
The Optimization of a Dimenhydrinate Transdermal Patch Formulation Based on the Quantitative Analysis of In Vitro Release Data by DDSolver through Skin Penetration Studies



Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
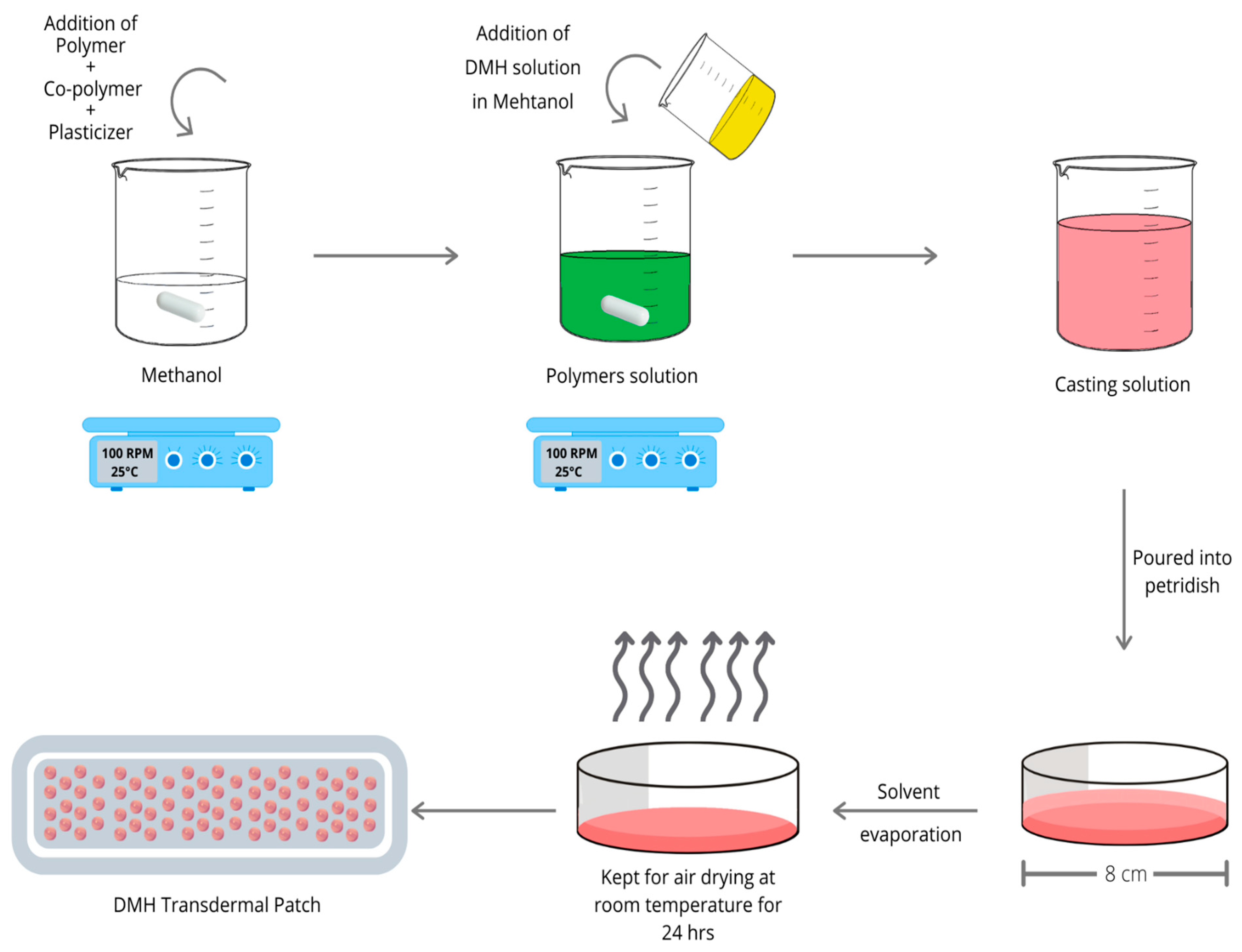
2.2. Preparation of DMH Transdermal Patches
2.3. Preliminary Screening
2.4. DMH Transdermal Patch Characterization
2.4.1. Patch Thickness and Surface pH
2.4.2. Weight Uniformity
2.4.3. Folding Endurance
2.4.4. Drug Content
2.4.5. Moisture Content
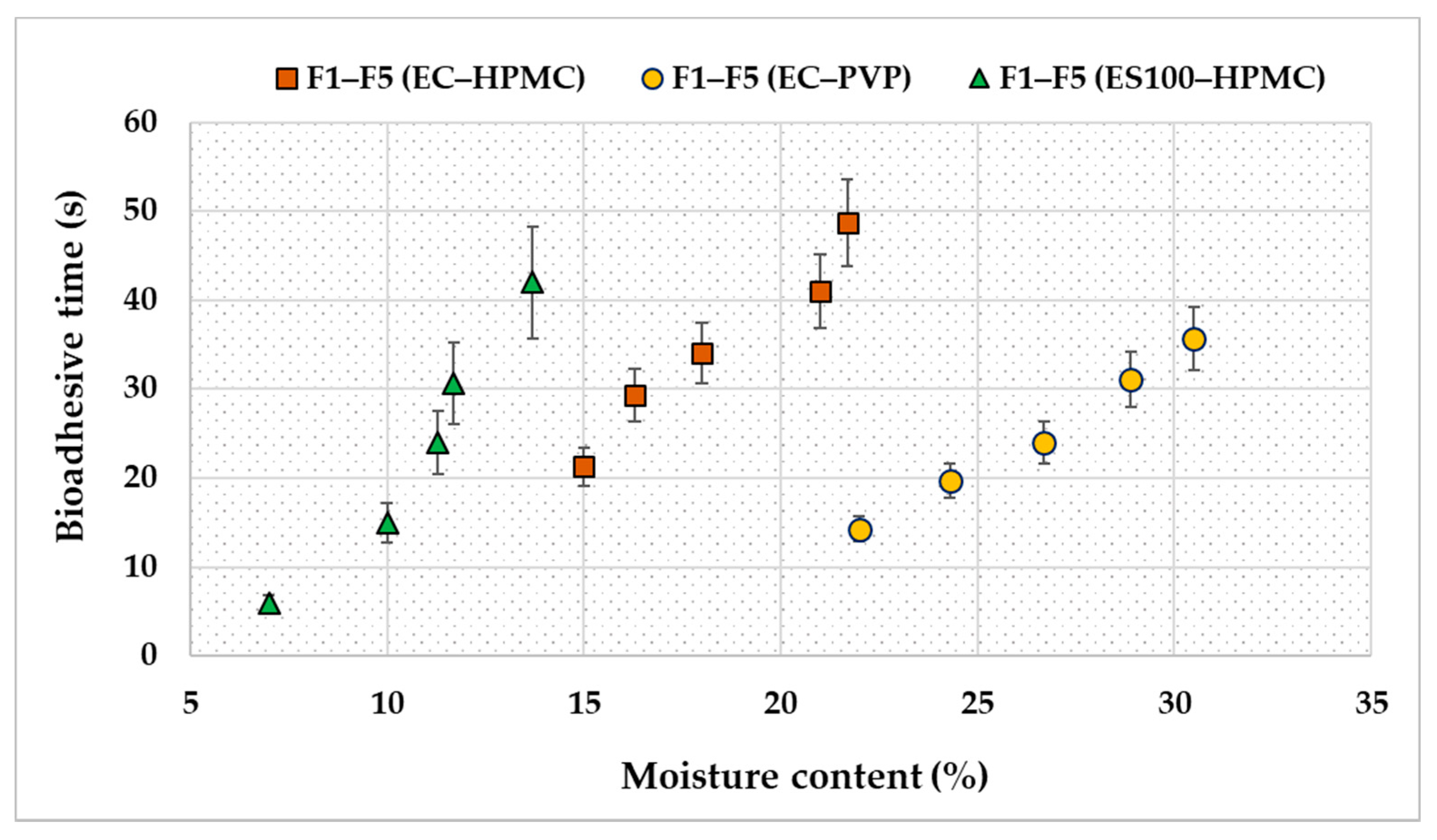
2.4.6. Adhesive Properties
2.5. In Vitro Drug Release
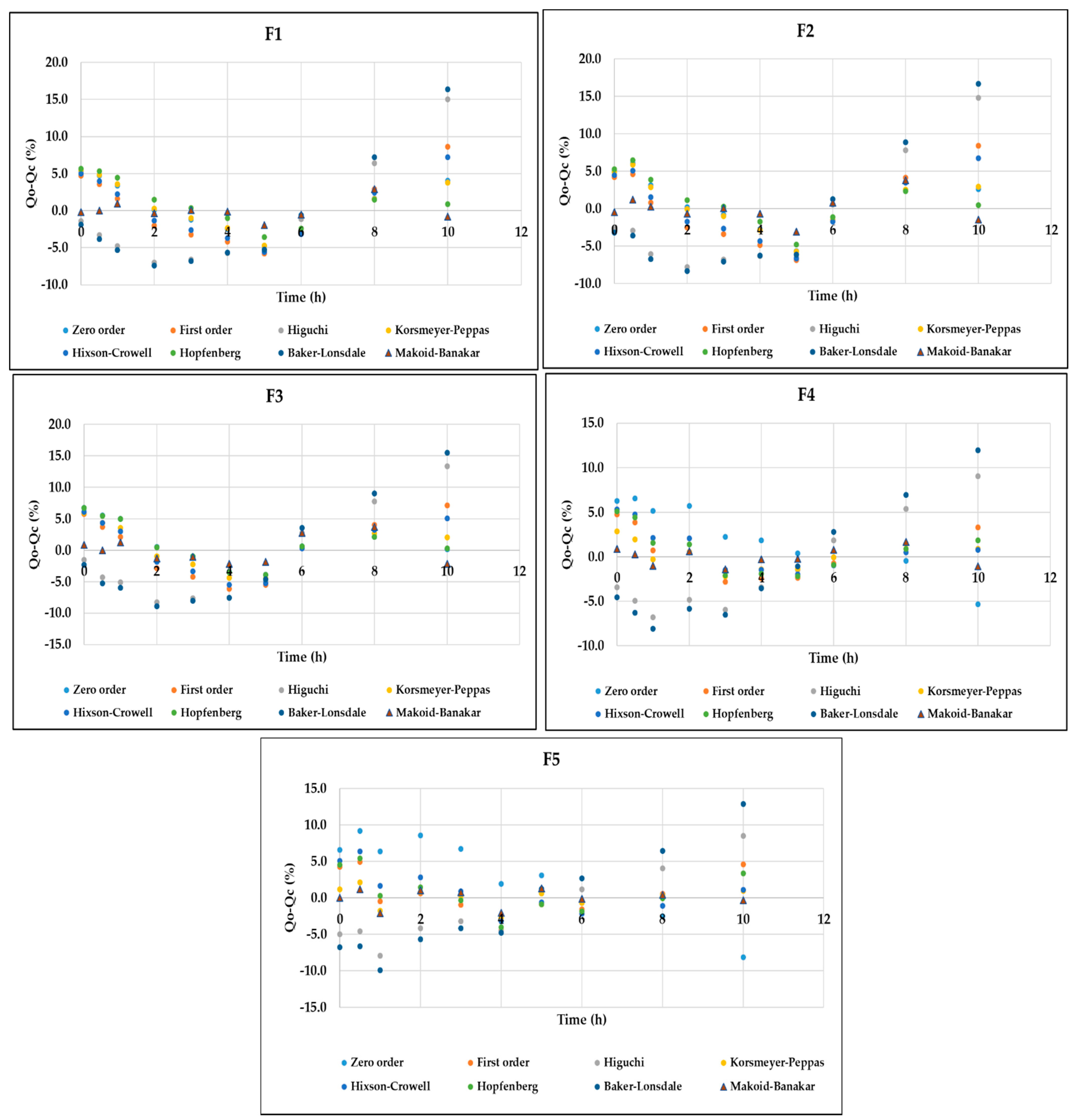
2.6. Mathematical Analysis of DMH Release Data
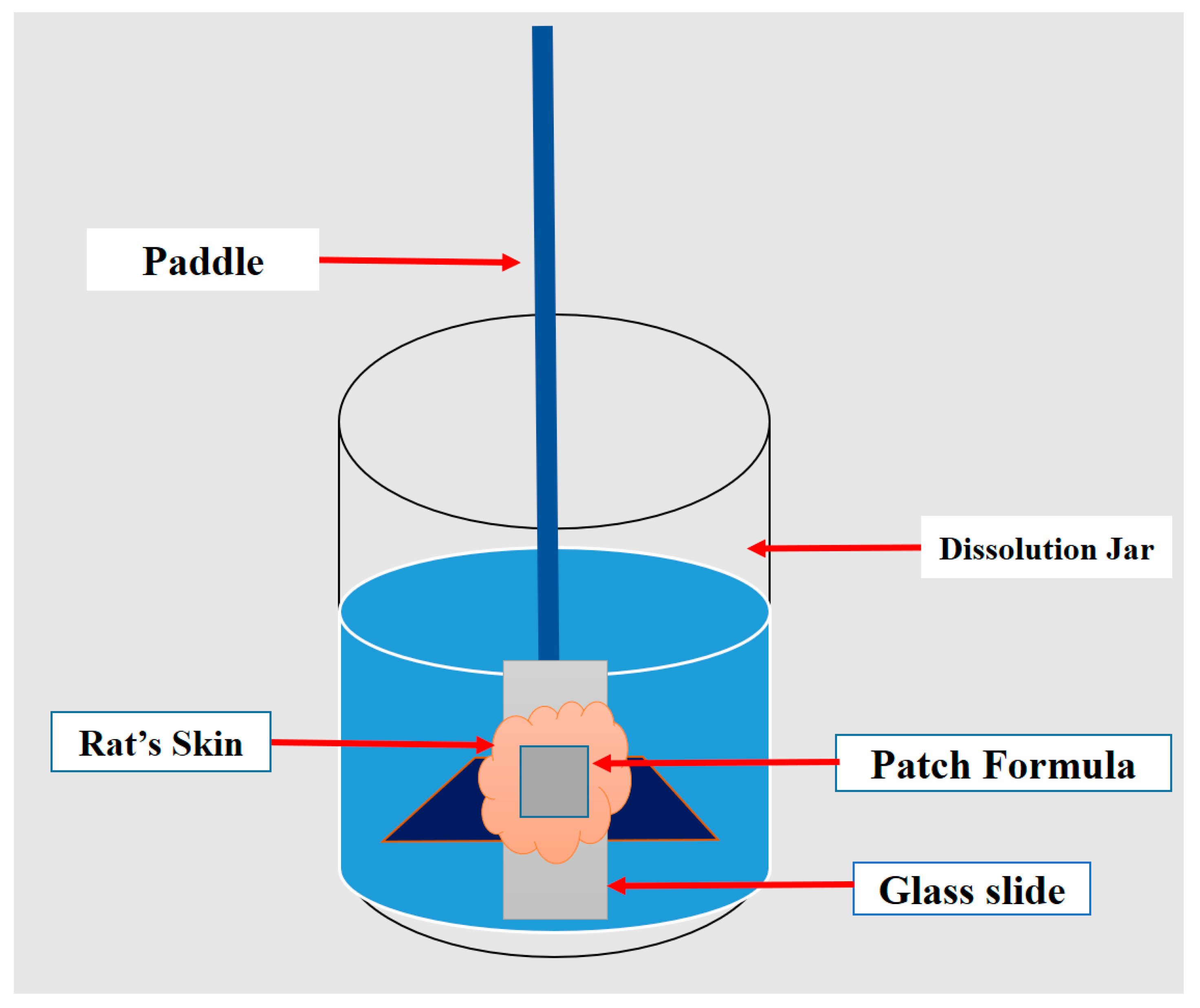
2.7. Ex Vivo Skin Permeation Study
2.7.1. Skin Preparation
2.7.2. Experimental Design
2.7.3. Data Analysis
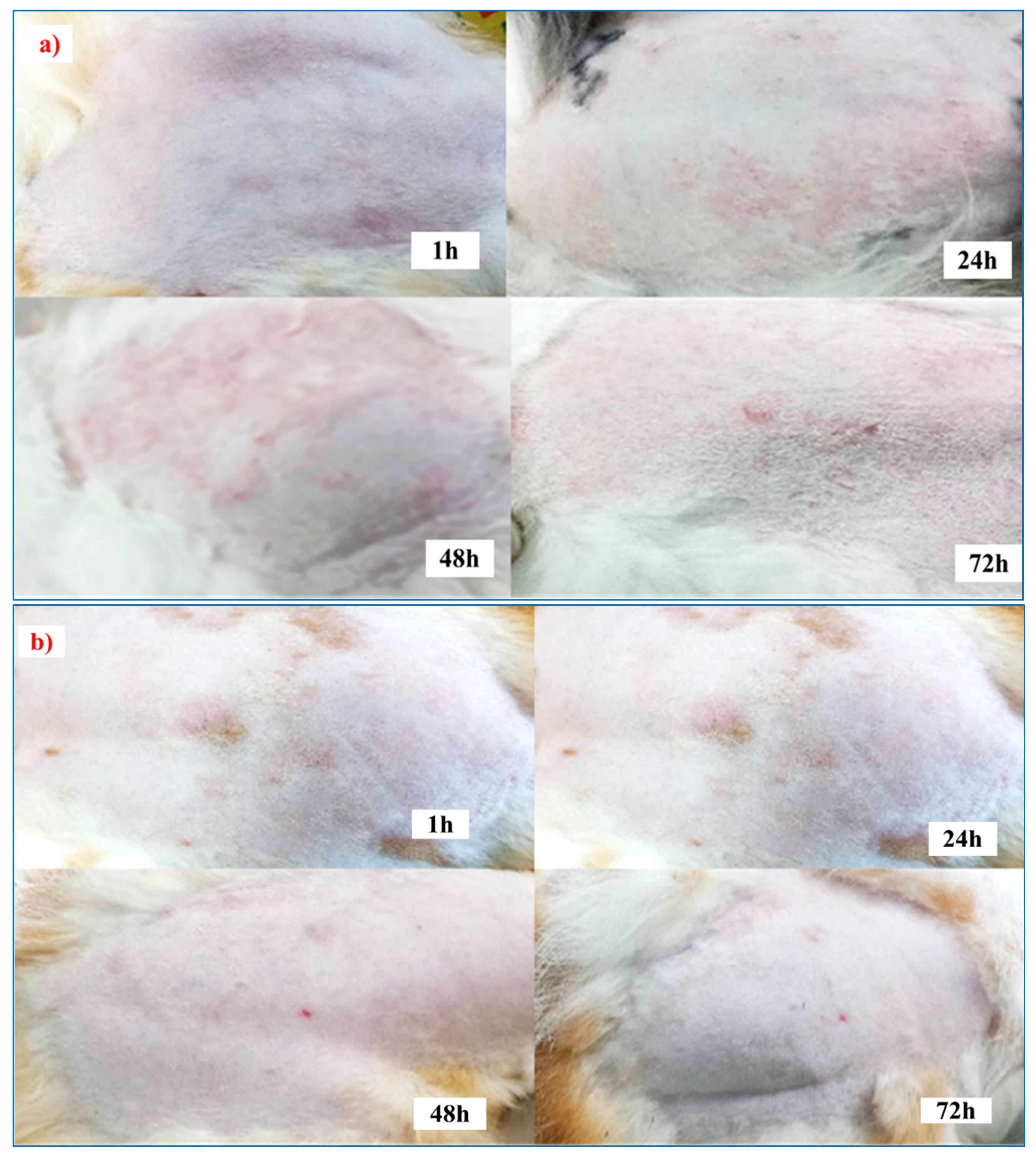
2.8. Skin-Irritation Study
2.9. FTIR Spectroscopy Analysis
2.10. Statistical Analysis
3. Results and Discussion
3.1. Preliminary Studies
3.2. Evaluation of the Transdermal Patches
