, 214, 69–83. [Google Scholar] [CrossRef]
- Esther, C.R.; Turkovic, L.; Rosenow, T.; Muhlebach, M.S.; Boucher, R.C.; Ranganathan, S.; Stick, S.M. Metabolomic biomarkers predictive of early structural lung disease in cystic fibrosis. Eur. Respir. J. 2016, 48, 1612–1621. [Google Scholar] [CrossRef]
- Neeland, I.J.; Boone, S.C.; Mook-Kanamori, D.O.; Ayers, C.; Smit, R.A.J.; Tzoulaki, I.; Karaman, I.; Boulange, C.; Vaidya, D.; Punjabi, N.; et al. Metabolomics Profiling of Visceral Adipose Tissue: Results From MESA and the NEO Study. J. Am. Heart Assoc. 2019, 8, e010810. [Google Scholar] [CrossRef]
- Cambiaghi, A.; Díaz, R.; Martinez, J.B.; Odena, A.; Brunelli, L.; Caironi, P.; Masson, S.; Baselli, G.; Ristagno, G.; Gattinoni, L.; et al. An Innovative Approach for the Integration of Proteomics and Metabolomics Data in Severe Septic Shock Patients Stratified for Mortality. Sci. Rep. 2018, 8, 6681. [Google Scholar] [CrossRef]
- Huang, Y.; Hui, Q.; Walker, D.I.; Uppal, K.; Goldberg, J.; Jones, D.P.; Vaccarino, V.; Sun, Y. V Untargeted metabolomics reveals multiple metabolites influencing smoking-related DNA methylation. Epigenomics 2018, 10, 379–393. [Google Scholar] [CrossRef] [PubMed]
- McGuire, J.L.; DePasquale, E.A.K.; Watanabe, M.; Anwar, F.; Ngwenya, L.B.; Atluri, G.; Romick-Rosendale, L.E.; McCullumsmith, R.E.; Evanson, N.K. Chronic Dysregulation of Cortical and Subcortical Metabolism After Experimental Traumatic Brain Injury. Mol. Neurobiol. 2019, 56, 2908–2921. [Google Scholar] [CrossRef] [PubMed]
- Gao, N.; Ding, L.; Pang, J.; Zheng, Y.; Cao, Y.; Zhan, H.; Shi, Y. Metabonomic-Transcriptome Integration Analysis on Osteoarthritis and Rheumatoid Arthritis. Int. J. Genomics 2020. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Zhao, X.; Sui, Z.; Niu, H.; Chen, L.; Hu, C.; Xuan, Q.; Hou, X.; Zhang, R.; Zhou, L.; et al. A multi-omics investigation of the molecular characteristics and classification of six metabolic syndrome relevant diseases. Theranostics 2020, 10, 2029–2046. [Google Scholar] [CrossRef] [PubMed]
- Piening, B.D.; Zhou, W.; Contrepois, K.; Röst, H.; Gu Urban, G.J.; Mishra, T.; Hanson, B.M.; Bautista, E.J.; Leopold, S.; Yeh, C.Y.; et al. Integrative Personal Omics Profiles during Periods of Weight Gain and Loss. Cell Syst. 2018, 6, 157–170. [Google Scholar] [CrossRef] [PubMed]
- Acharjee, A.; Kloosterman, B.; Visser, R.G.F.; Maliepaard, C. Integration of multi-omics data for prediction of phenotypic traits using random forest. BMC Bioinform. 2016, 17, 180. [Google Scholar] [CrossRef] [PubMed]
- Hubbard, A.H.; Zhang, X.; Jastrebski, S.; Singh, A.; Schmidt, C. Understanding the liver under heat stress with statistical learning: An integrated metabolomics and transcriptomics computational approach. BMC Genomics 2019, 20, 502. [Google Scholar] [CrossRef]
- Auslander, N.; Yizhak, K.; Weinstock, A.; Budhu, A.; Tang, W.; Wang, X.W.; Ambs, S.; Ruppin, E. A joint analysis of transcriptomic and metabolomic data uncovers enhanced enzyme-metabolite coupling in breast cancer. Sci. Rep. 2016, 6, 29662. [Google Scholar] [CrossRef]
- Kouznetsova, V.L.; Kim, E.; Romm, E.L.; Zhu, A.; Tsigelny, I.F. Recognition of early and late stages of bladder cancer using metabolites and machine learning. Metabolomics 2019, 15, 1–15. [Google Scholar] [CrossRef]
- Guo, Y.; Yu, H.; Chen, D.; Zhao, Y.Y. Machine learning distilled metabolite biomarkers for early stage renal injury. Metabolomics 2020, 16. [Google Scholar] [CrossRef]
- Kim, M.; Rai, N.; Zorraquino, V.; Tagkopoulos, I. Multi-omics integration accurately predicts cellular state in unexplored conditions for Escherichia coli. Nat. Commun. 2016, 7, 13090. [Google Scholar] [CrossRef] [PubMed]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Jafari, M.; Ansari-Pour, N. Why, when and how to adjust your P values? Cell J. 2019, 20, 604–607. [Google Scholar] [PubMed]
- Karathanasis, N.; Tsamardinos, I.; Lagani, V. omicsNPC: Applying the Non-Parametric Combination Methodology to the Integrative Analysis of Heterogeneous Omics Data. PLoS ONE 2016, 11, e0165545. [Google Scholar] [CrossRef] [PubMed]
- Jiang, B.; Zhang, X.; Zuo, Y.; Kang, G. A powerful truncated tail strength method for testing multiple null hypotheses in one dataset. J. Theor. Biol. 2011, 277, 67–73. [Google Scholar] [CrossRef] [PubMed]
- Taylor, J.; Tibshirani, R. A tail strength measure for assessing the overall univariate significance in a dataset. Biostatistics 2006, 7, 167–181. [Google Scholar] [CrossRef]
- Efron, B.; Tibshirani, R. Empirical Bayes methods and false discovery rates for microarrays. Genet. Epidemiol. 2002, 23, 70–86. [Google Scholar] [CrossRef]
- Baker, M. Statisticians issue warning over misuse of P values. Nature 2016, 531, 151. [Google Scholar] [CrossRef]
- Guo, H.; Chen, J.; Huang, Y.; Zhang, W.; Xu, F.; Zhang, Z. A pseudo-kinetics approach for time-series metabolomics investigations: More reliable and sensitive biomarkers revealed in vincristine-induced paralytic ileus rats. RSC Adv. 2016, 6, 54471–54478. [Google Scholar] [CrossRef]
- Abadie, C.; Blanchet, S.; Carroll, A.; Tcherkez, G. Metabolomics analysis of postphotosynthetic effects of gaseous O2 on primary metabolism in illuminated leaves. Funct. Plant Biol. 2017, 44, 929. [Google Scholar] [CrossRef]
- Yates, F. The Analysis of Multiple Classifications with Unequal Numbers in the Different Classes. J. Am. Stat. Assoc. 1934, 29, 51. [Google Scholar] [CrossRef]
- Xia, J.; Sinelnikov, I.V.; Wishart, D.S. MetATT: A web-based metabolomics tool for analyzing time-series and two-factor datasets. Bioinformatics 2011, 27, 2455–2456. [Google Scholar] [CrossRef] [PubMed]
