Novel Amino Acid Derivatives of Quinolines as Potential Antibacterial and Fluorophore Agents


 ,
,  ,
, 
Abstract
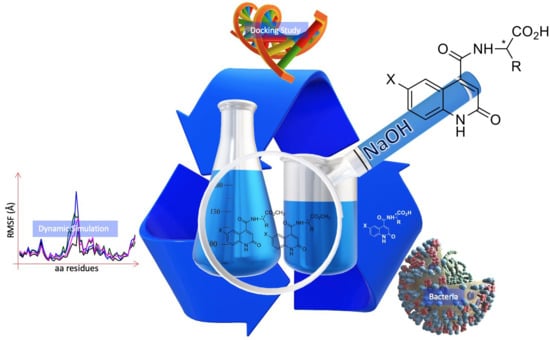
1. Introduction
2. Materials and Methods
2.1. Experimental
2.2. Computational Methods
3. Results and Discussion
3.1. Synthesis of Quinoline Amino Acids Derivatives
3.2. Characterisation of the Synthesized Compounds
3.3. Antibacterial Activity
3.4. Fluorometry
3.5. Docking and MD Simulation Studies
3.5.1. Fluroquinolone Binding Site at GyrA
3.5.2. Coumarin Binding Site at GyrB and ParE
3.5.3. NBTI Binding Site at GyrA Dimer Interface
3.5.4. Comparison of Key Interactions of Ligands at the Fluoroquinolone Binding Site
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Theuretzbacher, U.; Gottwalt, S.; Beyer, P.; Butler, M.; Czaplewski, L.; Lienhardt, C.; Moja, L.; Paul, M.; Paulin, S.; Rex, J.H.; et al. Analysis of the clinical antibacterial and antituberculosis pipeline. Lancet Infect. Dis. 2019, 19, 40–50. [Google Scholar] [CrossRef]
- Lesher, G.Y.; Froelich, E.J.; Gruett, M.D.; Bailey, J.H.; Brundage, R.P. 1,8-Naphthyridine Derivatives. A New Class of Chemotherapeutic Agents. J. Med. Chem. 1962, 5, 1063–1065. [Google Scholar] [CrossRef] [PubMed]
- Aldred, K.J.; Kerns, R.J.; Osheroff, N. Mechanism of quinolone action and resistance. Biochemistry 2014, 53, 1565–1574. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, A.; Sanchez, M.B.; Martínez, J.L. Quinolone resistance: Much more than predicted. Front. Microbiol. 2011, 2, 22. [Google Scholar] [CrossRef] [PubMed]
- Andersson, M.I. Development of the quinolones. J. Antimicrob. Chemother. 2003, 51, 1–11. [Google Scholar] [CrossRef]
- Emmerson, A.M. The quinolones: Decades of development and use. J. Antimicrob. Chemother. 2003, 51, 13–20. [Google Scholar] [CrossRef] [PubMed]
- Hooper, D.C. Mode of action of fluoroquinolones. Drugs 1999, 58, 6–10. [Google Scholar] [CrossRef]
- Davis, R.L.; Kelly, H.W.; Quenzer, R.W.; Standefer, J.; Steinberg, B.; Gallegos, J. Effect of norfloxacin on theophylline metabolism. Antimicrob. Agents Chemother. 1989, 33, 212–214. [Google Scholar] [CrossRef]
- Lange, R.P.; Locher, H.H.; Wyss, P.C.; Then, R.L. The targets of currently used antibacterial agents: Lessons for drug discovery. Curr. Pharm. Des. 2007, 13, 3140–3154. [Google Scholar] [CrossRef]
- Pham, T.D.; Ziora, Z.M.; Blaskovich, M.A. Quinolone antibiotics. Medchemcomm 2019, 10, 1719–1739. [Google Scholar] [CrossRef]
- Silver, L.L. Appropriate Targets for Antibacterial Drugs. Cold Spring Harb. Perspect. Med. 2016, 6, a030239. [Google Scholar] [CrossRef] [PubMed]
- Neuhauser, M.M.; Weinstein, R.A.; Rydman, R.; Danziger, L.H.; Karam, G.; Quinn, J.P. Antibiotic resistance among gram-negative bacilli in US intensive care units: Implications for fluoroquinolone use. JAMA 2003, 289, 885–888. [Google Scholar] [CrossRef] [PubMed]