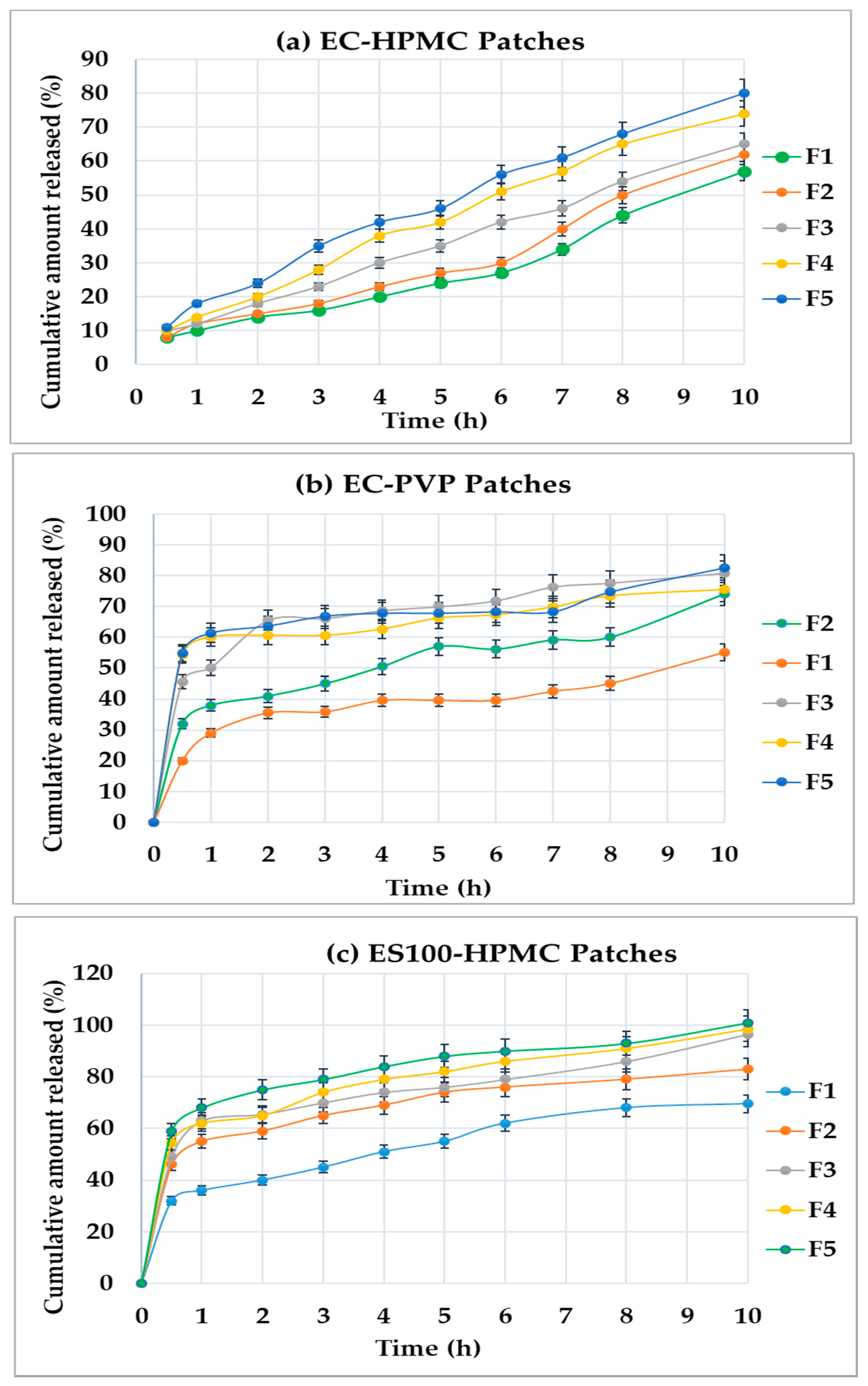
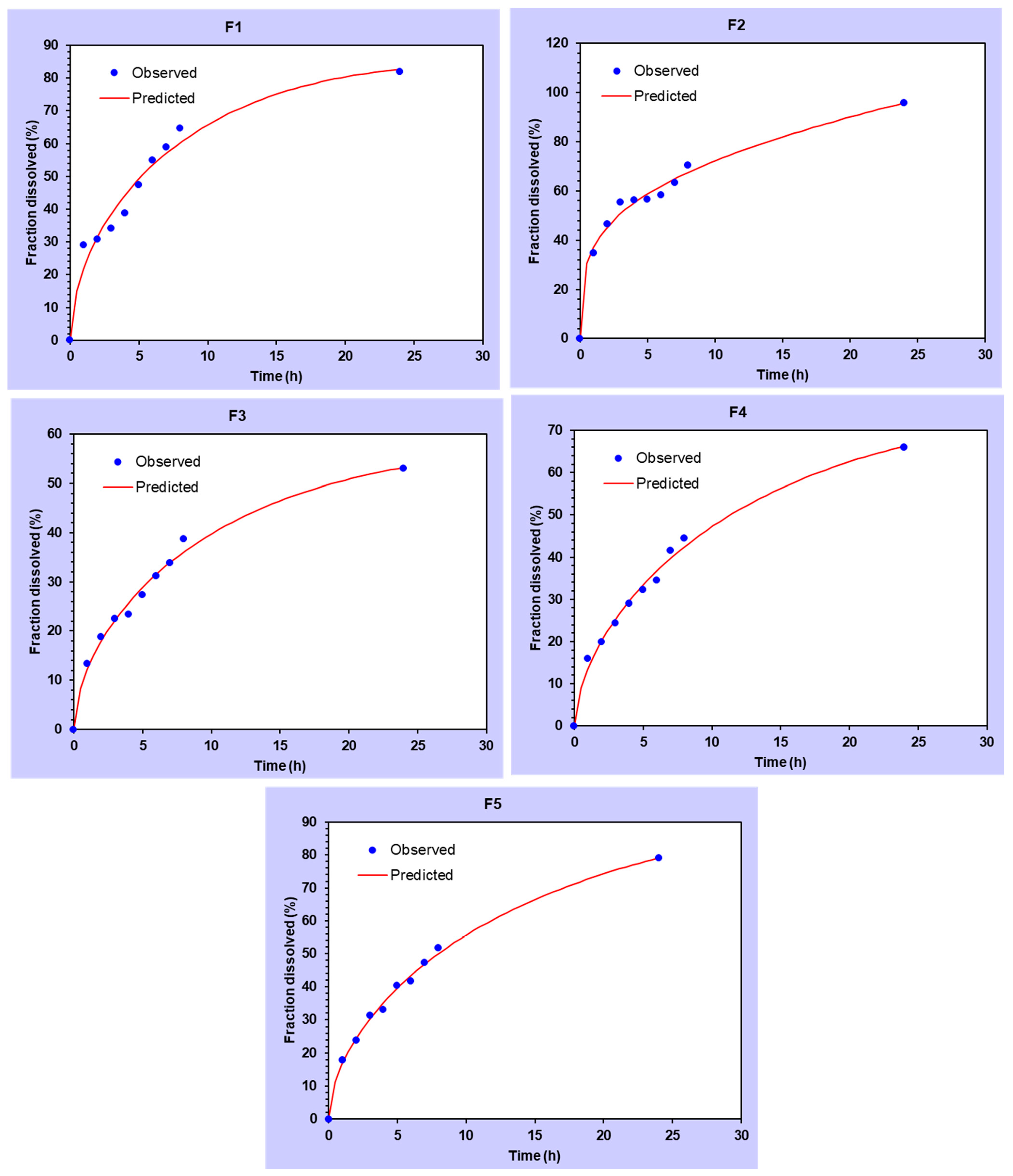
3.3. In Vitro Drug Release
- The amount released of DMH was significantly (p < 0.05) highest from the patches prepared from ES100-HPMC compared to other formulae with EC-HPMC and EC-PVP.
- There were insignificant (p > 0.05) differences (Q10) between Set-1 and Set-2 patches.
- Patches containing the EC-HPMC displayed desirable sustained drug-release profiles from their matrices. In contrast, the Set-1 and Set-2 patches showed an unwanted initial burst drug release, followed by a sustained pattern.
3.4. In Vitro Release Analysis of DMH from F1–F5 (EC-HPMC)
3.5. Ex Vivo Skin Permeation Study
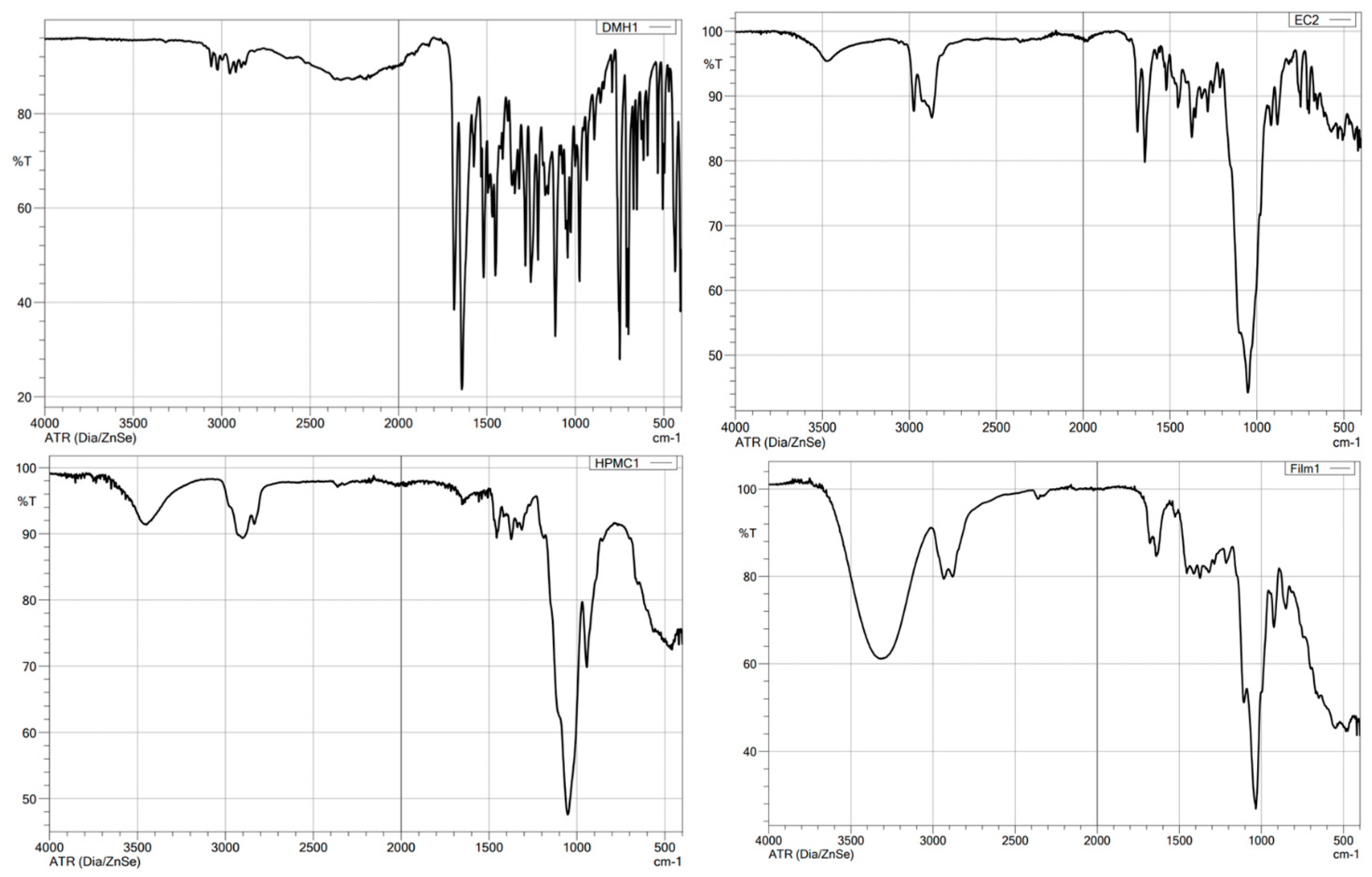
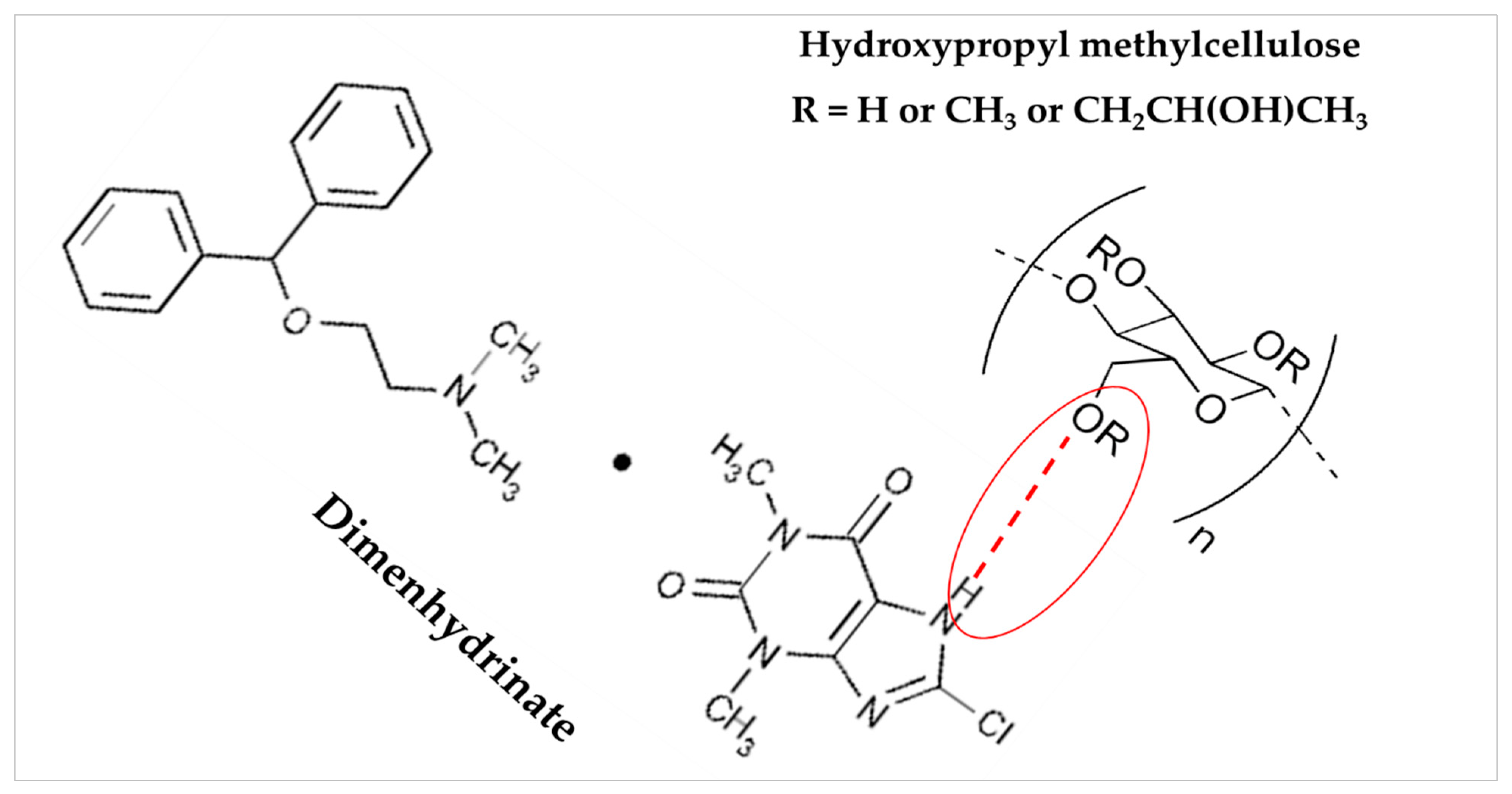
3.6. FTIR Spectroscopy Analysis
3.7. Skin-Irritation Study
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Marwah, H.; Garg, T.; Goyal, A.K.; Rath, G. Permeation enhancer strategies in transdermal drug delivery. Drug Deliv. 2016, 23, 564–578. [Google Scholar] [CrossRef] [PubMed]
