

Quantitative 1H NMR Spectroscopy Method for Determination of Anthraquinone Derivatives in Extracts from Rubia tinctorum L. Roots and Rhizomes

 , , , , , ,
, , , , , ,  ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Reagents
2.3. Sample Preparation
2.3.1. NMR Spectroscopy
2.3.2. HPLC-UV-MS
2.4. Experimental
2.4.1. NMR Spectroscopy
2.4.2. HPLC-UV-MS
2.5. Statistical Analysis
3. Results and Discussion
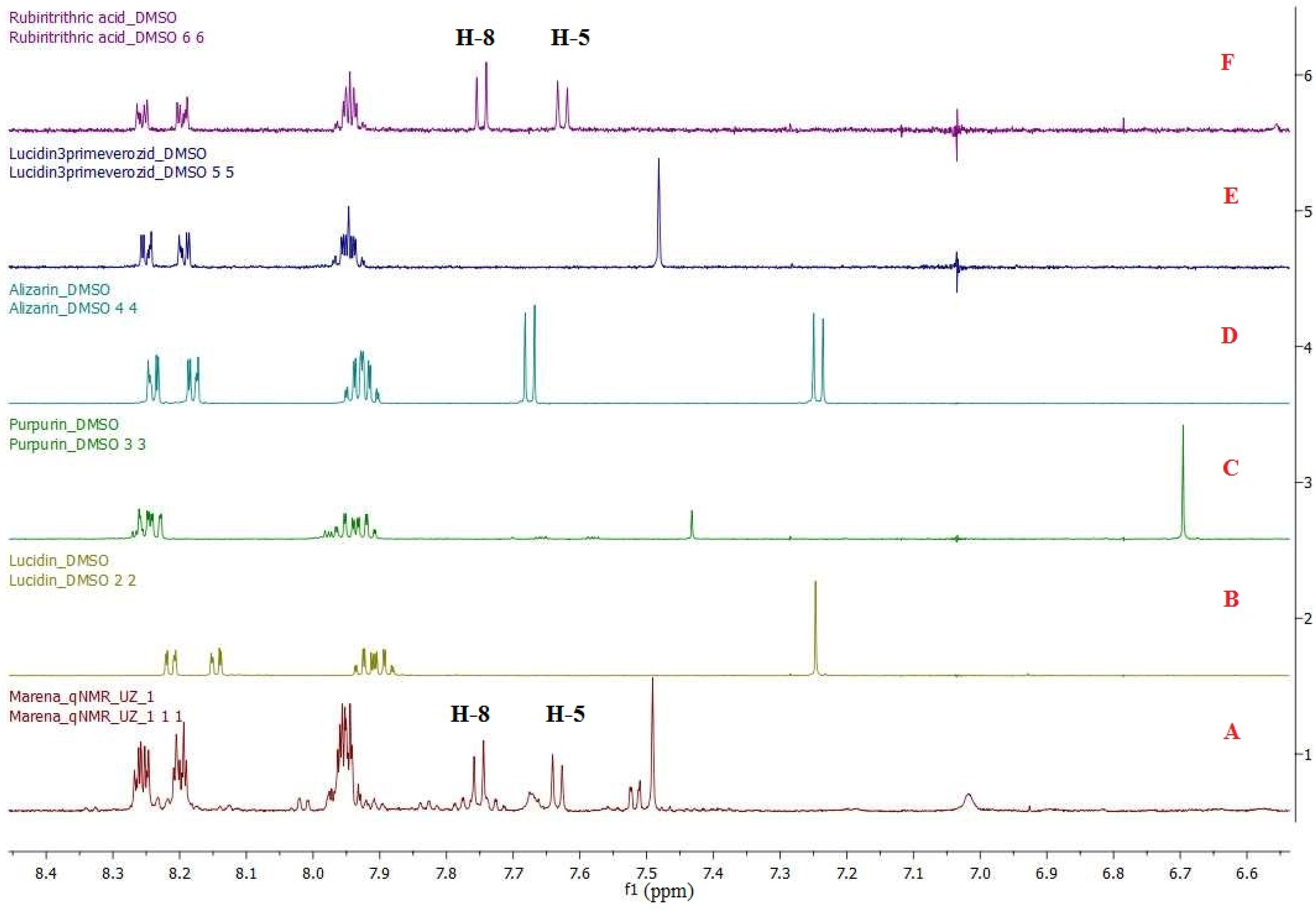
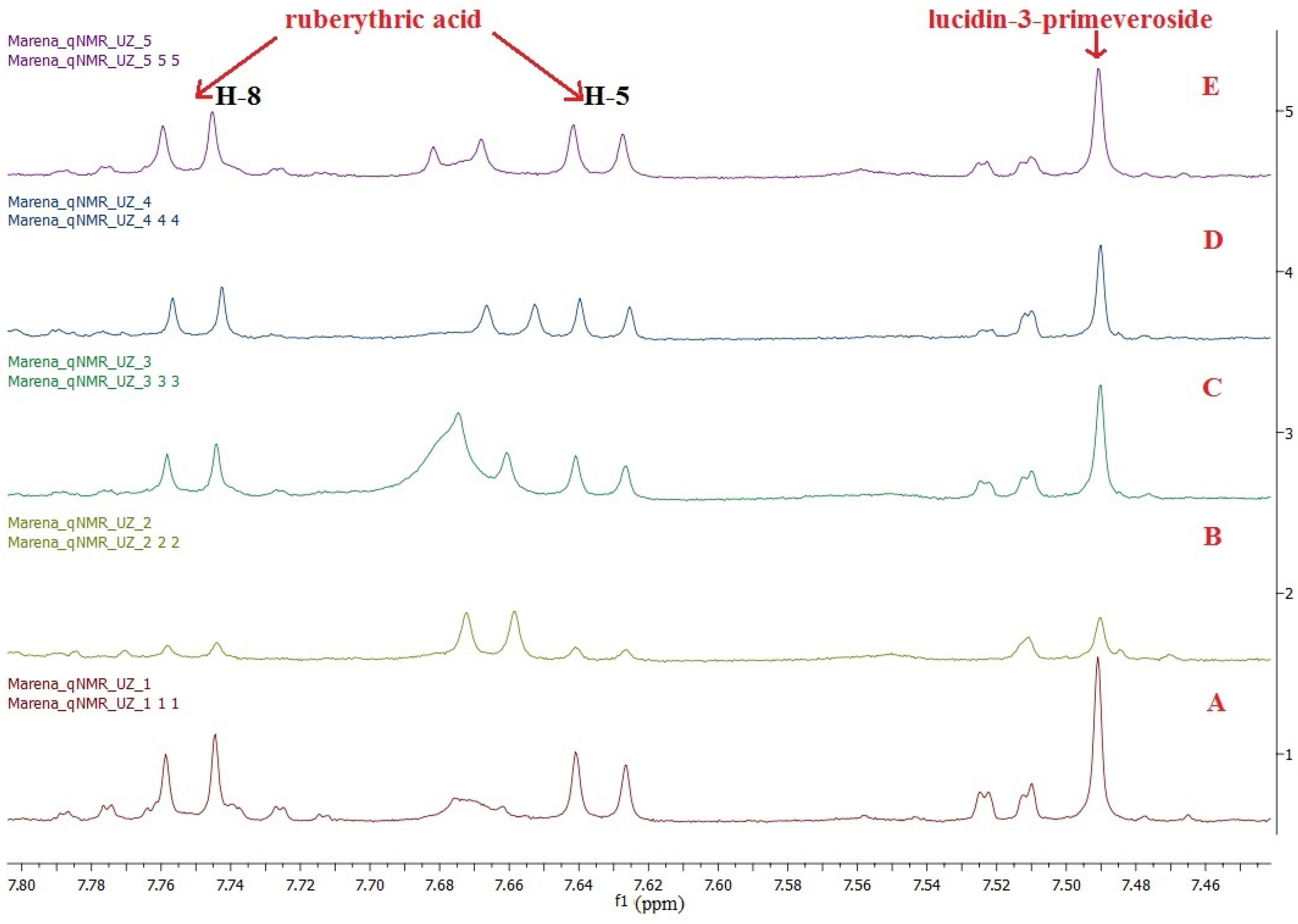
3.1. Selection of Quantification Signals of Analytes
3.2. Solvent Selection
3.3. Selecting a Reference Sample
3.4. Optimization of Experimental Parameters
3.5. Sample Preparation Conditions
3.6. Determination of RA and LP in Crude Extracts
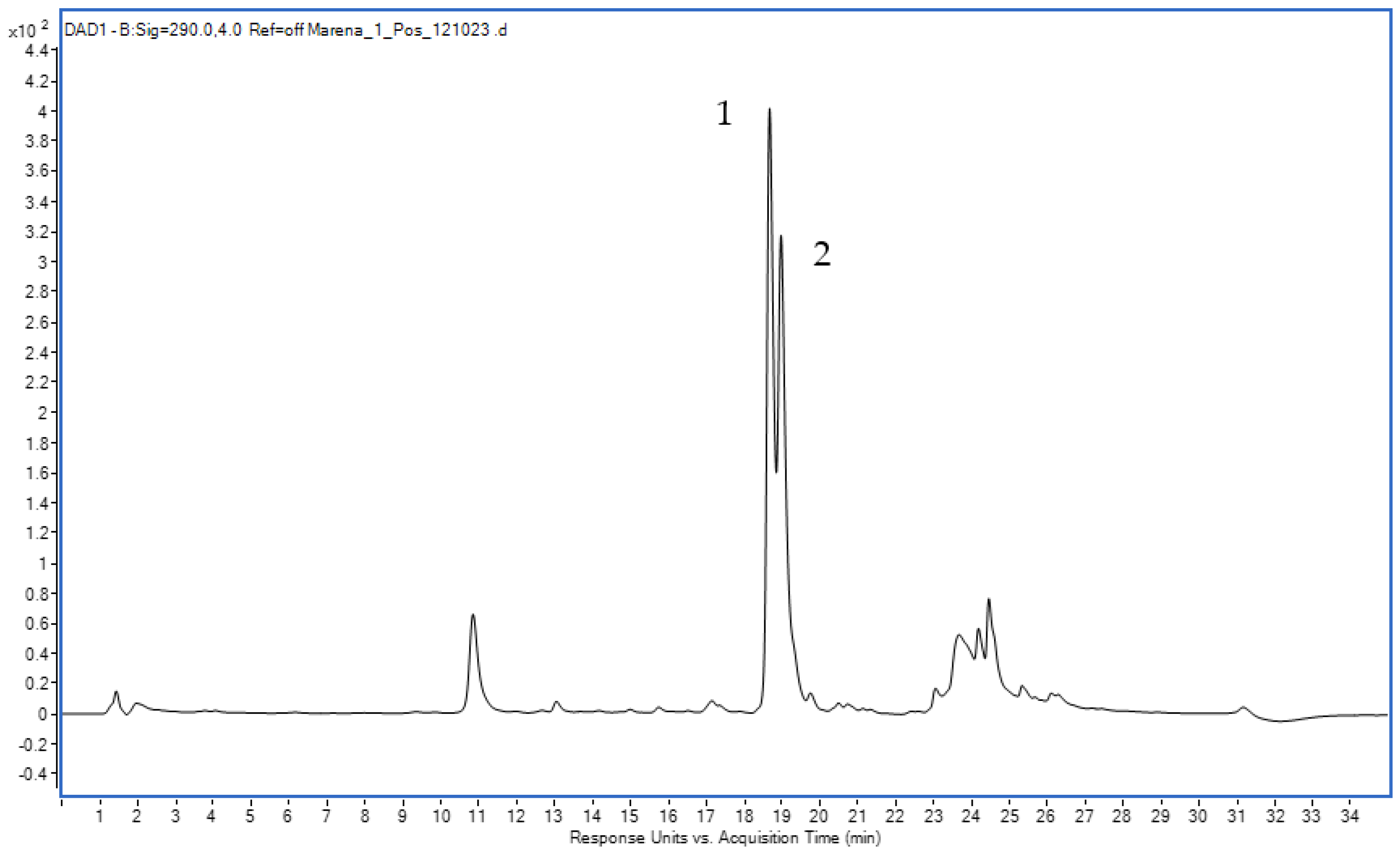
3.7. HPLC-UV-MS Experiment
3.8. Method Validation
3.8.1. Specificity
3.8.2. Linearity
3.8.3. Accuracy
3.8.4. Reproducibility
3.8.5. Sensitivity
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ford, L.L. Chemical Analysis and Elucidation of Anthraquinone and Flavonoid Type Compounds with Applications to Historical Artefacts and Sustainability. Ph.D. Thesis, University of Leeds, Leeds, UK, April 2017. [Google Scholar]
- Newman, R.; Gates, G.A. The Matter of madder in the ancient world. In Mummy Portraits of Roman Egypt: Emerging Research from the APPEAR Project; Svoboda, M., Cartwright, C.R., Eds.; Getty Publications: Los Angeles, NY, USA, 2020; pp. 24–33. [Google Scholar]
- Dyer, J.; Tamburini, D.; Sotiropoulou, S. The Identification of Lac as a Pigment in Ancient Greek Polychrome: The Case of a Hellenistic Oinochoe from Canosa di Puglia. Dye. Pigment. 2018, 149, 122–132. [Google Scholar] [CrossRef]