- Berk, M.; Ebbels, T.; Montana, G. A statistical framework for biomarker discovery in metabolomic time course data. Bioinformatics 2011, 27, 1979–1985. [Google Scholar] [CrossRef] [PubMed]
- Gromski, P.S.; Muhamadali, H.; Ellis, D.I.; Xu, Y.; Correa, E.; Turner, M.L.; Goodacre, R. A tutorial review: Metabolomics and partial least squares-discriminant analysis—A marriage of convenience or a shotgun wedding. Anal. Chim. Acta 2015, 879, 10–23. [Google Scholar] [CrossRef] [PubMed]
- Brereton, R.G.; Lloyd, G.R. Partial least squares discriminant analysis: Taking the magic away. J. Chemom. 2014, 28, 213–225. [Google Scholar] [CrossRef]
- Rodríguez-Pérez, R.; Fernández, L.; Marco, S. Overoptimism in cross-validation when using partial least squares-discriminant analysis for omics data: A systematic study. Anal. Bioanal. Chem. 2018, 410, 5981–5992. [Google Scholar] [CrossRef]
- Szymańska, E.; Saccenti, E.; Smilde, A.K.; Westerhuis, J.A. Double-check: Validation of diagnostic statistics for PLS-DA models in metabolomics studies. Metabolomics 2012, 8, 3–16. [Google Scholar] [CrossRef]
- Bylesjö, M.; Rantalainen, M.; Cloarec, O.; Nicholson, J.K.; Holmes, E.; Trygg, J. OPLS discriminant analysis: Combining the strengths of PLS-DA and SIMCA classification. J. Chemom. 2006, 20, 341–351. [Google Scholar] [CrossRef]
- Chun, H.; Keleş, S. Sparse partial least squares regression for simultaneous dimension reduction and variable selection. J. R. Stat. Soc. Ser. B Stat. Methodol. 2010, 72, 3–25. [Google Scholar] [CrossRef]
- Smyth, G.K. Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat. Appl. Genet. Mol. Biol. 2004, 3. [Google Scholar] [CrossRef]
- Li, Y.; Fan, T.W.M.; Lane, A.N.; Kang, W.-Y.; Arnold, S.M.; Stromberg, A.J.; Wang, C.; Chen, L. SDA: A semi-parametric differential abundance analysis method for metabolomics and proteomics data. BMC Bioinform. 2019, 20, 501–510. [Google Scholar] [CrossRef] [PubMed]
- Gross, S.M.; Tibshirani, R. Collaborative regression. Biostatistics 2015, 16, 326–338. [Google Scholar] [CrossRef] [PubMed]
- Hoerl, A.E.; Kennard, R.W. Ridge Regression: Biased Estimation for Nonorthogonal Problems. Technometrics 1970, 12, 55–67. [Google Scholar] [CrossRef]
- Tibshirani, R. Regression Shrinkage and Selection via the Lasso. J. R. Stat. Soc. 1996, 58, 267–288. [Google Scholar] [CrossRef]
- Zou, H.; Zou, H.; Hastie, T. Regularization and variable selection via the Elastic Net. J. R. Stat. Soc. Ser. B 2005, 67, 301–320. [Google Scholar] [CrossRef]
- Fukushima, A. DiffCorr: An R package to analyze and visualize differential correlations in biological networks. Gene 2013, 518, 209–214. [Google Scholar] [CrossRef]
- Siska, C.; Bowler, R.; Kechris, K. The discordant method: A novel approach for differential correlation. Bioinformatics 2016, 32, 690–696. [Google Scholar] [CrossRef]
- Ma, J.; Karnovsky, A.; Afshinnia, F.; Wigginton, J.; Rader, D.J.; Natarajan, L.; Sharma, K.; Porter, A.C.; Rahman, M.; He, J.; et al. Differential network enrichment analysis reveals novel lipid pathways in chronic kidney disease. Bioinformatics 2019, 35, 3441–3452. [Google Scholar] [CrossRef]
- Shi, W.J.; Zhuang, Y.; Russell, P.H.; Hobbs, B.D.; Parker, M.M.; Castaldi, P.J.; Rudra, P.; Vestal, B.; Hersh, C.P.; Saba, L.M.; et al. Unsupervised discovery of phenotype-specific multi-omics networks. Bioinformatics 2019, 35, 4336–4343. [Google Scholar] [CrossRef]
- Siddiqui, J.K.; Baskin, E.; Liu, M.; Cantemir-Stone, C.Z.; Zhang, B.; Bonneville, R.; McElroy, J.P.; Coombes, K.R.; Mathé, E.A. IntLIM: Integration using linear models of metabolomics and gene expression data. BMC Bioinform. 2018, 19, 81. [Google Scholar] [CrossRef]
- Fleming, R.M.T.; Vlassis, N.; Thiele, I.; Saunders, M.A. Conditions for duality between fluxes and concentrations in biochemical networks. J. Theor. Biol. 2016, 409, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Pandey, V.; Hadadi, N.; Hatzimanikatis, V. Enhanced flux prediction by integrating relative expression and relative metabolite abundance into thermodynamically consistent metabolic models. PLoS Comput. Biol. 2019, 15, e1007036. [Google Scholar] [CrossRef] [PubMed]
- Angione, C. Human Systems Biology and Metabolic Modelling: A Review-From Disease Metabolism to Precision Medicine. Biomed Res. Int. 2019, 2019, 8304260. [Google Scholar] [CrossRef] [PubMed]
- Lakshmanan, M.; Koh, G.; Chung, B.K.S.; Lee, D. Software applications for flux balance analysis. Brief. Bioinform. 2012, 15, 108–122. [Google Scholar] [CrossRef]
- Rätsch, G.; Sonnenburg, S.; Schäfer, C. Learning interpretable SVMs for biological sequence classification. BMC Bioinform. 2006, 7, S9. [Google Scholar] [CrossRef]
- Guyon, I.; Weston, J.; Barnhill, S.; Vapnik, V. Gene selection for cancer classification using support vector machines. Mach. Learn. 2002, 46, 389–422. [Google Scholar] [CrossRef]
- Rasmussen, P.M.; Madsen, K.H.; Lund, T.E.; Hansen, L.K. Visualization of nonlinear kernel models in neuroimaging by sensitivity maps. Neuroimage 2011, 55, 1120–1131. [Google Scholar] [CrossRef]
- Eicher, T.; Sinha, K. A support vector machine approach to identification of proteins relevant to learning in a mouse model of Down Syndrome. In Proceedings of the International Joint Conference on Neural Networks, Anchorage, AK, USA, 14–19 May 2017. [Google Scholar]
- Gaonkar, B.; Davatzikos, C. Analytic estimation of statistical significance maps for support vector machine based multi-variate image analysis and classification. Neuroimage 2013, 78, 270–283. [Google Scholar] [CrossRef]