- Waghule, T.; Singhvi, G.; Dubey, S.K.; Pandey, M.M.; Gupta, G.; Singh, M.; Dua, K. Microneedles: A smart approach and increasing potential for transdermal drug delivery system. Biomed. Pharmacother. 2019, 109, 1249–1258. [Google Scholar] [CrossRef] [PubMed]
- Wen, H.; Jung, H.; Li, X. Drug delivery approaches in addressing clinical pharmacology-related issues: Opportunities and challenges. AAPS J. 2015, 17, 1327–1340. [Google Scholar] [CrossRef]
- Eberhart, L.H.; Seeling, W.; Bopp, T.I.; Morin, A.M.; Georgieff, M. Dimenhydrinate for prevention of post-operative nausea and vomiting in female in-patients. Eur. J. Anaesthesiol. 1999, 16, 284–289. [Google Scholar] [CrossRef] [PubMed]
- Akhtar, N.; Singh, V.; Yusuf, M.; Khan, R.A. Non-invasive drug delivery technology: Development and current status of transdermal drug delivery devices, techniques and biomedical applications. Biomed. Eng. Biomed. Tech. 2020, 65, 243–272. [Google Scholar] [CrossRef]
- Singh, I.; Dastidar, D.G.; Ghosh, D.; Sengupta, A.; Ajala, T.O.; Odeku, O.A.; Singh, B.P.; Sharma, M. Bioadhesive films as drug delivery systems. Drug Deliv. Lett. 2021, 11, 2–15. [Google Scholar] [CrossRef]
- Abd, E.; Benson, H.A.E.; Roberts, M.S.; Grice, J.E. Minoxidil skin delivery from nanoemulsion formulations containing eucalyptol or oleic acid: Enhanced diffusivity and follicular targeting. Pharmaceutics 2018, 10, 19. [Google Scholar] [CrossRef]
- Wiedersberg, S.; Guy, R.H. Transdermal drug delivery: 30+ years of war and still fighting! J. Control. Release 2014, 190, 150–156. [Google Scholar] [CrossRef] [PubMed]
- Zsikó, S.; Csányi, E.; Kovács, A.; Budai-Szűcs, M.; Gácsi, A.; Berkó, S. Methods to evaluate skin penetration in vitro. Sci. Pharm. 2019, 87, 19. [Google Scholar] [CrossRef]
- Naz, Z.; Ahmad, F.J. Curcumin-loaded colloidal carrier system: Formulation optimization, mechanistic insight, ex vivo and in vivo evaluation. Int. J. Nanomed. 2015, 10, 4293. [Google Scholar] [CrossRef]
- Yu, Y.Q.; Yang, X.; Wu, X.F.; Fan, Y.B. Enhancing permeation of drug molecules across the skin via delivery in nanocarriers: Novel strategies for effective transdermal applications. Front. Bioeng. Biotechnol. 2021, 9, 646554. [Google Scholar] [CrossRef]
- Opatha, S.A.T.; Titapiwatanakun, V.; Chutoprapat, R. Transfersomes: A promising nanoencapsulation technique for transdermal drug delivery. Pharmaceutics 2020, 12, 855. [Google Scholar] [CrossRef]
- Shaker, D.S.; Ishak, R.A.H.; Ghoneim, A.; Elhuoni, M.A. Nanoemulsion: A review on mechanisms for the transdermal delivery of hydrophobic and hydrophilic drugs. Sci. Pharm. 2019, 87, 17. [Google Scholar] [CrossRef]
- Sae Yoon, A.; Sakdiset, P. Development of microemulsions containing Glochidion wallichianum leaf extract and potential for transdermal and topical skin delivery of gallic acid. Sci. Pharm. 2020, 88, 53. [Google Scholar] [CrossRef]
- Jahan, N.; Archie, S.R.; Shoyaib, A.A.; Kabir, N.; Cheung, K. Recent approaches for solid dose vaccine delivery. Sci. Pharm. 2019, 87, 27. [Google Scholar] [CrossRef]
- Dimenhydrinate, Drugs.Com. Available online: https://www.drugs.com/mtm/dimenhydrinate.html (accessed on 25 June 2021).