- Derksen, G.C.H.; Van Beek, T.A. Rubia tinctorum L. In Studies in Natural Products Chemistry, 1st ed.; Atta-ur-Rahman, Ed.; Elsevier: Amsterdam, The Netherlands, 2002; Volume 26 (part G), pp. 629–684. [Google Scholar] [CrossRef]
- Eltamany, E.E.; Nafie, M.S.; Khodeer, D.M.; El-Tanahy, A.H.H.; Abdel-Kader, M.S.; Badr, J.M.; Abdelhameed, R.F.A. Rubia tinctorum root extracts: Chemical profile and management of type II diabetes mellitus. RSC Adv. 2020, 10, 24159. [Google Scholar] [CrossRef] [PubMed]
- Hao, J.N.; Huang, M.P.; Lee, H. Structure-activity relationships of anthraquinones as inhibitors of 7-ethoxycoumarin O-deethylase and mutagenicity of 2-amino-3-methylimidazox [4,5-f] quinolone. Mutat. Res. 1995, 328, 183–191. [Google Scholar] [CrossRef] [PubMed]
- Marczylo, T.H.; Hayatsu, T.; Arimoto-Kobayashi, S.; Tada, M.; Fujita, K.; Kamataki, T.; Nakayama, K.; Hayatsu, H. Protection against the bacterial mutagenicity of heterocyclic amines by purpurin, a natural anthraquinone pigment. Mutat. Res. 1999, 444, 451–461. [Google Scholar] [CrossRef] [PubMed]
- Marczylo, T.H.; Arimoto-Kobayashi, S.; Hayatsu, H. Protection against Trp-P-2 mutagenicity by purpurin: Mechanism of in vitro antimutagenesis. Mutagenesis 2000, 15, 223–228. [Google Scholar] [CrossRef] [PubMed]
- Manojlović, N.T.; Solujić, S.; Sukdolak, S.; Milošev, M. Antifungal activity of Rubia tinctorum, Rhamnus frangula and Caloplaca cerina. Fitoterapia 2005, 76, 244–246. [Google Scholar] [CrossRef] [PubMed]