- Hyatt, J.M.; McKinnon, P.S.; Zimmer, G.S.; Schentag, J.J. The importance of pharmacokinetic/ pharmacodynamic surrogate markers to outcome. Focus on antibacterial agents. Clin. Pharmacokinet. 1995, 28, 143–160. [Google Scholar] [CrossRef] [PubMed]
- Hooper, D.C.; Jacoby, G.A. Mechanisms of drug resistance: Quinolone resistance. Ann. N. Y. Acad. Sci. 2015, 1354, 12–31. [Google Scholar] [CrossRef]
- Correia, S.; Poeta, P.; Hébraud, M.; Capelo, J.L.; Igrejas, G. Mechanisms of quinolone action and resistance: Where do we stand? J. Med. Microbiol. 2017, 66, 551–559. [Google Scholar] [CrossRef]
- Katagi, M.; Bolakatti, G.; Badiger, A.; Satyanarayana, D.; Mamledesai, S.; Sujatha, M. Synthesis, Spectral Characterization and Antimicrobial Activity of Substituted Thiazolyl Derivatives of 2-Quinolones. Drug Res. 2013, 63, 53–59. [Google Scholar] [CrossRef]
- Khamkhenshorngphanuch, T.; Kulkraisri, K.; Janjamratsaeng, A.; Plabutong, N.; Thammahong, A.; Manadee, K.; Na Pombejra, S.; Khotavivattana, T. Synthesis and Antimicrobial Activity of Novel 4-Hydroxy-2-quinolone Analogs. Molecules 2020, 25, 3059. [Google Scholar] [CrossRef]
- Jayashree, B.S.; Thomas, S.; Nayak, Y. Design and synthesis of 2-quinolones as antioxidants and antimicrobials: A rational approach. Med. Chem. Res. 2010, 19, 193–209. [Google Scholar] [CrossRef]
- Dhiman, P.; Arora, N.; Thanikachalam, P.V.; Monga, V. Recent advances in the synthetic and medicinal perspective of quinolones: A review. Bioorg. Chem. 2019, 92, 103291. [Google Scholar] [CrossRef]
- Arya, K.; Agarwal, M. Microwave prompted multigram synthesis, structural determination, and photo-antiproliferative activity of fluorinated 4-hydroxyquinolinones. Bioorg. Med. Chem. Lett. 2007, 17, 86–93. [Google Scholar] [CrossRef]
- Valadbeigi, E.; Ghodsi, S. Synthesis and Study of Some New Quinolone Derivatives Containing a 3-acetyl Coumarin for Their Antibacterial and Antifungal Activities. IJPR 2017, 16, 554–564. [Google Scholar] [PubMed]
- Wenjie, X.; Xueyao, L.; Guixing, M.; Hongmin, Z.; Ya, C.; Johannes, K.; Jie, X.; Song, W. N-thiadiazole-4-hydroxy-2-quinolone-3-carboxamides bearing heteroaromatic rings as novel antibacterial agents: Design, synthesis, biological evaluation and target identification. Eur. J. Med. Chem. 2020, 188, 112022. [Google Scholar]
- Townsend, C.A.; Brown, A.M. Nocardicin A: Biosynthetic experiments with amino acid precursors. J. Am. Chem. Soc. 1983, 105, 913–918. [Google Scholar] [CrossRef]
- Moloney, M.G. Excitatory amino acids. Nat. Prod. Rep. 1999, 16, 485–498. [Google Scholar] [CrossRef] [PubMed]
- Hughes, A.B. Amino Acids, Peptides and Proteins in Organic Chemistry: Building Blocks, Catalysis and Coupling Chemistry; Wiley-VCH: Weinheim, Germany, 2011; pp. 83–114. [Google Scholar]