- Dimenhydrinate, DrugBank Online. Available online: https://go.drugbank.com/drugs/DB00985 (accessed on 25 June 2021).
- Akram, M.R.; Ahmad, M.; Abrar, A.; Sarfraz, R.M.; Mahmood, A. Formulation design and development of matrix diffusion controlled transdermal drug delivery of glimepiride. Drug Des. Dev. Ther. 2018, 12, 349–364. [Google Scholar] [CrossRef]
- Al Hanbali, O.A.; Khan, H.M.S.; Sarfraz, M.; Arafat, M.; Ijaz, S.; Hameed, A. Transdermal patches: Design and current approaches to painless drug delivery. Acta Pharm. 2019, 69, 197–215. [Google Scholar] [CrossRef] [PubMed]
- Telange, D.R.; Nirgulkar, S.B.; Umekar, M.J.; Patil, A.T.; Pethe, A.M.; Bali, N.R. Enhanced transdermal permeation and anti-inflammatory potential of phospholipids complex-loaded matrix film of umbelliferone: Formulation development, physico-chemical and functional characterization. Eur. J. Pharm. Sci. 2019, 131, 23–38. [Google Scholar] [CrossRef]
- Zhang, Y.; Huo, M.; Zhou, J.; Zou, A.; Li, W.; Yao, C.; Xie, S. DDSolver: An add-in program for modeling and comparison of drug dissolution profiles. AAPS J. 2010, 12, 263–271. [Google Scholar] [CrossRef] [PubMed]
- Saoji, S.D.; Atram, S.C.; Dhore, P.W.; Deole, P.S.; Raut, N.A.; Dave, V.S. Influence of the component excipients on the quality and functionality of a transdermal film formulation. AAPS Pharm. Sci. Tech. 2015, 16, 1344–1356. [Google Scholar] [CrossRef]
- Organisation for Economic Cooperation and Development. Guidance document on integrated approaches to testing and assessment for skin irritation/corrosion. In OECD Series on Testing and Assessment. No. 203; Environment, Health and Safety Publications: Paris, France, 2014. [Google Scholar]
- Almazan, E.A.; Castañeda, P.S.; Torres, R.D.; Escobar-Chavez, J.J. Design and evaluation of losartan transdermal patch by using solid microneedles as a physical permeation enhancer. Iran. J. Pharm. Res. 2020, 19, 138–152. [Google Scholar] [CrossRef]
- Namjoshi, S.; Dabbaghi, M.; Roberts, M.S.; Grice, J.E.; Mohammed, Y. Quality by design: Development of the quality target product profile (QTPP) for semisolid topical products. Pharmaceutics 2020, 12, 287. [Google Scholar] [CrossRef]
- Cherukuri, S.; Batchu, U.R.; Mandava, K.; Cherukuri, V.; Ganapuram, K.R. Formulation and evaluation of transdermal drug delivery of topiramate. Int. J. Pharm. Investig. 2017, 7, 10–17. [Google Scholar] [CrossRef]
- Ali, S.M.; Yosipovitch, G. Skin pH: From basic science to basic skin care. Acta Derm. Venereol. 2013, 93, 261–267. [Google Scholar] [CrossRef] [PubMed]
- Prajapati, S.T.; Patel, C.G.; Patel, C.N. Formulation and Evaluation of Transdermal Patch of Repaglinide. ISRN Pharm. 2011, 2011, 651909. [Google Scholar] [CrossRef] [PubMed]
- Oshiro Junior, J.A.; Carvalho, F.C.; Soares, C.P.; Chorilli, M.; Chiavacci, L.A. Development of cutaneous bioadhesive ureasil-polyether hybrid films. Int. J. Polym. Sci. 2015, 2015, 1–7. [Google Scholar] [CrossRef]
- Cho, C.W.; Kim, D.B.; Shin, S.C. Development of bioadhesive transdermal bupivacaine gels for enhanced local anaesthetic action. Iran. J. Pharm. Res. 2012, 11, 423–431. [Google Scholar] [PubMed]
- Oh, C.M.; Heng, P.W.; Chan, L.W. Influence of hydroxypropyl methylcellulose on metronidazole crystallinity in spray-congealed polyethylene glycol microparticles and its impact with various additives on metronidazole release. AAPS Pharm. Sci. Tech. 2015, 16, 1357–1367. [Google Scholar] [CrossRef] [PubMed]
- Mohd, F.; Bontha, L.S.; Bontha, V.K.; Vemula, S.K. Formulation and evaluation of transdermal films of ondansetron hydrochloride. MOJ Bioequiv. Bioavailab. 2017, 3, 86–92. [Google Scholar] [CrossRef][Green Version]
- Papadimitriou, S.; Bikiaris, D. Dissolution rate enhancement of the poorly water-soluble drug tibolone using PVP, SiO2, and their nanocomposites as appropriate drug carriers. Drug Dev. Ind. Pharm. 2009, 35, 1128–1138. [Google Scholar] [CrossRef]
- Abdul Rasool, B.K.; Khalifa, A.; Abu-Gharbieh, E.; Khan, R. Employment of alginate floating in situ gel for controlled delivery of celecoxib: Solubilization and formulation studies. BioMed Res. Int. 2020, 2020, 1879125. [Google Scholar] [CrossRef]
- Gambhire, M.N.; Ambade, K.W.; Kurmi, S.D.; Kadam, V.J.; Jadhav, K.R. Development and in vitro evaluation of an oral floating matrix tablet formulation of diltiazem hydrochloride. AAPS Pharm. Sci. Tech. 2007, 8, E166–E174. [Google Scholar] [CrossRef] [PubMed]
- Papadopoulou, V.; Kosmidis, K.; Vlachou, M.; Macheras, P. On the use of the weibull function for the discernment of drug release mechanisms. Int. J. Pharm. 2006, 309, 44–50. [Google Scholar] [CrossRef]
- Costa, P.; Sousa Lobo, J.M. Modeling and comparison of dissolution profiles. Eur. J. Pharm. Sci. 2001, 13, 123–133. [Google Scholar] [CrossRef]
- Kosmidis, K.; Argyrakis, P.; Macheras, P. Fractal kinetics in drug release from finite fractal matrices. J. Chem. Phys. 2003, 119, 6373–6377. [Google Scholar] [CrossRef]
- Simancas-Herbad, R.; Fernández-Carballido, A.; Aparicio-Blanco, J.; Slowing, K.; Rubio-Retama, J.; López-Cabarcos, E.; Torres-Suárez, A.I. Controlled release of highly hydrophilic drugs from novel poly(magnesium acrylate) matrix tablets. Pharmaceutics 2020, 12, 174. [Google Scholar] [CrossRef]
- Tewari, D.; Zong, Y.; Harcum, W.W.; Hood, C.; Dürig, T. Impact of Molecular Weight and Molecular Weight Distribution of Hypromellose in Reducing Drug Release Variability from Erosion Dependent Matrix Systems. 2012. Available online: https://www.ashland.com/file_source/Ashland/links/PTR-082-CR_Benecel_HPMC-Impact_of_MW_and_MW_Distribution_of_HPMC_Custom_Grades-K250,K750,K1500.pdf (accessed on 27 May 2021).