- Breiman, L. Bagging Predictors. Machin. Learn. 1996, 24, 123–140. [Google Scholar] [CrossRef]
- Quinlan, R. C4.5: Programs for Machine Learning. Machin. Learn. 1993, 16, 235–240. [Google Scholar] [CrossRef]
- Archer, K.J.; Kimes, R.V. Empirical characterization of random forest variable importance measures. Comput. Stat. Data Anal. 2008, 52, 2249–2260. [Google Scholar] [CrossRef]
- Taufik, W.M. Minimizing False Negatives of Measles Prediction Model: An Experimentation of Feature Selection Based On Domain Knowledge and Random Forest Classifier. Int. J. Eng. Adv. Technol. 2019, 2249–8958. [Google Scholar]
- Calle, M.L.; Urrea, V. Letter to the Editor: Stability of Random Forest importance measures. Brief. Bioinform. 2011, 12, 86–89. [Google Scholar] [CrossRef] [PubMed]
- Altmann, A.; Toloşi, L.; Sander, O.; Lengauer, T. Permutation importance: A corrected feature importance measure. Bioinformatics 2010, 26, 1340–1347. [Google Scholar] [CrossRef] [PubMed]
- Van Rijsbergen, C.J. Foundation of evaluation. J. Doc. 1974, 30, 365–373. [Google Scholar] [CrossRef]
- Bradley, A.P. The use of the area under the ROC curve in the evaluation of machine learning algorithms. Pattern Recognit. 1997, 30, 1145–1159. [Google Scholar] [CrossRef]
- Minsky, M.; Papert, S. Perceptrons; an introduction to computational geometry; MIT Press: Cambridge, MA, USA, 1969; ISBN 9780262130431. [Google Scholar]
- Lecun, Y.; Eon Bottou, L.; Bengio, Y.; Haaner, P. Gradient-Based Learning Applied to Document Recognition. Proc. IEEE 1998, 86, 2278–2324. [Google Scholar] [CrossRef]
- Rumelhart, D.E.; Hinton, G.E.; Williams, R.J. Learning representations by back-propagating errors. Nature 1986, 323, 533–536. [Google Scholar] [CrossRef]
- Alwosheel, A.; van Cranenburgh, S.; Chorus, C.G. Is your dataset big enough? Sample size requirements when using artificial neural networks for discrete choice analysis. J. Choice Model. 2018, 28, 167–182. [Google Scholar] [CrossRef]
- Mirza, B.; Wang, W.; Wang, J.; Choi, H.; Chung, N.C.; Ping, P. Machine learning and integrative analysis of biomedical big data. Genes (Basel) 2019, 10, 87. [Google Scholar] [CrossRef]
- Yu, H.; Samuels, D.C.; Zhao, Y.Y.; Guo, Y. Architectures and accuracy of artificial neural network for disease classification from omics data. BMC Genomics 2019, 20, 167. [Google Scholar] [CrossRef] [PubMed]
- Tang, B.; Pan, Z.; Yin, K.; Khateeb, A. Recent Advances of Deep Learning in Bioinformatics and Computational Biology. Front. Genet. 2019, 10, 214. [Google Scholar] [CrossRef] [PubMed]
- Ren, S.; Shao, Y.; Zhao, X.; Hong, C.S.; Wang, F.; Lu, X.; Li, J.; Ye, G.; Yan, M.; Zhuang, Z.; et al. Integration of metabolomics and transcriptomics reveals major metabolic pathways and potential biomarker involved in prostate cancer. Mol. Cell. Proteomics 2016, 15, 154–163. [Google Scholar] [CrossRef] [PubMed]
- Torres, E.R.S.; Hall, R.; Bobe, G.; Choi, J.; Impey, S.; Pelz, C.; Lindner, J.R.; Stevens, J.F.; Raber, J. Integrated Metabolomics-DNA Methylation Analysis Reveals Significant Long-Term Tissue-Dependent Directional Alterations in Aminoacyl-tRNA Biosynthesis in the Left Ventricle of the Heart and Hippocampus Following Proton Irradiation. Front. Mol. Biosci. 2019, 6, 77. [Google Scholar] [CrossRef]
- Yu, J.; Chen, J.; Zhao, H.; Gao, J.; Li, Y.; Li, Y.; Xue, J.; Dahan, A.; Sun, D.; Zhang, G.; et al. Integrative proteomics and metabolomics analysis reveals the toxicity of cationic liposomes to human normal hepatocyte cell line L02. Mol. Omi. 2018, 14, 362–372. [Google Scholar] [CrossRef]
- Cao, H.; Zhang, A.; Sun, H.; Zhou, X.; Guan, Y.; Liu, Q.; Kong, L.; Wang, X. Metabolomics-proteomics profiles delineate metabolic changes in kidney fibrosis disease. Proteomics 2015, 15, 3699–3710. [Google Scholar] [CrossRef]
- Erawijantari, P.P.; Mizutani, S.; Shiroma, H.; Shiba, S.; Nakajima, T.; Sakamoto, T.; Saito, Y.; Fukuda, S.; Yachida, S.; Yamada, T. Influence of gastrectomy for gastric cancer treatment on faecal microbiome and metabolome profiles. Gut 2020. [Google Scholar] [CrossRef]
- O’Donovan, C.M.; Madigan, S.M.; Garcia-Perez, I.; Rankin, A.; O’ Sullivan, O.; Cotter, P.D. Distinct microbiome composition and metabolome exists across subgroups of elite Irish athletes. J. Sci. Med. Sport 2020, 23, 63–68. [Google Scholar] [CrossRef]
- Cronin, O.; Barton, W.; Skuse, P.; Penney, N.C.; Garcia-Perez, I.; Murphy, E.F.; Woods, T.; Nugent, H.; Fanning, A.; Melgar, S.; et al. A Prospective Metagenomic and Metabolomic Analysis of the Impact of Exercise and/or Whey Protein Supplementation on the Gut Microbiome of Sedentary Adults. mSystems 2018, 3. [Google Scholar] [CrossRef]
- Zachariou, M.; Minadakis, G.; Oulas, A.; Afxenti, S.; Spyrou, G.M. Integrating multi-source information on a single network to detect disease-related clusters of molecular mechanisms. J. Proteomics 2018, 188, 15–29. [Google Scholar] [CrossRef]
- Maifiah, M.H.M.; Cheah, S.E.; Johnson, M.D.; Han, M.L.; Boyce, J.D.; Thamlikitkul, V.; Forrest, A.; Kaye, K.S.; Hertzog, P.; Purcell, A.W.; et al. Global metabolic analyses identify key differences in metabolite levels between polymyxin-susceptible and polymyxin-resistant Acinetobacter baumannii. Sci. Rep. 2016, 6, 22287. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.D.; Luo, W.; Lin, Y.; Zhang, J.; Zhang, L.; Zhang, W.; Huang, S.M. Discovery of potential therapeutic targets for non-small cell lung cancer using high-throughput metabolomics analysis based on liquid chromatography coupled with tandem mass spectrometry. RSC Adv. 2019, 9, 10905–10913. [Google Scholar] [CrossRef]