- Houari, F.Z.; Erenler, R.; Bakir, S.; Capanoglu, E.; Hariri, A. LC-ESI-MS/MS analysis, toxicity and anti-anaemic activity of Rubia tinctorum L. aqueous extract. Nova Biotechnol. Chim. 2022, 21, e978. [Google Scholar] [CrossRef]
- Derksen, G.C.H.; Niederlander, H.A.G.; van Beek, T.A. Analysis of anthraquinones in Rubia tinctorum L. by liquid chromatography coupled with diode-array UV and mass spectrometric detection. J. Chromatogr. A 2002, 978, 119–127. [Google Scholar] [CrossRef] [PubMed]
- Ford, L.; Rayner, C.M.; Blackburn, R.S. Isolation and extraction of ruberythric acid from Rubia tinctorum L. and crystal structure elucidation. Phytochemistry 2015, 117, 168–173. [Google Scholar] [CrossRef]
- Shukla, V.; Asthana, S.; Gupta, P.; Dwivedi, P.D.; Tripathi, A.; Das, M. Toxicity of Naturally Occurring Anthraquinones. In Advances in Molecular Toxicology; Fishbein, J.C., Heilman, J.M., Eds.; Elsevier: Amsterdam, The Netherlands, 2017; Volume 11, pp. 1–50. [Google Scholar] [CrossRef]
- Cpmp List of Herbal Drugs with Serious Risks; EMEA: London, UK, 1992.
- Nguyen, T.T.H.; Han, J.M.; Jung, H.J.; Pandey, R.P.; Park, Y.I.; Sohng, J.K. Regio-specific biotransformation of alizarin to alizarin methoxide with enhanced cytotoxicity against proliferative cells. J. Ind. Microbiol. Biotechnol. 2020, 47, 537–542. [Google Scholar] [CrossRef]
- Russian Pharmacopoeia, 14th ed.; Emshanova, S.V., Potanina, O.G., Budanova, E.V., Chistyakov, V.V., Goryun, M.I., Eds.; Ministry of Health of the Russian Federation: Moscow, Russia, 2018; Volume 4, pp. 6243–6252. Available online: https://docs.rucml.ru/feml/pharma/v14/vol4/1061/ (accessed on 17 March 2024).
