1. Introduction
Clarithromycin (CLA) is a semi-synthetic macrolide derivative of erythromycin A comprised of 14-cladisone and desosamine residues attached at positions 3 and 5 (
Figure 1) [
1,
2]. CLA is a biopharmaceutical classification system (BCS) class II molecule that is poorly soluble and highly permeable. CLA exhibits an aqueous solubility of approximately 0.342 µg/mL at 25 °C, and has a pKa of 8.8 [
3]. CLA is acid-stable and has a broad spectrum of antimicrobial activity, inhibiting a range of Gram-positive and Gram-negative organisms, atypical pathogens and some anaerobic organisms [
4,
5]. CLA lacks a chromophore as it has no conjugated double bond in the lactone ring therefore, significant UV absorbance is only observed at wavelengths < 210 nm [
6]. Detection at these wavelengths is suitable for most in vitro samples but lack the necessary sensitivity for the quantitation of low concentrations of CLA, such as those observed in biological matrices [
2,
7] and nanoparticles [
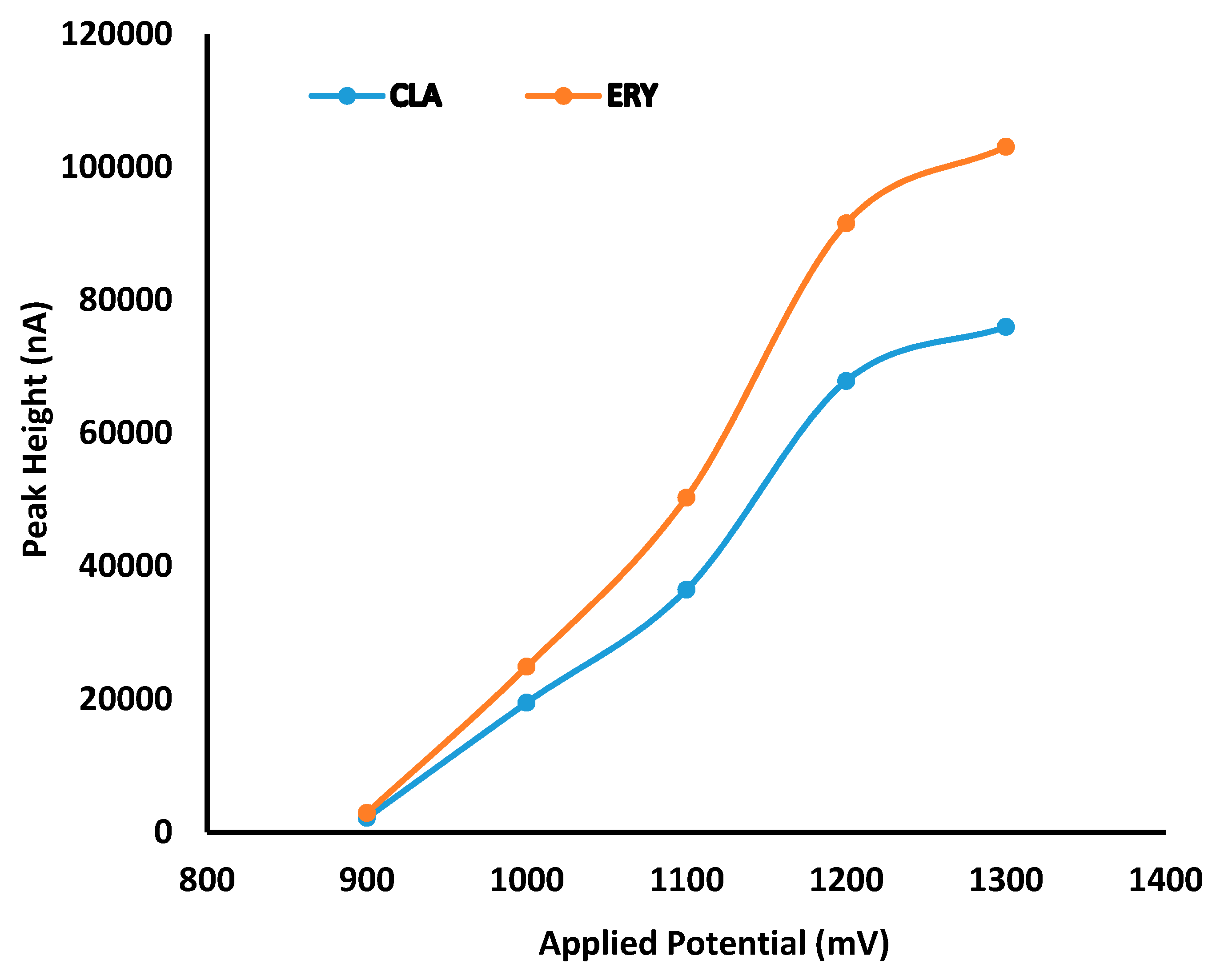
8]. CLA has a tertiary amino group which is reactive for electrochemical oxidation [
9], making electrochemical detection (ECD) a potentially useful tool for ensuring the accurate determination of CLA in dosage forms. Quantitative analysis of CLA has been achieved in biological samples using high-performance liquid chromatography (HPLC) with ECD [
10,
11,
12], UV [
2,
13], LCMS [
14] and fluorescence detection [
15]. Published HPLC-ECD methods have reported detection limits of 10.03 µg/mL [
16], 0.5 µg/mL [
17], 0.03 µg/mL [
10], 0.1 µg/mL [
18], 0.01 µg/mL [
19], 0.15 µg/mL [
20] and 0.02 µg/mL [
21]. In addition, characterization of degradation products of CLA have been reported using HPLC-MS [
22,
23] and HPLC-UV [
5,
13].