- Nguyen, T.M.; Shafi, A.; Nguyen, T.; Draghici, S. Identifying significantly impacted pathways: A comprehensive review and assessment. Genome Biol. 2019, 20, 203. [Google Scholar] [CrossRef] [PubMed]
- Johnson, C.H.; Ivanisevic, J.; Siuzdak, G. Metabolomics: Beyond biomarkers and towards mechanisms. Nat. Rev. Mol. Cell Biol. 2016, 17, 451–459. [Google Scholar] [CrossRef]
- Ewald, J.D.; Soufan, O.; Crump, D.; Hecker, M.; Xia, J.; Basu, N. EcoToxModules: Custom Gene Sets to Organize and Analyze Toxicogenomics Data from Ecological Species. Environ. Sci. Technol. 2020. [Google Scholar] [CrossRef]
- Lee, J.; Jo, K.; Lee, S.; Kang, J.; Kim, S. Prioritizing biological pathways by recognizing context in time-series gene expression data. BMC Bioinform. 2016, 17, 477. [Google Scholar] [CrossRef]
- Falcon, S.; Gentleman, R. Using GOstats to test gene lists for GO term association. Bioinformatics 2007, 23, 257–258. [Google Scholar] [CrossRef]
- Maere, S.; Heymans, K.; Kuiper, M. BiNGO: A Cytoscape plugin to assess overrepresentation of gene ontology categories in biological networks. Bioinformatics 2005, 21, 3448–3449. [Google Scholar] [CrossRef]
- Koelmel, J.P.; Ulmer, C.Z.; Jones, C.M.; Yost, R.A.; Bowden, J.A. Common cases of improper lipid annotation using high-resolution tandem mass spectrometry data and corresponding limitations in biological interpretation. Biochim. et Biophys. Acta (BBA) - Mol. Cell Boil. Lipids 2017, 1862, 766–770. [Google Scholar] [CrossRef]
- Fisher, R.A. Statistical Methods for Research Workers, 5th ed.; Oliver and Boyd: Edinburgh, UK, 1934. [Google Scholar]
- Stouffer, S.A.; Suchman, E.A.; Devinney, L.C.; Star, S.A.; Williams, R.M. The American soldier: Adjustment during army life. In Studies in social psychology in World War II; Princeton University Press: Princeton, NJ, USA, 1949; Volume 1. [Google Scholar]
- Zhang, B.; Hu, S.; Baskin, E.; Patt, A.; Siddiqui, J.K.; Mathé, E.A. RaMP: A Comprehensive Relational Database of Metabolomics Pathways for Pathway Enrichment Analysis of Genes and Metabolites. Metabolites 2018, 8, 16. [Google Scholar] [CrossRef]
- Kamburov, A.; Cavill, R.; Ebbels, T.M.D.; Herwig, R.; Keun, H.C. Integrated pathway-level analysis of transcriptomics and metabolomics data with IMPaLA. Bioinformatics 2011, 27, 2917–2918. [Google Scholar] [CrossRef] [PubMed]
- Kaever, A.; Landesfeind, M.; Feussner, K.; Morgenstern, B.; Feussner, I.; Meinicke, P. Meta-analysis of pathway enrichment: Combining independent and dependent omics data sets. PLoS ONE 2014, 9, e89297. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U.S.A. 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed]
- Xia, J.; Wishart, D.S. MSEA: A web-based tool to identify biologically meaningful patterns in quantitative metabolomic data. Nucleic Acids Res. 2010, 38, W71–W77. [Google Scholar] [CrossRef] [PubMed]
- Molenaar, M.R.; Jeucken, A.; Wassenaar, T.A.; Van De Lest, C.H.A.; Brouwers, J.F.; Helms, J.B. LION/web: A web-based ontology enrichment tool for lipidomic data analysis. Gigascience 2019, 8. [Google Scholar] [CrossRef]
- Tarca, A.; Draghici, S.; Khatri, P. A novel signaling pathway impact analysis. Bioinformatics 2009, 25, 75–82. [Google Scholar] [CrossRef]
- Ibrahim, M.A.H.; Jassim, S.; Cawthorne, M.A.; Langlands, K. A topology-based score for pathway enrichment. J. Comput. Biol. 2012, 19, 563–573. [Google Scholar] [CrossRef]
- Gu, Z.; Wang, J. CePa: An R package for finding significant pathways weighted by multiple network centralities. Bioinformatics 2013, 29, 658–660. [Google Scholar] [CrossRef]
- Gao, S.; Wang, X. TAPPA: Topological analysis of pathway phenotype association. Bioinformatics 2007, 23, 3100–3102. [Google Scholar] [CrossRef]
- Massa, M.S.; Chiogna, M.; Romualdi, C. Gene set analysis exploiting the topology of a pathway. BMC Syst. Biol. 2010, 4, 121. [Google Scholar] [CrossRef]
- Martini, P.; Sales, G.; Massa, M.S.; Chiogna, M.; Romualdi, C. Along signal paths: An empirical gene set approach exploiting pathway topology. Nucleic Acids Res. 2013, 41, e19. [Google Scholar] [CrossRef] [PubMed]
- Jacob, L.; Neuvial, P.; Dudoit, S. Gains in Power from Structured Two-Sample Tests of Means on Graphs. Ann. Appl. Stat. 2010, 6, 561–600. [Google Scholar] [CrossRef]
- Ihnatova, I.; Popovici, V.; Budinska, E. A critical comparison of topology-based pathway analysis methods. PLoS ONE 2018, 13, e0191154. [Google Scholar] [CrossRef] [PubMed]
- Picart-Armada, S.; Fernández-Albert, F.; Vinaixa, M.; Yanes, O.; Perera-Lluna, A. FELLA: An R package to enrich metabolomics data. BMC Bioinform. 2018, 19, 538. [Google Scholar] [CrossRef]
- Paley, S.M.; Karp, P.D. The Pathway Tools cellular overview diagram and Omics Viewer. Nucleic Acids Res. 2006, 34, 3771–3778. [Google Scholar] [CrossRef]
- Junker, B.H.; Klukas, C.; Schreiber, F. Vanted: A system for advanced data analysis and visualization in the context of biological networks. BMC Bioinform. 2006, 7, 109. [Google Scholar] [CrossRef]
- Hernández-de-Diego, R.; Tarazona, S.; Martínez-Mira, C.; Balzano-Nogueira, L.; Furió-Tarí, P.; Pappas, G.J.; Conesa, A. PaintOmics 3: A web resource for the pathway analysis and visualization of multi-omics data. Nucleic Acids Res. 2018, 46, W503–W509. [Google Scholar] [CrossRef]