- Marković, Z.S.; Manojlović, N.T.; Jeremić, S.R.; Živić, M. HPLC, UV-vis and NMR spectroscopic and DFT characterization of purpurin isolated from Rubia tinctorum L. Hem. Ind. 2013, 67, 77–88. [Google Scholar] [CrossRef]
- Angelini, L.G.; Pistelli, L.; Belloni, P.; Bertoli, A.; Panconesi, S. Rubia tinctorum a source of natural dyes: Agronomic evaluation, quantitative analysis of alizarin and industrial assays. Ind. Crop. Prod. 1997, 6, 303–311. [Google Scholar] [CrossRef]
- Hu, Y.Y.; Zhang, X.J.; Zhang, Z.H. Qualitative and quantitative analyses of quinones in multi-origin Rubia species by ultra-performance liquid chromatography-tandem mass spectrometry combined with chemometrics. J. Pharm. Biomed. Anal. 2020, 189, 113471. [Google Scholar] [CrossRef] [PubMed]
- Langa-Lomba, N.; Sánchez-Hernández, E.; Buzón-Durán, L.; González-García, V.; Casanova-Gascón, J.; Martín-Gil, J.; Martín-Ramos, P. Activity of Anthracenediones and Flavoring Phenols in Hydromethanolic Extracts of Rubia tinctorum against Grapevine Phytopathogenic Fungi. Plants 2021, 10, 1527. [Google Scholar] [CrossRef] [PubMed]
- Pierens, G.K.; Carroll, A.R.; Davis, R.A.; Palframan, M.E.; Ronald, J. Determination of Analyte Concentration Using the Residual Solvent Resonance in 1H NMR Spectroscopy. Quinn J. Nat. Prod. 2008, 71, 810–813. [Google Scholar] [CrossRef] [PubMed]
- AbouZid, S.F.; Chen, S.N.; Pauli, G.F. Silymarin content in Silybum marianum populations growing in Egypt. Ind. Crops Prod. 2016, 83, 729–737. [Google Scholar] [CrossRef] [PubMed]
- Imai, A.; Lankin, D.C.; Godecke, T.; Chen, S.N.; Pauli, G.F. NMR based quantitation of cycloartane triterpenes in black cohosh extracts. Fitoterapia 2020, 141, 104846. [Google Scholar] [CrossRef]