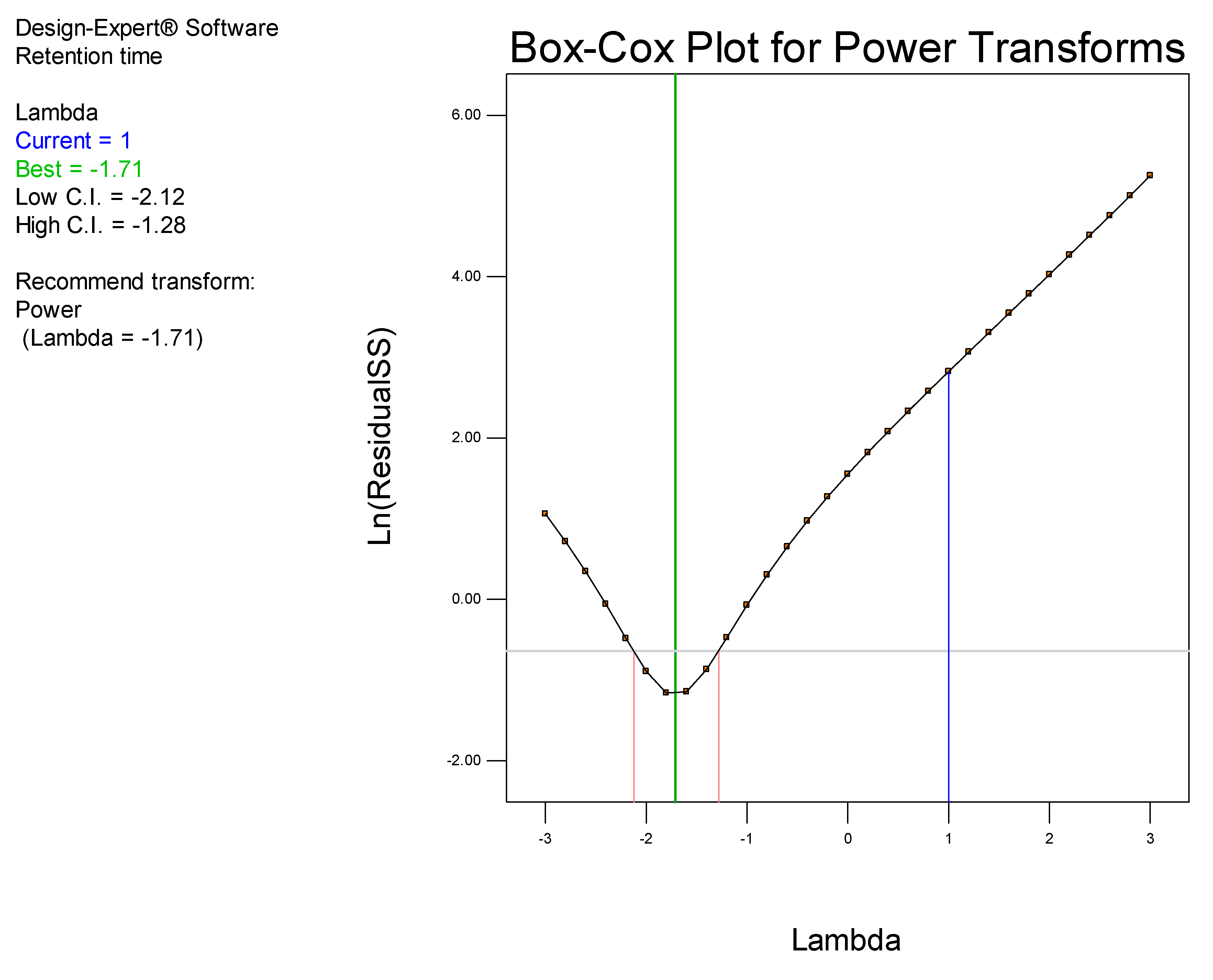
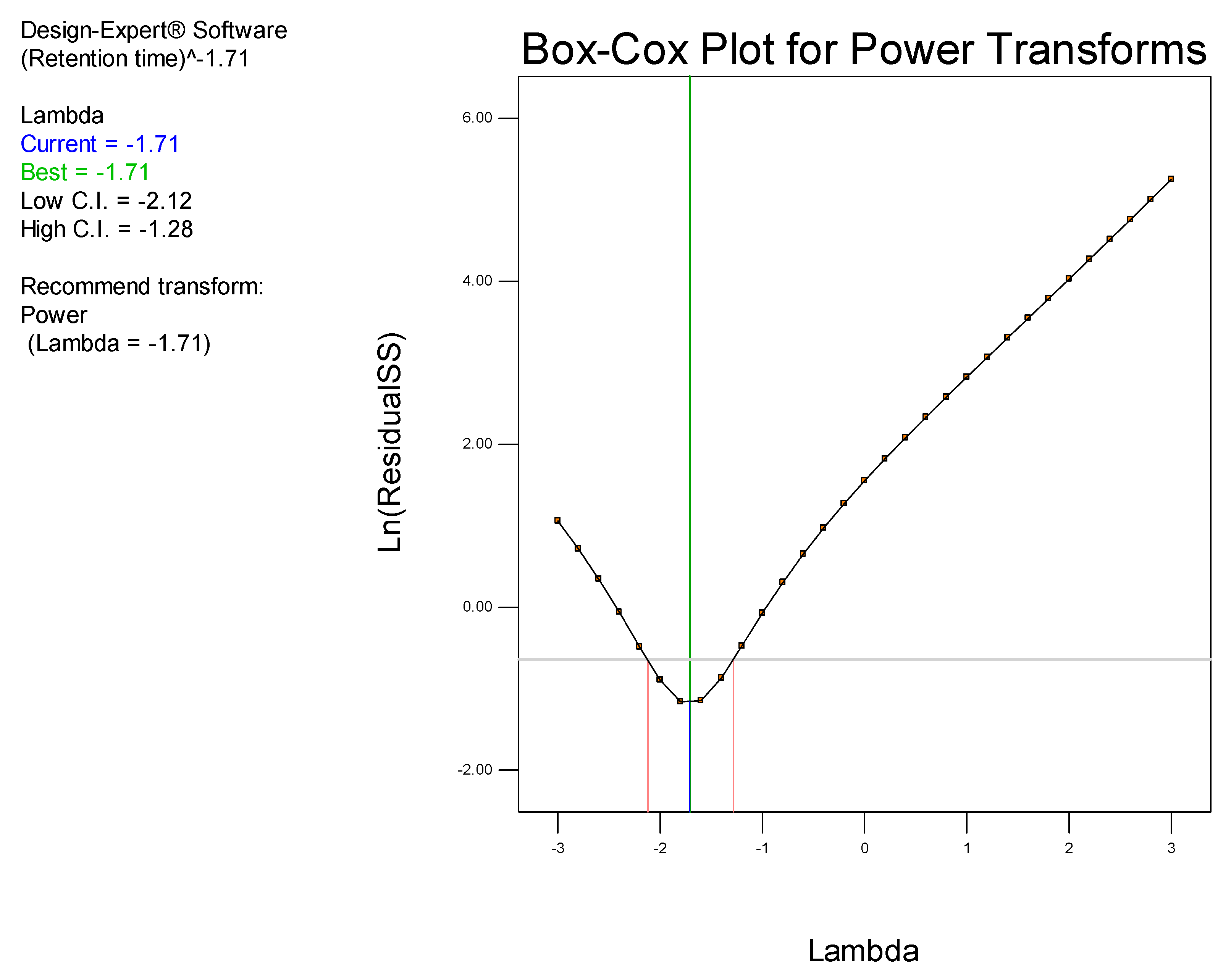
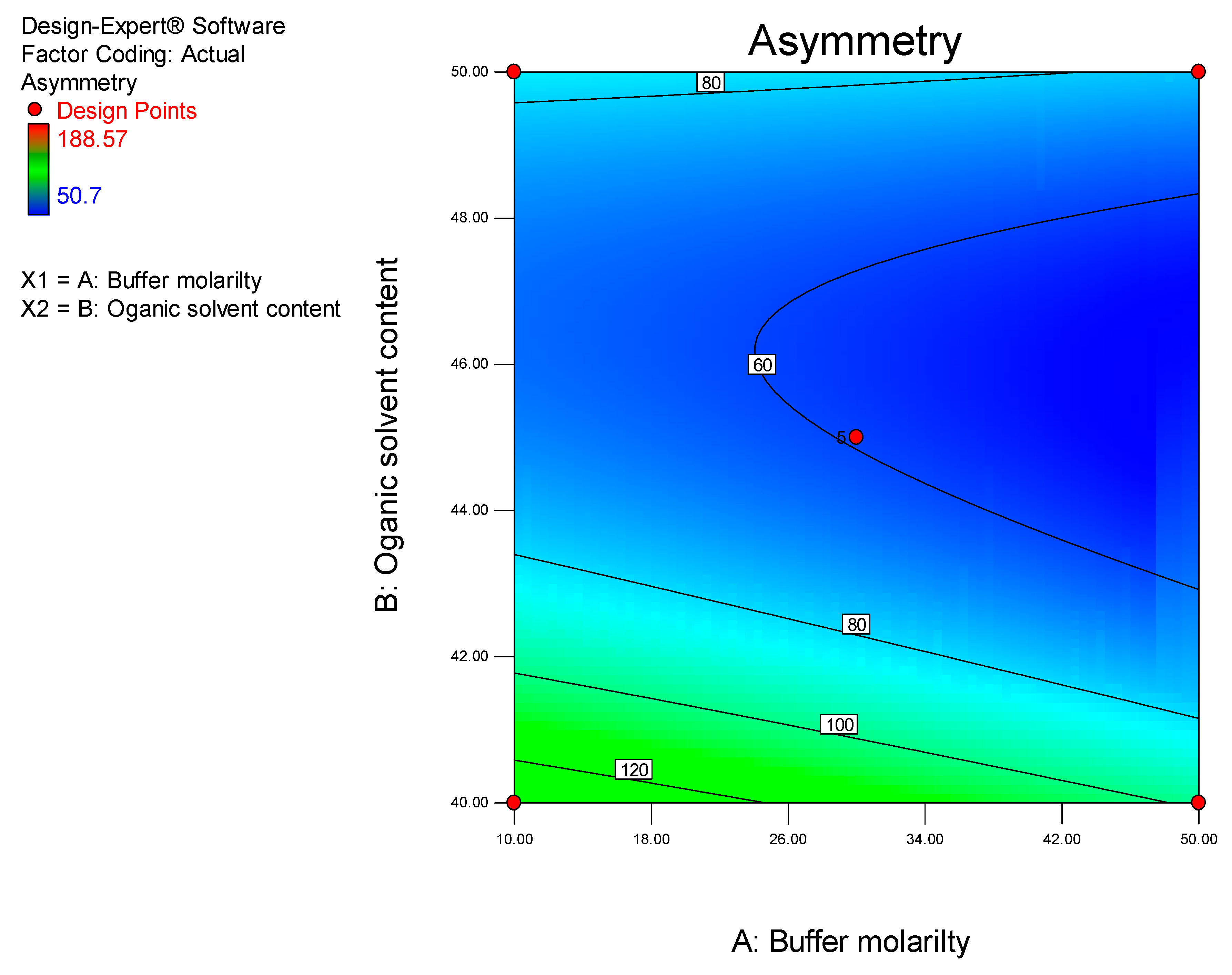
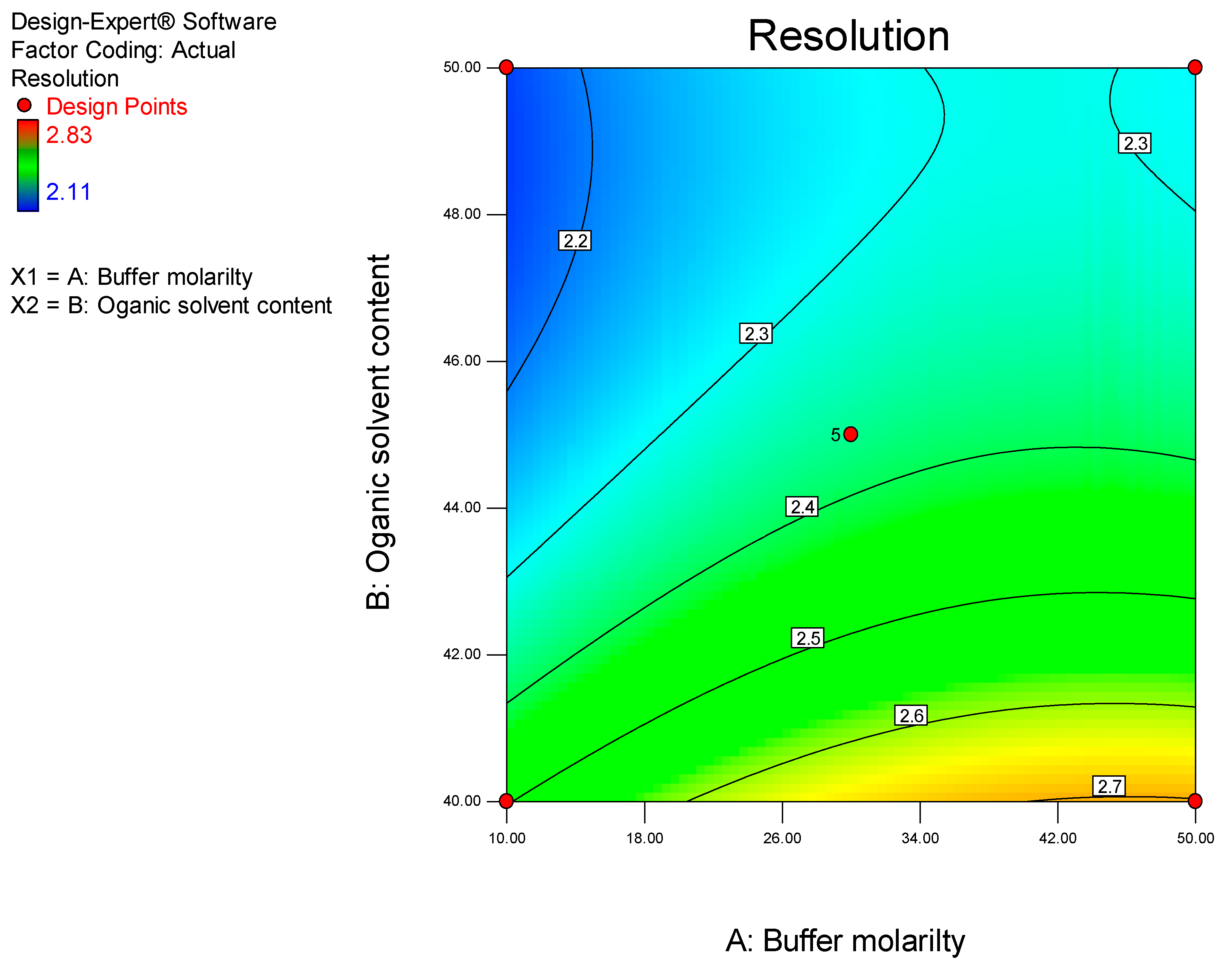
It has been established that step-by-step optimization procedures for any reliable HPLC method requires good peak resolution with acceptable retention time(s), the absence of ‘ghost’ peaks, stable sensitivity with minimal peak tailing [
25]. The initial aim of these studies was to screen factors that would hinder achievement of the appropriate chromatographic conditions and performance, with the ultimate goal of developing a rapid, simple and sensitive analytical method using a minimum number of experimental runs.