- Domingo-Fernández, D.; Hoyt, C.T.; Bobis-Álvarez, C.; Marín-Llaó, J.; Hofmann-Apitius, M. ComPath: An ecosystem for exploring, analyzing, and curating mappings across pathway databases. NPJ Syst. Biol. Appl. 2019, 5. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software Environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Gansner, E.R.; North, S.C. An open graph visualization system and its applications to software engineering. Softw. Pract. Exper. 2000, 11, 1203–1233. [Google Scholar] [CrossRef]
- Csardi, G.; Nepusz, T. The igraph software package for complex network research. InterJournal Complex Sy. 2006. [Google Scholar]
- Kutmon, M.; van Iersel, M.P.; Bohler, A.; Kelder, T.; Nunes, N.; Pico, A.R.; Evelo, C.T. PathVisio 3: An Extendable Pathway Analysis Toolbox. PLoS Comput. Biol. 2015, 11, e1004085. [Google Scholar] [CrossRef] [PubMed]
- Zhou, G.; Xia, J. OmicsNet: A web-based tool for creation and visual analysis of biological networks in 3D space. Nucleic Acids Res. 2018, 46, W514–W522. [Google Scholar] [CrossRef] [PubMed]
- Rougny, A.; Touré, V.; Moodie, S.; Balaur, I.; Czauderna, T.; Borlinghaus, H.; Dogrusoz, U.; Mazein, A.; Dräger, A.; Blinov, M.L.; et al. Systems Biology Graphical Notation: Process Description language Level 1 Version 2.0. J. Integr. Bioinform. 2019, 16. [Google Scholar] [CrossRef] [PubMed]
- Klyne, G.; Carroll, J.; McBride, B. Resource Description Framework (RDF): Concepts and Abstract Syntax. Available online: https://www.w3.org/TR/rdf-concepts/ (accessed on 25 March 2020).
- Frainay, C.; Schymanski, E.; Neumann, S.; Merlet, B.; Salek, R.; Jourdan, F.; Yanes, O. Mind the Gap: Mapping Mass Spectral Databases in Genome-Scale Metabolic Networks Reveals Poorly Covered Areas. Metabolites 2018, 8, 51. [Google Scholar] [CrossRef]
- Mubeen, S.; Hoyt, C.T.; Gemünd, A.; Hofmann-Apitius, M.; Fröhlich, H.; Domingo-Fernández, D. The Impact of Pathway Database Choice on Statistical Enrichment Analysis and Predictive Modeling. Front. Genet. 2019, 10. [Google Scholar] [CrossRef]
- Slenter, D.N.; Kutmon, M.; Hanspers, K.; Riutta, A.; Windsor, J.; Nunes, N.; Mélius, J.; Cirillo, E.; Coort, S.L.; Digles, D.; et al. WikiPathways: A multifaceted pathway database bridging metabolomics to other omics research. Nucleic Acids Res. 2018, 46, D661–D667. [Google Scholar] [CrossRef]
- Caspi, R.; Billington, R.; Keseler, I.M.; Kothari, A.; Krummenacker, M.; Midford, P.E.; Ong, W.K.; Paley, S.; Subhraveti, P.; Karp, P.D. The MetaCyc database of metabolic pathways and enzymes - a 2019 update. Nucleic Acids Res. 2020, 48, D445–D453. [Google Scholar] [CrossRef]
- Cerami, E.G.; Gross, B.E.; Demir, E.; Rodchenkov, I.; Babur, O.; Anwar, N.; Schultz, N.; Bader, G.D.; Sander, C. Pathway Commons, a web resource for biological pathway data. Nucleic Acids Res. 2011, 39, D685–D690. [Google Scholar] [CrossRef]
- Tran, V.D.T.; Moretti, S.; Coste, A.T.; Amorim-Vaz, S.; Sanglard, D.; Pagni, M. Condition-specific series of metabolic sub-networks and its application for gene set enrichment analysis. Bioinformatics 2019, 35, 2258–2266. [Google Scholar] [CrossRef]
- Wishart, D.S.; Feunang, Y.D.; Marcu, A.; Guo, A.C.; Liang, K.; Vázquez-Fresno, R.; Sajed, T.; Johnson, D.; Li, C.; Karu, N.; et al. HMDB 4.0: The human metabolome database for 2018. Nucleic Acids Res. 2018, 46, D608–D617. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Krieger, C.J. MetaCyc: A multiorganism database of metabolic pathways and enzymes. Nucleic Acids Res. 2004, 32, 438–442. [Google Scholar] [CrossRef] [PubMed]
- Le Boulch, M.; Déhais, P.; Combes, S.; Pascal, G. MACADAM database: A MetAboliC pAthways DAtabase for Microbial taxonomic groups for mining potential metabolic capacities of archaeal and bacterial taxonomic groups. Database 2019, 2019. [Google Scholar] [CrossRef]
- Wishart, D.; Li, C.; Marcu, A.; Badran, H.; Pon, A.; Budinski, Z.; Patron, J.; Lipton, D.; Cao, X.; Oler, E.O.; et al. PathBank: A comprehensive pathway database for model organisms. Nucleic Acids Res. 2019, 48, 470–478. [Google Scholar] [CrossRef]
- Barbarino, J.M.; Whirl-Carrillo, M.; Altman, R.B.; Klein, T.E. PharmGKB: A worldwide resource for pharmacogenomic information. Wiley Interdiscip. Rev. Syst. Biol. Med. 2018, 10, 1417. [Google Scholar] [CrossRef]
- Heller, S.R.; McNaught, A.; Pletnev, I.; Stein, S.; Tchekhovskoi, D. InChI, the IUPAC International Chemical Identifier. J. Cheminform. 2015, 7, 23. [Google Scholar] [CrossRef]
- Hastings, J.; de Matos, P.; Dekker, A.; Ennis, M.; Harsha, B.; Kale, N.; Muthukrishnan, V.; Owen, G.; Turner, S.; Williams, M.; et al. The ChEBI reference database and ontology for biologically relevant chemistry: Enhancements for 2013. Nucleic Acids Res. 2012, 41, 456–463. [Google Scholar] [CrossRef]
- Salek, R.M.; Steinbeck, C.; Viant, M.R.; Goodacre, R.; Dunn, W.B. The role of reporting standards for metabolite annotation and identification in metabolomic studies. Gigascience 2013, 2, 13. [Google Scholar] [CrossRef]
- Jamil, H.M. Improving Integration Effectiveness of ID Mapping Based Biological Record Linkage. IEEE/ACM Trans. Comput. Biol. Bioinform. 2015, 12, 473–486. [Google Scholar] [CrossRef]
- Rocca-Serra, P.; Salek, R.M.; Arita, M.; Correa, E.; Dayalan, S.; Gonzalez-Beltran, A.; Ebbels, T.; Goodacre, R.; Hastings, J.; Haug, K.; et al. Data standards can boost metabolomics research, and if there is a will, there is a way. Metabolomics 2015, 12, 14. [Google Scholar] [CrossRef] [PubMed]
- Wohlgemuth, G.; Haldiya, P.K.; Willighagen, E.; Kind, T.; Fiehn, O. The Chemical Translation Service--a web-based tool to improve standardization of metabolomic reports. Bioinformatics 2010, 26, 2647–2648. [Google Scholar] [CrossRef] [PubMed]