- Ohashi, Y.; Onuma, R.; Nagakuma, T.; Ogawa, T.; Naude, R.; Nokihara, K.; Muramoto, K. Antioxidant properties tripeptides revealed by a comparison of six different assays. Food Sci. Technol. Res. 2016, 21, 695–704. [Google Scholar] [CrossRef]
- Wei, Q.Y.; Jiang, H.; Zhang, J.X.; Zhang, C.; Guo, P.F. Antimicrobial activities of the cinnamoyl amide of amino acid derivatives. Asian J. Chem. 2012, 24, 2383–2388. [Google Scholar]
- Ghalehshahi, H.G.; Balalaie, S.; Aliahmadi, A.; Moghimi, R. Synthesis of 4-N-α-coumaryl amino acids and investigation of their antioxidant, antimicrobial activities and fluorescence spectra. Amino Acids 2018, 50, 1461–1470. [Google Scholar] [CrossRef]
- Shivaraj, Y.; Naveen, H.M.; Vijayakumar, R.G.; Kumar, D.B. Design, Synthesis and Antibacterial Activity Studies of Novel Quinoline Carboxamide Derivatives. J. Korean Chem. Soc. 2013, 57, 241–324. [Google Scholar] [CrossRef][Green Version]
- Khalid, M.; Ali, A.; Jawaria, R.; Asghar, M.A.; Asim, S.; Khan, M.U.; Hussain, R.; ur Rehman, M.F.; Ennise, C.J.; Akram, M.S. First Principles Study of Electronic and Nonlinear Optical Properties of A-D−π–A and D-A-D−π–A Configured Compounds Containing Novel Quinoline-Carbazole Derivatives. RSC Adv. 2020, 10, 22273–22283. [Google Scholar] [CrossRef]
- Basri, D.F.; Luoi, C.K.; Azmi, A.M.; Latip, J. Evaluation of the Combined Effects of Stilbenoid from Shorea gibbosa and Vancomycin against Methicillin-Resistant Staphylococcus aureus (MRSA). Pharmaceuticals 2012, 5, 1032–1043. [Google Scholar] [CrossRef]
- Ouedrhiri, W.; Bouhdid, S.; Balouiri, M.; Lalami, A.E.O.; Moja, S.; Chahdi, F.O.; Greche, H. Chemical composition of Citrus aurantium L. leaves and zest essential oils, their antioxidant, antibacterial single and combined effects. J. Chem. Pharm. 2015, 7, 78–84. [Google Scholar]
- Keiser, M.J.; Roth, B.L.; Armbruster, B.N.; Ernsberger, P.; Irwin, J.J.; Shoichet, B.K. Relating protein pharmacology by ligand chemistry. Nat. Biotech. 2007, 25, 197–206. [Google Scholar] [CrossRef] [PubMed]
- The UniProt Consortium. UniProt: A worldwide hub of protein knowledge. Nucleic Acids Research 2019, 47, 506–515. [Google Scholar] [CrossRef]
- Madeira, F.; Park, Y.M.; Lee, J.; Buso, N.; Gur, T.; Madhusoodanan, N.; Basutkar, P.; Tivey, A.R.N.; Potter, S.C.; Finn, R.D.; et al. The EMBL-EBI search and sequence analysis tools APIs in 2019. Nucleic Acids Res. 2019, 47, 636–641. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef]
- Hu, Y.; Shi, H.; Zhou, M.; Ren, Q.; Zhu, W.; Zhang, W.; Zhang, Z.; Zhou, C.; Liu, Y.; Ding, X.; et al. Discovery of Pyrido[2,3-b]indole Derivatives with Gram-Negative Activity Targeting Both DNA Gyrase and Topoisomerase IV. J. Med. Chem. 2020, 17, 9623–9649. [Google Scholar] [CrossRef]