- Feng, Y.; Li, Q.; Yang, L.; Zhang, Y.; Qiu, D. The Use of H-1-qNMR Method for Simultaneous Determination of Osthol, Columbianadin, and Isoimperatorin in Angelicae Pubescentis Radix. J. AOAC Int. 2020, 103, 851–856. [Google Scholar] [CrossRef]
- Yang, L.; Li, Q.; Feng, Y.; Qiu, D. Simultaneous Determination of Three Coumarins in Angelica dahurica by H-1-qNMR Method: A Fast and Validated Method for Crude Drug Quality Control. J. Anal. Methods Chem. 2020, 2020, 8987560. [Google Scholar] [CrossRef]
- Dong, J.-W.; Li, X.-J.; Shi, J.-Y.; Liu, K.-Q. Application of a proton quantitative nuclear magnetic resonance spectroscopy method for the determination of actinodaphnine in Illigera aromatica and Illigera henryi. J. Nat. Med. 2019, 73, 312–317. [Google Scholar] [CrossRef]
- Napolitano, J.G.; Godecke, T.; Lankin, D.C.; Jaki, B.U.; McAlpine, J.B.; Chen, S.N.; Pauli, G.F. Orthogonal analytical methods for botanical standardization: Determination of green tea catechins by qNMR and LC-MS/MS. J. Pharm. Biomed. Anal. 2014, 93, 59–67. [Google Scholar] [CrossRef]
- Kurkin, V.A.; Shmygareva, A.A.; Rybalko, M.V.; Daeva, E.D.; Kadentse, V.I. Xanthopurposide, A New Anthraglycoside from Rubia tinctorum Rhizomes. Chem. Nat. Compd. 2021, 57, 14–15. [Google Scholar] [CrossRef]
- Henderson, R.L.; Rayner, C.M.; Blackburn, R.S. Isolation and extraction of lucidin primeveroside from Rubia tinctorum L. and crystal structure elucidation. Phytochemistry 2013, 95, 105–108. [Google Scholar] [CrossRef] [PubMed]