- Ravikumar, K.E.; Wagholikar, K.B.; Li, D.; Kocher, J.-P.; Liu, H. Text mining facilitates database curation - extraction of mutation-disease associations from Bio-medical literature. BMC Bioinform. 2015, 16, 185. [Google Scholar] [CrossRef] [PubMed]
- Ruch, P. Text Mining to Support Gene Ontology Curation and Vice Versa. Methods Mol. Biol. 2017, 1446, 69–84. [Google Scholar] [PubMed]
- Galeota, E.; Kishore, K.; Pelizzola, M. Ontology-driven integrative analysis of omics data through Onassis. Sci. Rep. 2020, 10, 1–9. [Google Scholar] [CrossRef]
- Mohanraj, K.; Karthikeyan, B.S.; Vivek-Ananth, R.P.; Chand, R.P.B.; Aparna, S.R.; Mangalapandi, P.; Samal, A. IMPPAT: A curated database of Indian Medicinal Plants, Phytochemistry and Therapeutics. Sci. Rep. 2018, 8, 4329. [Google Scholar] [CrossRef]
- Liu, Y.; Liang, Y.; Wishart, D. PolySearch2: A significantly improved text-mining system for discovering associations between human diseases, genes, drugs, metabolites, toxins and more. Nucleic Acids Res. 2015, 43, W535–W542. [Google Scholar] [CrossRef]
- Tutubalina, E.V.; Miftahutdinov, Z.S.; Nugmanov, R.I.; Madzhidov, T.I.; Nikolenko, S.I.; Alimova, I.S.; Tropsha, A.E. Using semantic analysis of texts for the identification of drugs with similar therapeutic effects. Russ. Chem. Bull. 2017, 66, 2180–2189. [Google Scholar] [CrossRef]
- Kulkarni, C.; Xu, W.; Ritter, A.; Machiraju, R. An Annotated Corpus for Machine Reading of Instructions in Wet Lab Protocols. In Proceedings of the NAACL-HLT 2018, New Orleans, LA, USA, 1–6 June 2018; pp. 97–106. [Google Scholar]
- Westergaard, D.; Staerfeldt, H.-H.; Tønsberg, C.; Jensen, L.J.; Brunak, S. A comprehensive and quantitative comparison of text-mining in 15 million full-text articles versus their corresponding abstracts. PLOS Comput. Biol. 2018, 14, e1005962. [Google Scholar] [CrossRef]
- Ben Abdessalem Karaa, W.; Mannai, M.; Dey, N.; Ashour, A.S.; Olariu, I. Gene-disease-food relation extraction from biomedical database. Adv. Intell. Syst. Comput. 2018, 633, 394–407. [Google Scholar]
- Nikfarjam, A.; Ransohoff, J.D.; Callahan, A.; Jones, E.; Loew, B.; Kwong, B.Y.; Sarin, K.Y.; Shah, N.H. Early detection of adverse drug reactions in social health networks: A natural language processing pipeline for signal detection. J. Med. Internet Res. 2019, 5, e11264. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Zhang, R. Using natural language processing methods to classify use status of dietary supplements in clinical notes. BMC Med. Inform. Decis. Mak. 2018, 18, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Huan, C.-C.; Lu, Z. Community challenges in biomedical text mining over 10 years: Success, failure and the future. Brief. Bioinform. 2015, 17, 132–144. [Google Scholar] [CrossRef] [PubMed]
- Cohen, K.B.; Lanfranchi, A.; Choi, M.J.Y.; Bada, M.; Baumgartner, W.A.; Panteleyeva, N.; Verspoor, K.; Palmer, M.; Hunter, L.E. Coreference annotation and resolution in the Colorado Richly Annotated Full Text (CRAFT) corpus of biomedical journal articles. BMC Bioinform. 2017, 18, 372. [Google Scholar] [CrossRef] [PubMed]
- Pletscher-Frankild, S.; Pallejà, A.; Tsafou, K.; Binder, J.X.; Jensen, L.J. DISEASES: Text mining and data integration of disease-gene associations. Methods 2015, 74, 83–89. [Google Scholar] [CrossRef] [PubMed]
- Fdez-Glez, J.; Ruano-Ordás, D.; Méndez, J.R.; Fdez-Riverola, F.; Laza, R.; Pavón, R. Determining the Influence of Class Imbalance for the Triage of Biomedical Documents. Curr. Bioinform. 2017, 13, 592–605. [Google Scholar] [CrossRef]
- Wei, C.-H.; Allot, A.; Leaman, R.; Lu, Z. PubTator central: Automated concept annotation for biomedical full text articles. Nucleic Acids Res. 2019, 47, W587–W593. [Google Scholar] [CrossRef]
- Jiang, X.; Ringwald, M.; Blake, J.A.; Arighi, C.; Zhang, G.; Shatkay, H. An effective biomedical document classification scheme in support of biocuration: Addressing class imbalance. Database 2019, 2019. [Google Scholar] [CrossRef]
- Alshuwaier, F.; Areshey, A.; Poon, J. A comparative study of the current technologies and approaches of relation extraction in biomedical literature using text mining. In Proceedings of the 4th IEEE International Conference on Engineering Technologies and Applied Sciences, Salmabad, Bahrain, 29 November–1 December 2017; pp. 1–13. [Google Scholar]
- Sung, J.; Kim, S.; Cabatbat, J.J.T.; Jang, S.; Jin, Y.S.; Jung, G.Y.; Chia, N.; Kim, P.J. Global metabolic interaction network of the human gut microbiota for context-specific community-scale analysis. Nat. Commun. 2017, 8, 1–12. [Google Scholar]
- Griffith, M.; Griffith, O.L.; Coffman, A.C.; Weible, J.V.; McMichael, J.F.; Spies, N.C.; Koval, J.; Das, I.; Callaway, M.B.; Eldred, J.M.; et al. DGIdb: Mining the druggable genome. Nat. Methods 2013, 10, 1209–1210. [Google Scholar] [CrossRef]
- Chen, T.; Li, M.; He, Q.; Zou, L.; Li, Y.; Chang, C.; Zhao, D.; Zhu, Y. LiverWiki: A wiki-based database for human liver. BMC Bioinform. 2017, 18, 452. [Google Scholar] [CrossRef] [PubMed]
- Djoumbou Feunang, Y.; Eisner, R.; Knox, C.; Chepelev, L.; Hastings, J.; Owen, G.; Fahy, E.; Steinbeck, C.; Subramanian, S.; Bolton, E.; et al. ClassyFire: Automated chemical classification with a comprehensive, computable taxonomy. J. Cheminform. 2016, 8, 61. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Thiessen, P.A.; Bolton, E.E.; Chen, J.; Fu, G.; Gindulyte, A.; Han, L.; He, J.; He, S.; Shoemaker, B.A.; et al. PubChem Substance and Compound databases. Nucleic Acids Res. 2015, 44, 1202–1213. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, P.; Erehman, J.; Gohlke, B.O.; Wilhelm, T.; Preissner, R.; Dunkel, M. Super Natural II-a database of natural products. Nucleic Acids Res. 2015, 43, D935–D939. [Google Scholar] [CrossRef] [PubMed]