- Edwards, M.J.; Flatman, R.H.; Mitchenall, L.A.; Stevenson, C.E.; Le, T.B.; Clarke, T.A.; McKay, A.R.; Fiedler, H.P.; Buttner, M.J.; Lawson, D.M.; et al. A Crystal Structure of the Bifunctional Antibiotic Simocyclinone D8, Bound to DNA Gyrase. Science 2009, 5958, 1415–1418. [Google Scholar] [CrossRef]
- Singh, S.B.; Kaelin, D.E.; Wu, J.; Miesel, L.; Tan, C.M.; Black, T.; Nargund, R.; Meinke, P.T.; Olsen, D.B.; Lagrutta, A.; et al. Tricyclic 1,5-Naphthyridinone Oxabicyclooctane-Linked Novel Bacterial Topoisomerase Inhibitors as Broad-Spectrum Antibacterial Agents-SAR of Left-Hand-Side Moiety (Part-2). Bioorg. Med. Chem. Lett. 2015, 25, 1831–1835. [Google Scholar] [CrossRef]
- Singh, S.B.; Kaelin, D.E.; Wu, J.; Miesel, L.; Tan, C.M.; Meinke, P.T.; Olsen, D.; Lagrutta, D.; Bradley, P.; Lu, J.; et al. Oxabicyclooctane-Linked Novel Bacterial Topoisomerase Inhibitors as Broad Spectrum Antibacterial Agents. ACS Med. Chem. Lett. 2014, 5, 609–614. [Google Scholar] [CrossRef]
- Gibson, G.; Bax, B.; Chan, P.F.; Osheroff, N. Mechanistic and Structural Basis for the Actions of the Antibacterial Gepotidacin against Staphylococcus aureus Gyrase Elizabeth. ACS Infect. Dis. 2019, 4, 570–581. [Google Scholar] [CrossRef]
- Vanden Broeck, A.; Lotz, C.; Ortiz, J.; Lamour, V. Cryo-EM structure of the complete E. coli DNA gyrase nucleoprotein complex. Nat. Commun. 2019, 10, 4935. [Google Scholar] [CrossRef] [PubMed]
- Stanger, F.V.; Dehio, C.; Schirmer, T. Structure of the N-Terminal Gyrase B Fragment in Complex with ADP Pi Reveals Rigid-Body Motion Induced by ATP Hydrolysis. PLoS ONE 2014, 9, e107289. [Google Scholar] [CrossRef] [PubMed]
- Holdgate, G.A.; Tunnicliffe, A.; Ward, W.H.; Weston, S.A.; Rosenbrock, G.; Barth, P.T.; Taylor, I.W.; Pauptit, R.A.; Timms, D. The Entropic Penalty of Ordered Water Accounts for Weaker Binding of the Antibiotic Novobiocin to a Resistant Mutant of DNA Gyrase: A Thermodynamic and Crystallographic Study Geoffrey. Biochemistry 1997, 32, 9663–9673. [Google Scholar] [CrossRef] [PubMed]
- Vanden Broeck, A.; McEwen, A.G.; Chebaro, Y.; Potier, N.; Lamour, V. Structural Basis for DNA Gyrase Interaction with Coumermycin A1. J. Med. Chem. 2019, 62, 4225–4231. [Google Scholar] [CrossRef] [PubMed]
- Mesleh, M.F.; Cross, J.B.; Zhang, J.; Kahmann, J.; Andersen, O.A.; Barker, J.; Cheng, R.K.; Felicetti, B.; Wood, M.; Hadfield, A.T.; et al. Fragment-based discovery of DNA gyrase inhibitors targeting the ATPase subunit of GyrB. Bioorg. Med. Chem. Lett. 2016, 26, 1314–1318. [Google Scholar] [CrossRef] [PubMed]
- Sherer, B.A.; Hull, K.; Green, O.; Basarab, G.; Hauck, S.; Hill, P.; Loch, J.T.; Mullen, G.; Bist, S.; Bryant, J.; et al. Pyrrolamide DNA gyrase inhibitors: Optimization of antibacterial activity and efficacy. Bioorg. Med. Chem. Lett. 2011, 21, 7416–7420. [Google Scholar] [CrossRef]
- Brvar, M.; Perdih, A.; Renko, M.; Anderluh, G.; Turk, D.; Solmajer, T. Structure-Based Discovery of Substituted 4,5′-Bithiazoles as Novel DNA Gyrase Inhibitors. J. Med. Chem. 2012, 55, 6413–6426. [Google Scholar] [CrossRef]