Prior to using Design of Experiments (DoE), a conventional approach was used to establish the optimum operating conditions so as to eliminate factors that resulted in a long retention time, baseline drift and background noise. DoE was ultimately used to identify the optimum analytical conditions for this separation.
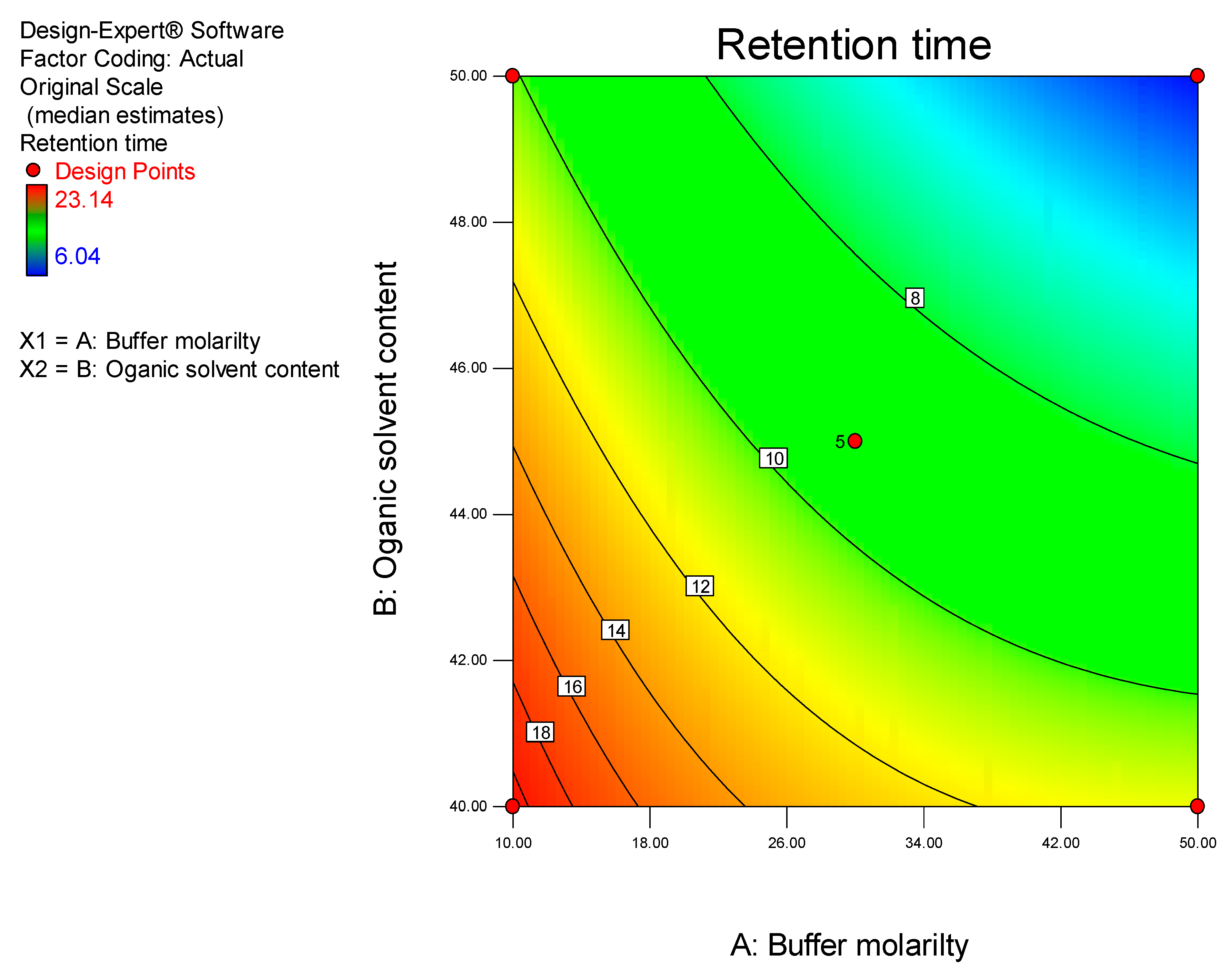
DoE is a chemometric approach that allows for a reduction in the number of experiments conducted associated with lower reagent consumption and less laboratory work [
26]. The use of statistical experimental design modelling methods and response optimization based on derived models reduces the time and effort required for the development of complex multi-variable analyses. Furthermore, optimization of HPLC methods are complex processes since, several variables including mobile phase pH and composition, buffer concentration, flow rate, injection volume, column temperature and detector settings must be controlled to achieve an appropriate separation [
27]. The approach facilitates the development of suitable polynomial regression models and the addition of valuable scientific information in support of an ability to assess the statistical significance of the influence of input variables on target chromatographic responses [
26]. In these studies, a Central Composite Design (CCD) was used to identify the optimum conditions for the HPLC-ECD method.
The development of an HPLC-UV analytical method for the quantification of CLA in pharmaceutical dosage forms using DoE has been reported [
28]. The CCD is a three-level full factorial design covering a symmetrical domain with a center point used to estimate experimental error [
29]. A CCD may include a two-level full factorial design (2
f experiments), a star design (2
f experiments) and a center point, requiring
N = 2
f + 2
f + 1 experiments to examine
f factors [
30,
31]. The points of the full factorial design are located at factor levels −1 and +1 and for the star design at factor levels 0, −
α and +
α, whereas for the center point at a factor level 0 [
32].