- Karp, P.D.; Paley, S.; Romero, P. The pathway tools software. Bioinformatics 2002, 18. [Google Scholar] [CrossRef]
- Fadason, T.; Schierding, W.; Kolbenev, N.; Liu, J.; Ingram, J.; O’Sullivan, J.M. Reconstructing the blood metabolome and genotype using long-range chromatin interactions. bioRxiv 2019, 656132. [Google Scholar] [CrossRef]
- Le, V.; Quinn, T.P.; Tran, T.; Venkatesh, S. Deep in the Bowel: Highly Interpretable Neural Encoder-Decoder Networks Predict Gut Metabolites from Gut Microbiome. bioRxiv 2019, 686394. [Google Scholar] [CrossRef]
- Mikolov, T.; Chen, K.; Corrado, G.; Dean, J. Distributed Representations of Words and Phrases and their Compositionality. In Proceedings of the 26th International Conference on Neural Information Processing Systems – Volume 2 (NIPS’13), New York, NY, USA, 5–10 December 2013; pp. 3111–3119. [Google Scholar]
- Morton, J.T.; Aksenov, A.A.; Nothias, L.F.; Foulds, J.R.; Quinn, R.A.; Badri, M.H.; Swenson, T.L.; Van Goethem, M.W.; Northen, T.R.; Vazquez-Baeza, Y.; et al. Learning representations of microbe–metabolite interactions. Nat. Methods 2019, 16, 1306–1314. [Google Scholar] [CrossRef]
- Romero, P.; Wagg, J.; Green, M.L.; Kaiser, D.; Krummenacker, M.; Karp, P.D. Computational prediction of human metabolic pathways from the complete human genome. Genome Biol. 2004, 6, R2. [Google Scholar] [CrossRef]
- Degtyarenko, K.; de Matos, P.; Ennis, M.; Hastings, J.; Zbinden, M.; McNaught, A.; Alcántara, R.; Darsow, M.; Guedj, M.; Ashburner, M. ChEBI: A database and ontology for chemical entities of biological interest. Nucleic Acids Res. 2007, 36, D344–D350. [Google Scholar] [CrossRef]
- Luscombe, N.M.; Greenbaum, D.; Gerstein, M. What is bioinformatics? A proposed definition and overview of the field. Methods Inf. Med. 2001, 40, 346–358. [Google Scholar] [CrossRef] [PubMed]
- Baggerly, K. Disclose all data in publications. Nature 2010, 467, 401. [Google Scholar] [CrossRef] [PubMed]
- Hüllermeier, E. Fuzzy methods in machine learning and data mining: Status and prospects. Fuzzy Sets Syst. 2005, 156, 387–406. [Google Scholar] [CrossRef]
- Bonneau, G.P.; Hege, H.C.; Johnson, C.R.; Oliveira, M.M.; Potter, K.; Rheingans, P.; Schultz, T. Overview and state-of-the-art of uncertainty visualization. In Scientific Visualization Uncertainty, Multifield, Biomedical, and Scalable Visualization; Springer: Berlin/Heidelberg, Germany, 2014; Volume 37, pp. 3–27. [Google Scholar]



{kind=link}
{kind=link}
| Type of Method | Functionality | Reference | |
|---|---|---|---|
| Dimensionality Reduction | t-Distributed Stochastic Neighbor Embedding (t-SNE) | Visualize gut microbial communities and serum metabolites by diet and supplements. | [46] |
| Visualize prefrontal cortex metabolites and lipids by human population group. | [47] † | ||
| Clustering | Hierarchical Clustering | Identify multi-omic molecular subtypes in hepatocellular carcinoma. | [48] ‡ |
| Identify multi-omic clusters in breast tumor tissue associated with prognosis. | [49] †‡ | ||
| k-means | Identify lipid–protein–metabolite clusters associated with diabetes and periodontal disease. | [50] | |
| Partitioning Around Medoids (PAM) | Identify microbial–metabolite clusters associated with diarrhea. | [51] *†‡ | |
| Gaussian Mixture Modeling (GMM) | Identify clinical depression score clusters associated with blood metabolomic and genomic data in blood to predict drug response. | [52] ‡ | |
| Density-Based Spatial Clustering of Applications with Noise (DBSCAN) | Evaluate the impact of bacterial metabolism on mucosal immunity. | [53] | |
| Other Machine Learning Methods | Random Forest | Identify clusters of histological stromal features associated with prognosis and metabolites in cancer-associated fibroblasts. | [54] ‡ |
| Autoencoder | Cluster plasma protein and metabolite levels to identify temporal trends in murine cardiac remodeling. | [55] | |
| Type of Method | Functionality | Reference | |
|---|---|---|---|
| Associative Networks | Correlation Networks | Find metabolite–metabolite associations specific to or shared across blood, urine, and saliva. | [91] † |
| Find modules of blood metabolites and genes associated with body weight change. | [92] ‡ | ||
| Find associations between serum, blood, and gut antibodies, metabolites, and microbiome and patient disease activity reports in inflammatory bowel disease. | [93] *†‡ | ||
| Find associations between metabolites, transcripts, cytokines, and cell frequencies in plasma and whole blood associated with adaptive immune response to Herpes zoster vaccine. | [94] †‡ | ||
| Partial Correlation Networks | Visualize associations between sleep survey responses and levels of serum cytokines, metabolites, lipids, proteins, and genes. | [95] *‡ | |
| Visualize associations between metabolites and lipids associated with metabolic disease treatment in rat liver tissue and clinical chemistry measurements from serum. | [96] † | ||
| Weighted Gene Co-Expression Network Analysis (WGCNA) | Characterize complex transcriptomic and metabolic traits in major depressive disorder. | [97] ‡ | |
| Identify co-regulated modules of blood metabolites and transcripts in children with asthma. | [98] ‡ | ||
| Identify co-regulated modules of metabolites and transcripts in glioblastoma multiforme. | [99] | ||