- Bellon, S.; Parsons, J.D.; Wei, Y.; Hayakawa, K.; Swenson, L.L.; Charifson, P.S.; Lippke, J.A.; Aldape, R.; Gross, C.H. Crystal Structures of Escherichia Coli Topoisomerase IV ParE Subunit (24 and 43 Kilodaltons): A Single Residue Dictates Differences in Novobiocin Potency against Topoisomerase IV and DNA Gyrase. Antimicrob. Agents Chemother. 2004, 5, 1856–1864. [Google Scholar] [CrossRef]
- Roos, K.; Wu, C.; Damm, W.; Reboul, M.; Stevenson, J.M.; Lu, C.; Dahlgren, M.K.; Mondal, S.; Chen, W.; Wang, L.; et al. OPLS3e: Extending Force Field Coverage for Drug-Like Small Molecules. J. Chem. Theory Comput. 2019, 3, 1863–1874. [Google Scholar] [CrossRef]
- Greenwood, J.R.; Calkins, D.; Sullivan, A.P.; Shelley, J.C. Towards the comprehensive, rapid, and accurate prediction of the favorable tautomeric states of drug-like molecules in aqueous solution. J. Comput. Aided Mol. Des. 2010, 24, 591–604. [Google Scholar] [CrossRef]
- Shelley, J.C.; Cholleti, A.; Frye, L.; Greenwood, J.R.; Timlin, M.R.; Uchimaya, M. Epik: A software program for pKa prediction and protonation state generation for drug-like molecules. J. Comput. Aided Mol. Des. 2007, 21, 681–691. [Google Scholar] [CrossRef] [PubMed]
- Sastry, G.M.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein and ligand preparation: Parameters, protocols, and influence on virtual screening enrichments. J. Comput. Aided Mol. Des. 2013, 3, 221–234. [Google Scholar] [CrossRef] [PubMed]
- Friesner, R.A.; Murphy, R.B.; Repasky, M.P.; Frye, L.L.; Greenwood, J.R.; Halgren, T.A.; Sanschagrin, P.C.; Mainz, D.T. Extra Precision Glide: Docking and Scoring Incorporating a Model of Hydrophobic Enclosure for Protein-Ligand Complexes. J. Med. Chem. 2006, 49, 6177–6196. [Google Scholar] [CrossRef] [PubMed]
- Halgren, T.A.; Murphy, R.B.; Friesner, R.A.; Beard, H.S.; Frye, L.L.; Pollard, W.T.; Banks, J.L. Glide: A New Approach for Rapid, Accurate Docking and Scoring. 2. Enrichment Factors in Database Screening. J. Med. Chem. 2004, 47, 1750–1759. [Google Scholar] [CrossRef]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shaw, D.E.; Shelley, M.; et al. Glide: A New Approach for Rapid, Accurate Docking and Scoring. 1. Method and Assessment of Docking Accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef]
- Jacobson, M.P.; Pincus, D.L.; Rapp, C.S.; Day, T.J.F.; Honig, B.; Shaw, D.E.; Friesner, R.A. A Hierarchical Approach to All-Atom Protein Loop Prediction. Proteins Struct. Funct. Bioinf. 2004, 55, 351–367. [Google Scholar] [CrossRef]
- Jacobson, M.P.; Friesner, R.A.; Xiang, Z.; Honig, B. On the Role of Crystal Packing Forces in Determining Protein Sidechain Conformations. J. Mol. Biol. 2002, 320, 597–608. [Google Scholar] [CrossRef]