- Siswanto, A.; Fudholi, A.; Nugroho, A.K.; Martono, S. In vitro release modeling of aspirin floating tablets using DDsolver. Indones. J. Pharm. 2015, 26, 94. [Google Scholar] [CrossRef]
- Blyden, G.T.; Greenblatt, D.J.; Scavone, J.M.; Shader, R.I. Pharmacokinetics of diphenhydramine and a demethylated metabolite following intravenous and oral administration. J. Clin. Pharmacol. 1986, 26, 529–533. [Google Scholar] [CrossRef] [PubMed]
- Williams, A.C.; Barry, B.W. Penetration enhancers. Adv. Drug Deliv. Rev. 2004, 56, 603–618. [Google Scholar] [CrossRef]
- Sarode, A.L.; Sandhu, H.; Shah, N.; Malick, W.; Zia, H. Hot melt extrusion (HME) for amorphous solid dispersions: Predictive tools for processing and impact of drug-polymer interactions on supersaturation. Eur. J. Pharm. Sci. 2013, 48, 371–384. [Google Scholar] [CrossRef]
- Zhang, J.; Yang, W.; Vo, A.Q.; Feng, X.; Ye, X.; Kim, D.W.; Repka, M.A. Hydroxypropyl methylcellulose-based controlled release dosage by melt extrusion and 3D printing: Structure and drug release correlation. Carbohydr. Polym. 2017, 177, 49–57. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Formula Code | Polymers (mg) | Copolymers (mg) | Polymer: Copolymer w/w Ratio | ||
|---|---|---|---|---|---|
| EC | ES100 | PVP | HPMC K100 | ||
| F0 (EC) | 100 | 0 | 1:0 | ||
| F1(EC-HPMC) | 70 | 30 | 7:3 | ||
| F2(EC-HPMC) | 60 | 40 | 3:2 | ||
| F3(EC-HPMC) | 50 | 50 | 1:1 | ||
| F4(EC-HPMC) | 40 | 60 | 2:3 | ||
| F5(EC-HPMC) | 30 | 70 | 3:7 | ||
| F1(EC-PVP) | 70 | 30 | 7:3 | ||
| F2(EC-PVP) | 60 | 40 | 3:2 | ||
| F3(EC-PVP) | 50 | 50 | 1:1 | ||
| F4(EC-PVP) | 40 | 60 | 2:3 | ||
| F5(EC-PVP) | 30 | 70 | 3:7 | ||
| F0 (ES) | 100 | 0 | 1:0 | ||
| F1(ES-HPMC) | 70 | 30 | 7:3 | ||
| F2(ES-HPMC) | 60 | 40 | 3:2 | ||
| F3(ES-HPMC) | 50 | 50 | 1:1 | ||
| F4(ES-HPMC) | 40 | 60 | 2:3 | ||
| F5(ES-HPMC) | 30 | 70 | 3:7 | ||
| F1(ES-PVP) | 70 | 30 | 7:3 | ||
| F2(ES-PVP) | 60 | 40 | 3:2 | ||
| F3(ES-PVP) | 50 | 50 | 1:1 | ||
| F4(ES-PVP) | 40 | 60 | 2:3 | ||
| F5(ES-PVP) | 30 | 70 | 3:7 | ||
| Formula Code | Penetration Enhancers (% w/w) | ||
|---|---|---|---|
| OA | PG | Euc | |
| F5(OA-3) | 3 | ||
| F5(OA-5) | 5 | ||
| F5(PG-5) | 5 | ||
| F5(PG-10) | 10 | ||
| F5(Euc-3) | 3 | ||
| F5(Euc-5) | 5 | ||
| Formulas Codes | Thickness (µm) | Folding Endurance | Surface pH | Drug Content % (SD) | Moisture Content (%) | Adhesion Time (s) |
|---|---|---|---|---|---|---|
| F0 (EC) | 0.09 ± 0.01 | 5 | 7.31 ± 0.05 | 96 (2.08) | 7.7 ± 2.5 | 5.7 ± 1.5 |
| F1 (EC-HPMC) | 0.10 ± 0.00 | 12 | 6.75 ± 0.05 | 96 (2.08) | 10.0 ± 1.0 | 21.3 ± 0.5 |
| F2 (EC-HPMC) | 0.13 ± 0.01 | 30 | 6.35 ± 0.05 | 97 (1.53) | 11.3 ± 1.5 | 29.3 ± 0.6 |
| F3 (EC-HPMC) | 0.11 ± 0.01 | 41 | 6.12 ± 0.21 | 98 (1.73) | 13.7 ± 1.5 | 34.0 ± 1.0 |
| F4 (EC-HPMC) | 0.13 ± 0.01 | 64 | 6.19 ± 0.03 | 99 (2.08) | 17.7 ± 1.5 | 41.0 ± 1.0 |
| F5 (EC-HPMC) | 0.11 ± 0.01 | >100 * | 6.71 ± 0.25 | 98 (1.01) | 22.0 ± 1.0 | 48.7 ± 1.5 * |
| F1 (EC-PVP) | 0.12 ± 0.01 | 10 | 6.71 ± 0.06 | 95 (1.02) | 21.7 ± 1.5 | 14.3 ± 2.1 |
| F2 (EC-PVP) | 0.11 ± 0.01 | 42 | 6.77 ± 0.07 | 96 (0.58) | 24.3 ± 1.5 | 19.7 ± 1.5 |
| F3 (EC-PVP) | 0.08 ± 0.01 | 47 | 6.55 ± 0.06 | 97 (0.58) | 26.7 ± 1.5 | 24.0 ± 1.0 |
| F4 (EC-PVP) | 0.09 ± 0.01 | 89 | 6.39 ± 0.03 | 98 (1.01) | 26.9 ± 0.4 | 31.0 ± 1.0 |
| F5 (EC-PVP) | 0.09 ± 0.01 | >100 * | 6.59 ± 0.05 | 95 (1.03) | 28.5 ± 0.62 * | 35.7 ± 2.1 |
| F0 (ES) | 0.07 ± 0.01 | 3 | 6.70 ± 0.02 | 95 (1.04) | 7.0 ± 1.01 | 6.0 ± 1.0 |
| F1 (ES-HPMC) | 0.09 ± 0.01 | 10 | 6.70 ± 0.10 | 99 (2.08) | 15.0 ± 1.8 | 15.0 ± 1.0 |
| F2 (ES-HPMC) | 0.11 ± 0.01 | 11 | 6.39 ± 0.03 | 98 (1.73) | 16.3 ± 0.6 | 24.0 ± 1.0 |
| F3 (ES-HPMC) | 0.09 ± 0.01 | 23 | 6.32 ± 0.02 | 97 (1.53) | 21.3 ± 1.5 | 30.7 ± 1.5 |
| F4 (ES-HPMC) | 0.11 ± 0.00 | 38 | 6.39 ± 0.05 | 95 (2.31) | 21.0 ± 1.7 | 38.3 ± 2.5 |
| F5 (ES-HPMC) | 0.09 ± 0.01 | 50 | 6.21 ± 0.01 | 96 (2.08) | 22.0 ± 1.0 | 42.0 ± 1.0 |
| Modeling | Corresponding Equation | Formulae | Parameters | R2 Adjusted | AIC | MSC |
| Zero-Order | f = k0 t f = amount of the drug release k0 = reaction rate coefficient t = time | F1 | k0 = 5.298 | 0.9452 | 49.9729 | 2.7036 |
| F2 | k0 = 5.939 | 0.9536 | 50.4320 | 2.8712 | ||
| F3 | k0 = 6.482 | 0.9560 | 51.0126 | 2.9237 | ||
| F4 | k0 = 7.435 | 0.9482 | 53.8607 | 2.7611 | ||
| F5 | k0 = 8.813 | 0.9180 | 61.2336 | 2.3015 | ||
| First-Order | f = 100 [1 − e −k1t] f = amount of the drug release k1 = rate constant t = time | F1 | k1 = 0.066 | 0.9118 | 54.7326 | 2.2277 |
| F2 | k1 = 0.077 | 0.9197 | 55.9275 | 2.3217 | ||
| F3 | k1 = 0.087 | 0.9305 | 55.5887 | 2.4661 | ||
| F4 | k1 = 0.107 | 0.9792 | 44.7324 | 3.6739 | ||
| F5 | k1 = 0.140 | 0.9808 | 46.7211 | 3.7527 | ||
| Higuchi | f = kH t0.5 f = amount of the drug release kH = dissolution constant t = time | F1 | kH = 13.282 | 0.7926 | 63.2796 | 1.3730 |
| F2 | kH = 14.908 | 0.8083 | 64.6256 | 1.4519 | ||
| F3 | kH = 16.338 | 0.8354 | 64.2098 | 1.6040 | ||
| F4 | kH = 18.966 | 0.9222 | 57.9372 | 2.3535 | ||