Method validation is a process that demonstrates that a method will successfully meet or exceed the minimum standards recommended by regulatory authorities for accuracy, precision, selectivity, sensitivity, reproducibility and stability [
33]. According to the USP [
34], certain parameters of a method may be adjusted or scaled on the condition that allowable limits and equivalency are maintained.
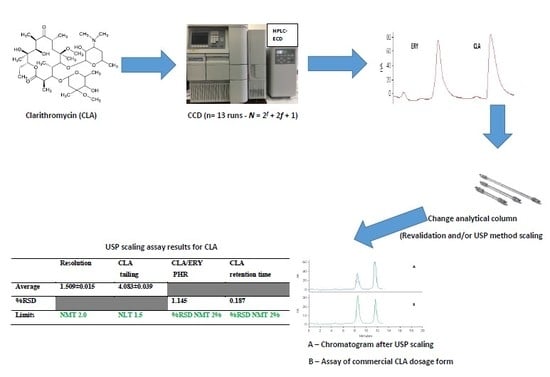
The objective of these studies was to develop a stability indicating the HPLC-ECD method for the quantitative determination of CLA in pharmaceutical formulations and monitoring CLA incorporation into lipid nano-carrier technologies. The method was developed and validated according to International Conference of Harmonization (ICH) guidelines [
35]. Method re-validation in addition to modified method scaling as per United States Pharmacopeia (USP) [
34] guidelines was also investigated in order to assess and assure the performance of the method when moving across analytical columns of different packing material (L-designation). To our knowledge, this is the first time an experimental design has been applied to the development of a simple, rapid, sensitive and reliable HPLC method using amperometric detection for the quantification of CLA in dosage forms. Furthermore, this is the first study to develop a stability indicating the HPLC-ECD method for CLA whilst investigating a modified USP scaling approach through use of a column of different L-designation.
2. Materials and Methods
2.1. Chemicals and Reagents
All reagents were at least of analytical reagent grade and used without further purification. CLA was purchased from Skyrun Industrial Co. Limited (Taizhou, China) and the internal standard, erythromycin (ERY) was purchased from Sigma Aldrich Chemical Co. (Milwaukee, WI, USA). Clarihexal®, Klarithran®, Klarithran® MR 500 mg tablets, Klarizon 250 mg tablets and Clarihexal®, Klarithran® 250 mg/mL granules for oral suspension were purchased from a local pharmacy. HPLC-grade water was prepared by reverse osmosis using a RephiLe® Direct-Pure UP ultrapure and RO water system (Microsep®, Johanessburg, South Africa), consisting of a deionization RephiDuO® H PAK cartridge and a polishing RephiDuO® PAK cartridge. The water was filtered through a 0.22 μm PES high-flux capsule filter (Microsep®, Johannesburg, South Africa) and used to prepare all buffer solutions. HPLC far UV-grade acetonitrile (ACN) and methanol (MeOH) was purchased from Anatech® Instruments Pty, Ltd. (Randburg, Johannesburg, South Africa). Potassium dihydrogen orthophosphate, sodium chloride and sodium hydroxide pellets were purchased from Merck® Laboratories (Merck®, Wadeville, South Africa).
2.2. Instrumentation and Analytical Conditions
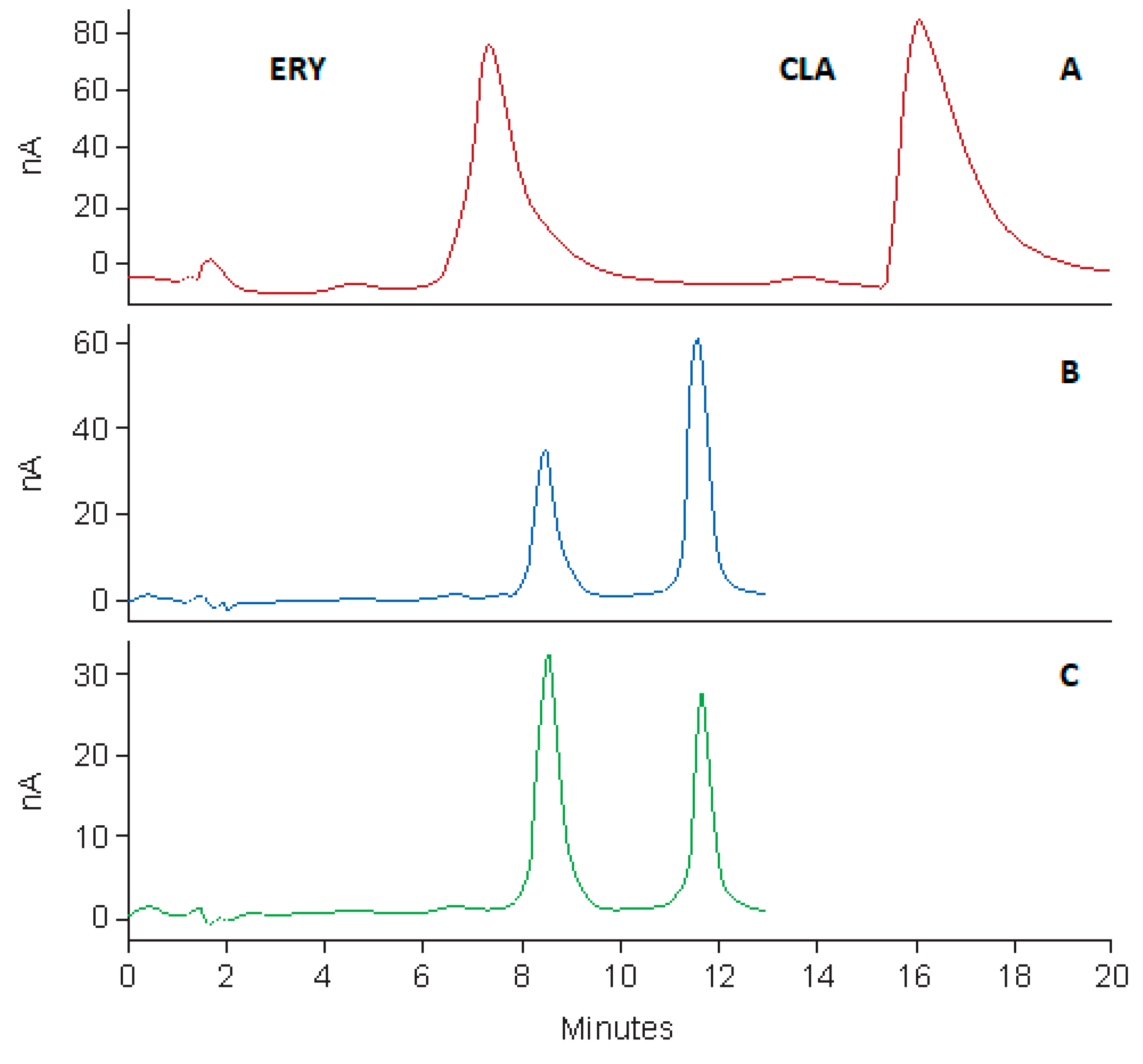