- Li, J.; Abel, R.; Zhu, K.; Cao, Y.; Zhao, S.; Friesner, R.A. The VSGB 2.0 model: A next generation energy model for high resolution protein structure modeling. Proteins 2011, 10, 2794–2812. [Google Scholar] [CrossRef]
- Bowers, K.J.; Chow, D.E.; Xu, H.; Dror, R.O.; Eastwood, M.P.; Gregersen, B.A.; Klepeis, J.L.; Kolossvary, I.; Moraes, M.A.; Sacerdoti, F.D.; et al. Scalable Algorithms for Molecular Dynamics Simulations on Commodity Clusters. In Proceedings of the 2006 ACM/IEEE Conference on Supercomputing, Tampa, FL, USA, 11–17 November 2006; Volume 84, pp. 11–17. [Google Scholar]
- Berendsen, H.J.C.; Postma, J.P.M.; van Gunsteren, W.F.; Hermans, J. Interaction Models for Water in Relation to Protein Hydration. In Intermolecular Forces: The Jerusalem Symposia on Quantum Chemistry and Biochemistry; Pullman, B., Ed.; Springer: Dordrecht, Holland, 1981; Volume 14, pp. 331–342. [Google Scholar]
- Nosé, S. A unified formulation of the constant temperature molecular dynamics methods. J. Chem. Phys. 1984, 81, 511–519. [Google Scholar] [CrossRef]
- Nosé, S. A molecular dynamics method for simulations in the canonical ensemble. Mol. Phys. 1984, 52, 255–268. [Google Scholar] [CrossRef]
- Hoover, W.G. Canonical dynamics: Equilibrium phase-space distributions. Aps Phys. 1985, 31, 1695–1697. [Google Scholar] [CrossRef] [PubMed]
- Martyna, G.J.; Tobias, D.J.; Klein, M.L. Constant pressure molecular dynamics algorithms. J. Chem. Phys. 1994, 101, 4177. [Google Scholar] [CrossRef]
- Moussaoui, O.; Touimi, G.B.; Bennani, B.; Tama, A.B.; Chakroune, S.; Boukir, A. Synthesis of a new serie of quinoline-carboxamides based on methylated aminoesters: NMR characterization and antimicrobial activity. Mediterr. J. Chem. 2019, 9, 326–336. [Google Scholar] [CrossRef]
- Jacob, F.; Bernner, S.; Cuzin, F. On the Regulation of DNA Replication in Bacteria. Cold Spring Harb. Symp. Quant. Biol. 1963, 28, 329–333. [Google Scholar] [CrossRef]
- Hawkey, P.M. Mechanisms of quinolone action and microbial response. J. Antimicrob. Chemother. 2003, 51, 29–35. [Google Scholar] [CrossRef]
- Horowitz, D.S.; Wang, J.C. Mapping the Active Site Tyrosine of Escherichia Coli DNA Gyrase. J. Biol. Chem. 1987, 262, 5339–5344. [Google Scholar]
- Gellert, M.; Mizuuchi, K.; O’Dea, M.H.; Nash, H.A. DNA Gyrase: An Enzyme That Introduces Superhelical Turns into DNA. Proc. Natl. Acad. Sci. USA 1976, 11, 3872–3876. [Google Scholar] [CrossRef]
- Nakada, N.; Gmunder, H.; Hirata, T.; Arisawa, M. Mechanism of Inhibition of DNA Gyrase by Cyclothialidine, a Novel DNA Gyrase Inhibitor. Antimicrob. Agents Chemother. 1994, 9, 1966–1973. [Google Scholar] [CrossRef][Green Version]
- Bax, B.D.; Chan, P.F.; Eggleston, D.S.; Fosberry, A.; Gentry, D.R.; Gorrec, F.; Giordano, I.; Hann, M.M.; Hennessy, A.; Hibbs, M.; et al. Type IIA Topoisomerase Inhibition by a New Class of Antibacterial Agents. Nature 2010, 7309, 935–940. [Google Scholar] [CrossRef]