- Ford, L.; Rayner, C.M. Degradation of lucidin: New insights into the fate of this natural pigment present in Dyer’s madder (Rubia tinctorum L.) during the extraction of textile artefacts. Dye. Pigment. 2018, 154, 290–295. [Google Scholar] [CrossRef]
- Kalabin, G.; Vasil’ev, V.; Ivlev, V.; Babkin, V. Fast screening of some flavonoids content in raw plant materials: Opportunities of 1H NMR spectroscopy. E3S Web Conf. 2020, 169, 02006. [Google Scholar] [CrossRef]
- Vasil’ev, V.G.; Prokop’ev, A.S.; Kalabin, G.A. Identification of Terpene Lactones and Flavonol Glycosides in Preparations Based on Ginkgo Biloba Extract and a New Way of Semi-Quantitative Determination of Flavonol Glycosides by 1H NMR Spectroscopy. Russ. J. Bioorg. Chem. 2017, 43, 776–782. [Google Scholar] [CrossRef]
- Santosh, K.B.; Raja, R. Quantitative 1H NMR spectroscopy. TrAC 2012, 35, 5–26. [Google Scholar] [CrossRef]
- Malz, F.; Jancke, H. Validation of quantitative NMR. J. Pharm. Biomed. Anal. 2005, 38, 813–823. [Google Scholar] [CrossRef]
- ICH Topic Q 2 (R1). Validation of Analytical Procedures: Text and Methodolog; EMEA: London, UK, 1995.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample ID | C (RA), % | C (LP), % | C (total), % | |||
|---|---|---|---|---|---|---|
| NMR | HPLC-UV | NMR | HPLC-UV | NMR | HPLC-UV | |
| 1 | 1.67 ± 0.02 | 1.65 ± 0.04 | 2.18 ± 0.04 | 1.86 ± 0.03 | 3.85 ± 0.05 | 3.51 ± 0.04 |
| 2 | 0.35 ± 0.02 | 0.30 ± 0.02 | 0.76 ± 0.02 | 0.72 ± 0.02 | 1.11 ± 0.02 | 1.02 ± 0.02 |
| 3 | 1.13 ± 0.03 | 1.07 ± 0.03 | 1.87 ± 0.03 | 1.76 ± 0.03 | 3.00 ± 0.04 | 2.83 ± 0.03 |
| 4 | 0.73 ± 0.02 | 0.50 ± 0.02 | 1.15 ± 0.02 | 1.18 ± 0.03 | 1.89 ± 0.03 | 1.68 ± 0.02 |