The HPLC system was a Waters® Alliance Model 2695 separation module equipped with a solvent delivery module, an autosampler, an online degasser and a Model 2465 Electrochemical Detector (Waters®, Milford, MA, USA). Data acquisition, processing and reporting were achieved using Waters® Empower 3 software (Waters®, Milford, MA, USA). The separation was achieved under isocratic conditions using a Beckman® C8, 4 μm (150 mm × 4.0 i.d) (Beckman Instruments, Inc., San Ramon, CA, USA) cartridge column with a mobile phase consisting of 50 mM phosphate buffer (pH 7.0), ACN and MeOH in a 58.5:36.5:5.0 v/v/v ratio. The flow rate of the mobile phase and the injection volume were 1.0 mL/min and 10 µL, respectively. The analytical column was maintained at 30 °C using an integral column heater (Waters®, Milford, MA, USA).
2.3. Preparation of Solutions
2.3.1. Stock solution and calibration standards
Standard stock solutions of CLA (100 µg/mL) and ERY (50 µg/mL) were prepared by accurately weighing approximately 10 mg and 5 mg of each API using a Model AE 163 Mettler® analytical balance (Mettler® Inc., Zurich, Switzerland) into 100 mL A-grade volumetric flasks and dissolving in a small volume of ACN. The stock solutions were sonicated using an ultrasonic bath (Ultrasonic Manufacturing Company (Pty), Ltd., Kenware, Krugersdorp, South Africa) until a clear solution formed, after which the solutions were made up to volume with ACN. Calibration standards of CLA over the concentration range 5–50 µg/mL were prepared by serial dilution of the standard stock solution on the day of analysis, using ACN as a diluent. A 0.75 mL aliquot of the 50 µg/mL ERY stock solution was added to all calibration standards and test samples prior to analysis.
2.3.2. Buffer and Mobile Phase
Phosphate buffer solutions (50 mM) were prepared by accurately weighing 6.0845 g potassium dihydrogen orthophosphate into a 1 L A-grade volumetric flask and making up to volume with HPLC grade water. A 0.11 g aliquot of sodium chloride was added to the buffer solution to produce 2 mM equivalent chloride ions. The pH of the buffers was monitored at 22 °C using a Model Basic 20+ Grison pH-meter (Crison Instruments, Barcelona, Spain) and was adjusted to 7.0 using sodium hydroxide pellets. The buffer was degassed under vacuum with the aid of a Model A-2S Eyela Aspirator degasser (Rikakikai Co., Ltd., Tokyo, Japan) and filtered through a 0.2 µm cellulose membrane filter (Sartorius Stedim Biotech GmbH, Goettingen, Germany) prior to being transferred into a 1 L Schott® Duran bottle (Schott Duran GmbH, Wertheim, Germany).
2.4. Statistical Analysis
Data generated were analyzed using Design Expert® version 8.0.2 statistical software (Stat-Ease Inc., Minneapolis, MN, USA). The significance of relevant factors was determined using Fisher’s statistical test for Analysis of Variance (ANOVA). Models were estimated and run to compare first-order interaction terms. ANOVA for linear regression, partitions the total variation of a sample into components that are then used to compute an F-ratio which is used to evaluate the effectiveness of the model. If the probability associated with the F-ratio is low, the model is considered to better fit the data statistically. In these calculations, the higher-order interaction terms are assumed not to contribute, to any great extent, to the behavior of the statistical model.