- Charrier, C.; Salisbury, A.M.; Savage, V.J.; Duffy, T.; Moyo, E.; Chaffer-Malam, N.; Ooi, N.; Newman, R.; Cheung, J.; Metzger, R.; et al. Novel bacterial topoisomerase inhibitors with potent broad-spectrum activity against drug-resistant bacteria. Antimicrob. Agents Chemother. 2017, 61, e02100–e02116. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | NMR 1H (ppm) | NMR 13C (ppm) |
|---|---|---|
| 3a | 3–4 Unstable with signal of H2O | 174.2 |
| 3b | 3–4 Unstable with signal of H2O | 173.1 |
| 3c | 13.04 | 172.0 |
| 3d | 3–4 Unstable with signal of H2O | 173.2 |
| 3e | 13.00 | 173.4 |
| 4a | 3–4 Unstable with signal of H2O | 173.0 |
| 4b | 3–4 Unstable with signal of H2O | 173.0 |
| 4c | 13.03 | 172.0 |
| 4d | 13.03 | 173.0 |
| 4e | 12.98 | 173.4 |
| Compound | Staphylococcus aureus ATCC 29213 | Pseudomonas aeruginosa ATCC 27853 | Bacillus subtilis ATCC 3366 | Escherichia coli ATCC 25922 |
|---|---|---|---|---|
| 1a | 10.0 | 10.0 | 2.5 | NA |
| 1b | NA | NA | 5.0 | 10 |
| 1c | NA | 10.0 | NA | NA |
| 1d | NA | 10.0 | NA | NA |
| 1e | NA | 10.0 | 10.0 | NA |
| 2a | NA | NA | NA | NA |
| 2b | NA | NA | NA | NA |
| 2c | NA | NA | NA | NA |
| 2d | 10.0 | 2.5 | 2.5 | NA |
| 2e | NA | NA | NA | NA |
| 3a | 0.62 | 0.62 | 0.62 | 0.62 |
| 3b | 2.5 | 2.5 | 2.5 | 1.25 |
| 3c | 2.5 | 2.5 | 2.5 | 1.25 |
| 3d | 5.0 | 2.5 | 1.25 | 1.25 |
| 3e | 0.31 | 2.5 | 2.5 | 2.5 |
| 4a | 5.0 | 1.25 | 2.5 | 1.25 |
| 4b | 1.25 | 2.5 | 1.25 | 2.5 |
| 4c | 5 | 1.25 | 2.5 | 1.25 |
| 4d | 5 | 1.25 | 2.5 | 1.25 |
| 4e | 0.62 | 2.5 | 2.5 | 1.25 |
| Compound | UV(λmax) | ε (M−1cm−1) | λexc | λem | Stokes Shift (nm) | ΦI |
|---|---|---|---|---|---|---|
| 3a | 390 | 9440 | 376 | 439 | 63 | 0.151 |
| 3c | 371 | 8530 | 374 | 431 | 57 | 0.179 |
| 3d | 392 | 9980 | 371 | 412 | 41 | 0.071 |
| 3e | 372 | 9900 | 379 | 422 | 43 | 0.100 |
| 4d | 382 | 10,100 | 377 | 425 | 48 | 0.180 |
| 4e | 394 | 10,130 | 376 | 429 | 53 | 0.037 |
| Ligands |
Fluoroquinolone Binding site at GyrA Subunit of S. aureus DNA Gyrase (PDB ID: 2XCT) |
Coumarin Binding Site at GyrB Subunit of E. coli DNA Gyrase (PDB ID: 4DUH) |
Coumarin Binding Site at ParE Subunit of E. coli Topoisomerase IV (PDB ID: 1S14) |
NBTI Binding Site at GyrA Subunit of S. aureus DNA gyrase (PDB ID: 5BS3) | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Glide SP Docking Score | Glide XP Docking Score | MMGBSA ΔGbind (kcal/mol) | Glide SP Docking Score | Glide XP Docking Score | MMGBSA ΔGbind (kcal/mol) | Glide SP Docking Score | Glide XP Docking Score | MMGBSA ΔGbind (kcal/mol) | Glide SP Docking Score | Glide XP Docking Score | MMGBSA ΔGbind (kcal/mol) | |
| Ciprofloxacin | −8.257 a | −8.096 a | −54.99/ −44.29 b | - | - | - | - | - | - | |||
| Inhibitor 18 (4,5′-bithiazole | - | - | −10.075 a | −8.834 a | −76.59/ −68.27 b | - | - | - | - | |||