| 5 | 1.44 ± 0.03 | 1.34 ± 0.03 | 1.66 ± 0.03 | 1.57 ± 0.03 | 3.10 ± 0.04 | 2.91 ± 0.04 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vasil’ev, V.; Sheremeta, A.; Ivlev, V.; Goriainov, S.; Hajjar, F.; Esparza, C.; Platonov, E.; Khromov, A.; Kolesnov, A.; Romashchenko, V.; et al. Quantitative 1H NMR Spectroscopy Method for Determination of Anthraquinone Derivatives in Extracts from Rubia tinctorum L. Roots and Rhizomes. Sci. Pharm. 2024, 92, 24. https://doi.org/10.3390/scipharm92020024
Vasil’ev V, Sheremeta A, Ivlev V, Goriainov S, Hajjar F, Esparza C, Platonov E, Khromov A, Kolesnov A, Romashchenko V, et al. Quantitative 1H NMR Spectroscopy Method for Determination of Anthraquinone Derivatives in Extracts from Rubia tinctorum L. Roots and Rhizomes. Scientia Pharmaceutica. 2024; 92(2):24. https://doi.org/10.3390/scipharm92020024
Chicago/Turabian StyleVasil’ev, Vasilii, Anzhelika Sheremeta, Vasilii Ivlev, Sergey Goriainov, Fadi Hajjar, Cesar Esparza, Evgeniy Platonov, Arkadiy Khromov, Alexandr Kolesnov, Victoria Romashchenko, and et al. 2024. "Quantitative 1H NMR Spectroscopy Method for Determination of Anthraquinone Derivatives in Extracts from Rubia tinctorum L. Roots and Rhizomes" Scientia Pharmaceutica 92, no. 2: 24. https://doi.org/10.3390/scipharm92020024
APA StyleVasil’ev, V., Sheremeta, A., Ivlev, V., Goriainov, S., Hajjar, F., Esparza, C., Platonov, E., Khromov, A., Kolesnov, A., Romashchenko, V., & Kalabin, G. (2024). Quantitative 1H NMR Spectroscopy Method for Determination of Anthraquinone Derivatives in Extracts from Rubia tinctorum L. Roots and Rhizomes. Scientia Pharmaceutica, 92(2), 24. https://doi.org/10.3390/scipharm92020024