| Topological Analysis of Networks | Subnetworks | Identify subnetworks of correlated proteins and metabolites in adrenocorticotropic hormone-secreting pituitary adenomas. | [100] |
| Identify subnetworks of correlated genetic, proteomic, metabolomic, clinical, and microbiome data from multiple biofluids in cardiometabolic disease. | [101] ‡ | ||
| Type of Method | Functionality | Reference | |
|---|---|---|---|
| Univariate Statistical Methods | Student’s t-test and effect size | Identify metabolites, miRNAs, mRNAs, and lncRNAs altered by exposure to benzo[a]pyrene to identify mechanisms of toxicity. | [119] |
| Multivariate Statistical Methods | Partial Least Squares Discriminant Analysis (PLS-DA) (and variants) | Identify breast tumor tissue metabolites that differentiate MRI features. | [120] ‡ |
| Identify metabolites that differentiate normal and tumor tissue in the prostate. | [121] ‡ | ||
| Identify differences between fibromyalgia and control groups in gut microbes, serum metabolites, miRNA, and cytokine levels. | [122] *‡ | ||
| Discover temporal changes in plasma lipid and metabolite patterns from normal and hyperlipidemic patients. | [123] † | ||
| Linear Models (and variants) | Identify metabolites from bronchial alveolar lavage associated with continuous CT scan features in cystic fibrosis. | [124] ‡ | |
| Identify serum metabolites associated with visceral adipose tissue features from MRI and tomography. | [125] ‡ | ||
| Identify plasma metabolites and proteins associated with prognosis in septic shock patients. | [126] ‡ | ||
| Find associations between blood DNA methylation and metabolite levels in smokers. | [127] ‡ | ||
| Identifying Analyte Relationships that Differ by Phenotype | DiffCorr | Identify differences in metabolite-metabolite correlations between traumatic brain injury and control groups. | [128] |
| IntLIM | Identify synovial fluid metabolites and blood and bone marrow transcripts that differentiate between osteoarthritis and rheumatoid arthritis. | [129] * | |
| Machine Learning Methods for Predicting Phenotype | Random Forest | Identify serum metabolites, proteins, and peptides differentiating between metabolic syndrome and control groups. | [130] |
| Identify metabolites and other analytes predictive of weight gain and loss. | [131] *‡ | ||
| Identify metabolites, transcripts, and proteins predictive of potato quality traits. | [132] † | ||
| Identify metabolites and transcripts predictive of heat stress in the liver. | [133] † | ||
| Support Vector Machine (SVM) | Predict metabolite levels using genes and metabolites in breast and hepatocellular carcinoma. | [134] | |
| Multilayer Perceptron (MLP) | Predict early and late stage bladder cancer using urinary metabolites and genes. | [135] | |
| Predict early renal injury using serum metabolites and lipids. | [136] †‡ | ||
| Convolutional Neural Network (CNN) | Predict early renal injury using serum metabolites and lipids. | [136] †‡ | |
| Recurrent Neural Network (RNN) | Integrate transcript and metabolite levels to predict cellular state in Escherichia coli. | [137] *† | |
| Type of Method | Functionality | Reference | |
|---|---|---|---|
| Pathway enrichment methods | Overrepresentation Analysis (ORA) | Identify dysregulated pathways in prostate tumor tissue using metabolite and transcript data. | [191] |
| Identify dysregulated pathways in the murine hippocampus and left ventricle during proton irradiation using metabolite and DNA methylation data. | [192] | ||
| Identify dysregulated pathways in cationic liposome treatment of human hepatocyte cells using metabolomic and proteomic data. | [193] | ||
| Identify dysregulated pathways in kidney disease in the rat serum metabolome and proteome. | [194] ‡ | ||
| Identify dysregulated gut microbial pathways in gastrectomy patients. | [195] *‡ | ||
| Identify dysregulated gut microbial pathways in sports classification groups of Irish athletes. | [196] *‡ | ||
| Identify dysregulated gut microbial pathways as a result of whey protein supplementation. | [197] *‡ | ||
| Topological Scoring | Identify functional connections between dysregulated pathways in Alzheimer’s using genes, metabolites, miRNA, and proteins from multiple sources. | [198] | |
| Visualization of biological pathways and networks | Visualize metabolic networks in drug-susceptible and drug-resistant strains of Acinetobacter baumannii. | [199] | |
| Visualize interactions between metabolites and genes in non-small cell lung cancer. | [200] | ||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Eicher, T.; Kinnebrew, G.; Patt, A.; Spencer, K.; Ying, K.; Ma, Q.; Machiraju, R.; Mathé, E.A. Metabolomics and Multi-Omics Integration: A Survey of Computational Methods and Resources. Metabolites 2020, 10, 202. https://doi.org/10.3390/metabo10050202
Eicher T, Kinnebrew G, Patt A, Spencer K, Ying K, Ma Q, Machiraju R, Mathé EA. Metabolomics and Multi-Omics Integration: A Survey of Computational Methods and Resources. Metabolites. 2020; 10(5):202. https://doi.org/10.3390/metabo10050202
Chicago/Turabian StyleEicher, Tara, Garrett Kinnebrew, Andrew Patt, Kyle Spencer, Kevin Ying, Qin Ma, Raghu Machiraju, and Ewy A. Mathé. 2020. "Metabolomics and Multi-Omics Integration: A Survey of Computational Methods and Resources" Metabolites 10, no. 5: 202. https://doi.org/10.3390/metabo10050202
APA StyleEicher, T., Kinnebrew, G., Patt, A., Spencer, K., Ying, K., Ma, Q., Machiraju, R., & Mathé, E. A. (2020). Metabolomics and Multi-Omics Integration: A Survey of Computational Methods and Resources. Metabolites, 10(5), 202. https://doi.org/10.3390/metabo10050202