| Novobiocin | - | - | - | - | −4.801 a | −4.857 a | −67.07/ −55.58 b | - | - | |||
| Compound 7 (NBTI) | - | - | - | - | - | - | −8.974 a | −7.902 a | −71.40/ −58.00 b | |||
| 3a | −8.743 | −7.209 | −18.53/ −19.36 b | −7.728 | −6.521 | −25.70 | −6.111 | −3.806 | −33.35 | −5.046 | −4.710 | −12.30 |
| 3b | −9.781 | −9.969 | 3.86/ −22.56 b | −8.111 | −6.771 | −17.81 | −5.529 | −4.587 | −35.53 | −5.494 | −6.636 | −15.69 |
| 3c | −9.802 | −8.117 | 2.10/ −24.63 b | −7.375 | −6.847 | −38.99 | −6.122 | −4.748 | −34.59 | −4.262 | −6.291 | −1131.43 c/ −18.55 b |
| 3d | −8.631 | −9.060 | −21.00/ −24.91 b | −7.440 | −7.368 | −40.05 | −6.028 | −4.394 | −34.63 | −5.011 | −5.986 | −1384.35 c/ −24.25 b |
| 3e | −10.203 | −10.826 | −18.99/ −27.88 b | −7.116 | −7.669 | −38.03/ −48.84 b | −6.481 | −6.765 | −34.60/ −33.52 b | −6.592 | −8.059 | −3.46 |
| 4a | −8.494 | −8.824 | −14.34/ −43.03 b | −7.464 | −7.125 | −24.59 | −5.026 | −3.806 | −30.38 | −5.595 | −5.645 | −11.44 |
| 4b | −8.915 | −9.346 | −4.66/ −20.28 b | −7.439 | −7.279 | −18.40 | −5.472 | −4.360 | −32.37 | −6.54 | −7.085 | −15.27 |
| 4c | −8.812 | −8.718 | −2.69/ −20.97 b | −3.053 | −6.364 | −32.44 | −6.015 | −3.743 | −33.53 | −5.823 | −6.663 | −12.12 |
| 4d | −7.311 | −9.068 | −20.88/ −35.56 b | −2.552 | −6.415 | −21.79 | −5.228 | −3.733 | −31.26 | −5.114 | −5.672 | −12.26 |
| 4e | −9.109 | −11.419 | −53.79/ −41.26 b | −4.405 | −7.064 | −37.62 | −5.688 | −4.487 | −30.26 | −6.684 | −8.479 | −18.14 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moussaoui, O.; Bhadane, R.; Sghyar, R.; El Hadrami, E.M.; El Amrani, S.; Ben Tama, A.; Kandri Rodi, Y.; Chakroune, S.; Salo-Ahen, O.M.H. Novel Amino Acid Derivatives of Quinolines as Potential Antibacterial and Fluorophore Agents. Sci. Pharm. 2020, 88, 57. https://doi.org/10.3390/scipharm88040057
Moussaoui O, Bhadane R, Sghyar R, El Hadrami EM, El Amrani S, Ben Tama A, Kandri Rodi Y, Chakroune S, Salo-Ahen OMH. Novel Amino Acid Derivatives of Quinolines as Potential Antibacterial and Fluorophore Agents. Scientia Pharmaceutica. 2020; 88(4):57. https://doi.org/10.3390/scipharm88040057
Chicago/Turabian StyleMoussaoui, Oussama, Rajendra Bhadane, Riham Sghyar, El Mestafa El Hadrami, Soukaina El Amrani, Abdeslem Ben Tama, Youssef Kandri Rodi, Said Chakroune, and Outi M. H. Salo-Ahen. 2020. "Novel Amino Acid Derivatives of Quinolines as Potential Antibacterial and Fluorophore Agents" Scientia Pharmaceutica 88, no. 4: 57. https://doi.org/10.3390/scipharm88040057
APA StyleMoussaoui, O., Bhadane, R., Sghyar, R., El Hadrami, E. M., El Amrani, S., Ben Tama, A., Kandri Rodi, Y., Chakroune, S., & Salo-Ahen, O. M. H. (2020). Novel Amino Acid Derivatives of Quinolines as Potential Antibacterial and Fluorophore Agents. Scientia Pharmaceutica, 88(4), 57. https://doi.org/10.3390/scipharm88040057