| F5 | kH = 22.604 | 0.9455 | 57.1595 | 2.7089 | ||
| Hixson–Crowell | Wo1/3 − Wt1/3 = KHC t Wo= amount of drug remaining at time 0 Wt = remaining amount of drug in the dosage form KHC = Hixson–Crowell constant | F1 | kHC = 0.021 | 0.9245 | 53.1731 | 2.3836 |
| F2 | kHC = 0.024 | 0.9335 | 54.0360 | 2.5108 | ||
| F3 | kHC = 0.026 | 0.9432 | 53.5611 | 2.6689 | ||
| F4 | kHC = 0.032 | 0.9798 | 44.4466 | 3.7025 | ||
| F5 | kHC = 0.040 | 0.9791 | 47.5570 | 3.6691 | ||
| Korsmeyer–Peppas | f = kKP tn kKP = constant depicting the experimental parameters based on geometry and dosage forms f = amount of the drug release n = release exponent | F1 | KKP = 5.154 n = 1.014 | 0.9384 | 51.9584 | 2.5051 |
| F2 | KKP = 6.143 n = 0.983 | 0.9480 | 52.4053 | 2.6739 | ||
| F3 | KKP = 7.673 n = 0.914 | 0.9546 | 52.1478 | 2.8102 | ||
| F4 | KKP = 12.040 n = 0.752 | 0.9923 | 35.6781 | 4.5794 | ||
| F5 | KKP = 15.887 n = 0.697 | 0.9953 | 33.5659 | 5.0682 | ||
| Hopfenberg | Mt/M∞ = 1 − [1 − (ko t)/(C0 a0)]n Mt = the amount of drug dissolved in time t M∞ = the amount of drug dissolved in time ∞ Ko = erosion rate constant | F1 | KHB = 0.072 n = 0.641 | 0.9466 | 50.5306 | 2.6479 |
| F2 | kHB = 0.071 n = 0.771 | 0.9511 | 51.7920 | 2.7352 | ||
| F3 | kHB = 0.064 n = 1.017 | 0.9505 | 53.0102 | 2.7240 | ||
| F4 | kHB = 0.020 n = 5.046 | 0.9780 | 46.1106 | 3.5361 | ||
| F5 | kHB = 0.017 n = 8.061 | 0.9792 | 48.3411 | 3.5907 | ||
| Baker–Lonsdale | kBL t = 3/2 [1 − (1 − f/100)2/3)] − f/100 kBL = release constant t = time f = amount of the drug release | F1 | kBL = 0.003 | 0.7610 | 64.6971 | 1.2312 |
| F2 | kBL = 0.004 | 0.7720 | 66.3583 | 1.2786 | ||
| F3 | kBL = 0.005 | 0.7954 | 66.3847 | 1.3865 | ||
| F4 | kBL = 0.008 | 0.8798 | 62.2832 | 1.9189 | ||
| F5 | kBL = 0.011 | 0.8953 | 63.6787 | 2.0570 | ||
| Makoid–Banakar | f = kMB tn e(−ct) f = amount of the drug release kMB = dissolution constant t = time n and c = empirical parameters | F1 | kMB = 8.569 n = 0.174 k = −0.151 | 0.9919 | 32.3633 | 4.4646 |
| F2 | kMB = 9.471 n = 0.263 k = −0.130 | 0.9862 | 39.7641 | 3.9380 | ||
| F3 | kMB = 10.800 n = 0.320 k = −0.109 | 0.9833 | 42.8041 | 3.7446 | ||
| F4 | kMB = 13.200 n = 0.560 k = −0.038 | 0.9967 | 27.9183 | 5.3553 | ||
| F5 | kMB = 16.533 n = 0.604 k = −0.019 | 0.9961 | 32.1616 | 5.2087 |
| Patch Code | Steady-State Flux Jss (μg·cm−2·h−1) | Permeability Coefficient × 10−3 (cm2/h) | Enhancement Ratio (ER) |
|---|---|---|---|
| F5EC-HPMC–oleic acid 3% | 136 ± 1.6 | 68 ± 1.1 | 1.81 |
| F5EC-HPMC–oleic acid 5% | 130 ± 1.2 | 65 ± 0.9 | 1.73 |
| F5EC-HPMC–PG 5% | 81 ± 3.8 | 41 ± 2.4 | 1.08 |
| F5EC-HPMC–PG10% | 97 ± 3.5 | 49 ± 3.1 | 1.29 |
| F5EC-HPMC–Euc3% | 112 ± 1.9 | 56 ± 2.7 | 1.49 |
| F5EC-HPMC–Euc5% | * 222 ± 2.7 | * 112 ± 1.5 | * 2.96 |
| F5EC-HPMC | 024 ± 2.7 | 55 ± 2.4 | 0.32 |
| Control | 75 ± 1.4 | 38 ± 1.3 | ..... |
| Compound | Chemical Functional Group | Peak Frequency (cm−1) | Chemical Structure |
|---|---|---|---|
| DMH | CH stretch -N (CH3)2 C=O stretching Aromatic rings skeletal vibrations | 3035 2400–2700 1650–1675 1592, 1407 |  |
| Ethylcellulose | –C–O–C– stretching C–H stretching C–H bending | 1052 2850, 2950 1375 |  |
| HPMC K100M | C–H stretching O–H stretching C–O stretching C–O–C | 2900 2898 1373 1053 |  |
| Animals | The Score Obtained at Various Periods | |||||||
|---|---|---|---|---|---|---|---|---|
| 1 h | 24 h | 48 h | 72 h | |||||
| E | O | E | O | E | O | E | O | |
| Control | ¾ | 2/0 | 4/3 | 2/3 | 4/3 | 3/2 | 0/2 | 2/0 |
| F5 (EC-HPMC) | 0/0 | 0/0 | 0/0 | 0/0 | 0/0 | 0/0 | 0/0 | 0/0 |
| F5 (Euc 5%) Treated-1 | 0/0 | 0/0 | 0/0 | 0/0 | 0/0 | 0/0 | 0/0 | 0/0 |
| F5 (Euc 5%) Treated-2 | 0/0 | 0/0 | 1/0 | 0/0 | 0/0 | 0/0 | 0/0 | 0/0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abdul Rasool, B.K.; Mohammed, A.A.; Salem, Y.Y. The Optimization of a Dimenhydrinate Transdermal Patch Formulation Based on the Quantitative Analysis of In Vitro Release Data by DDSolver through Skin Penetration Studies. Sci. Pharm. 2021, 89, 33. https://doi.org/10.3390/scipharm89030033
Abdul Rasool BK, Mohammed AA, Salem YY. The Optimization of a Dimenhydrinate Transdermal Patch Formulation Based on the Quantitative Analysis of In Vitro Release Data by DDSolver through Skin Penetration Studies. Scientia Pharmaceutica. 2021; 89(3):33. https://doi.org/10.3390/scipharm89030033
Chicago/Turabian StyleAbdul Rasool, Bazigha K., Amira A. Mohammed, and Yasmein Y. Salem. 2021. "The Optimization of a Dimenhydrinate Transdermal Patch Formulation Based on the Quantitative Analysis of In Vitro Release Data by DDSolver through Skin Penetration Studies" Scientia Pharmaceutica 89, no. 3: 33. https://doi.org/10.3390/scipharm89030033
APA StyleAbdul Rasool, B. K., Mohammed, A. A., & Salem, Y. Y. (2021). The Optimization of a Dimenhydrinate Transdermal Patch Formulation Based on the Quantitative Analysis of In Vitro Release Data by DDSolver through Skin Penetration Studies. Scientia Pharmaceutica, 89(3), 33. https://doi.org/10.3390/scipharm89030033