2.5. Method Validation
2.5.1. Linearity and Range
The linearity was determined using a minimum of five standards injected five times as per the ICH [
35] guidelines. The linearity of the method was evaluated over the concentration range of 5–50 µg/mL and least squares linear regression analysis of the peak height ratio (PHR) versus concentration data was used to evaluate the linearity of the method.
2.5.2. Precision
The precision of the method was evaluated at two different levels
viz., repeatability (intra-day precision) and intermediate precision (inter-day precision). The repeatability for CLA was determined by analysing a sample solution containing the target level of CLA. Ten replicates (
n = 10) of the sample solution were analysed as per the final method procedure [
36]. Repeatability was determined using a 50 µg/mL sample solution. The inter-day precision for CLA was determined by analysing sample solutions in replicate (
n = 5) at three different concentrations
viz., low, middle and high, within the range tested on three different days.
2.5.3. Accuracy
The accuracy of the method was determined by replicate analysis (n = 5) of samples containing known amounts of CLA at low (6.0 µg/mL), middle (25.0 µg/mL) and high (45.0 µg/mL) concentrations. The mean recovery was assessed for compliance according to the ICH guidelines.
2.5.4. LOQ and LOD
The limit of quantitation (LOQ) of the method was determined by evaluating the lowest concentration of CLA that resulted in a precision of < 5% RSD, and the limit of detection (LOD) was taken as the 0.3 × LOQ value. Alternatively, the LOD may be inferred from the concentration resulting in a % RSD of ≤ 20% when the LOQ results in a % RSD of ≤ 10% [
37].
2.6. Method Scaling and Re-Validation
Method scaling is used when an adjustment of the method is required and where parameters are changed within permitted limits, to preserve the chromatographic separation. When a change is made to the column, flow rates may need to be adjusted to maintain the separation and chromatographic performance of a method [
34,
38]. Scaling methods can be used to ensure the same quality of separation while reducing run times, with a reduction in solvent consumption thus ensuring environmental and financial sustainability. In addition, reduced run times result in increased throughput, efficiency of operation and overall profitability.
Chapter 621 of the USP [
34] defines permitted adjustments, for a method, for the purposes of scaling without the need for re-validation. These adjustments are permissible provided system suitability requirements, as described in the monograph, are met when the changes are implemented [
34,
38].
Any column changes must stay within the original method L-designation. For isocratic separations, the particle size and/or the length of the column may be modified provided that the ratio of column length (L) to particle size (dp) (the L/dp) remains constant or falls within the limits of −25%–+50%. When the particle size is changed, the flow rate may require adjustment and can be calculated using Equation (1) [
34].
where,
F1 and
F2 are the flow rates for the original and modified conditions,
dc1 and
dc2 are the respective column diameters and
dp1 and
dp2 are the particle sizes of the stationary phase used. Following method development and validation using a Beckman
® C
8, 4 μm (150 mm × 4.0 i.d) analytical column, further analysis using the column could not be performed. Attempts to regenerate and/or purchase an identical column were unsuccessful as the specific column has been phased out by the manufacturer. Consequently, re-validation and an investigation into the application of method scaling using a different L-designation column were undertaken using a Phenomenex Luna
® CN, 5 µm 150 mm × 4.6 mm i.d (Phenomenex
®, Torrace, CA, USA) which exhibited a −20% decrease in the L/dp value that fell within the range of −25% to + 50% as per the USP [
34] method scaling guidelines. Analysis was undertaken using a mobile phase flow rate of 1.058 mL/min that was calculated using Equation (1).
USP [
34] system suitability requirements for resolution, tailing factor and % RSD for peak height ratio (PHR) and retention time, were used to determine if the modified scaling for the HPLC-ECD method resulted in outcomes that were comparable to those observed using re-validation and thus assess, the applicability of the scaled method for the determination of CLA.
2.7. Assay of Clarithromycin Dosage Forms
2.7.1. Assay of Commercial Tablets
Briefly, 20 tablets were crushed using a mortar and pestle and an aliquot of powder equivalent to the mass of one tablet transferred quantitatively to a 100 mL A-grade volumetric flask. Approximately 50 mL ACN was then added to the volumetric flask and the mixture sonicated using a bath sonicator (Ultrasonic Manufacturing Company (Pty), Ltd., Kenware, Krugersdorp, South Africa) with regular shaking at 20 min intervals for 1 h. The solution was allowed to cool to room temperature (22 °C) prior to making up to volume with ACN. A 5 mL aliquot of the resultant mixture was filtered through a 0.45 µm Millipore® Millex-HV Hydrophilic PVDF filter membrane (Millipore® Co., Bedford, MA, USA) and a 25 µg/mL sample solution in ACN was analyzed using the validated HPLC method.
2.7.2. Assay of Commercial Suspensions
The granules for suspension were reconstituted using HPLC-grade water as per the label instructions. A 5 mL aliquot of the reconstituted suspension was transferred to a 100 mL A-grade volumetric flask. Approximately 20 mL 50 mM phosphate buffer (pH 7.00) was then added to the volumetric flask and the mixture sonicated using a bath sonicator (Ultrasonic Manufacturing Company (Pty), Ltd., Kenware, Krugersdorp, South Africa) with regular shaking at 10 min intervals for 30 min. Approximately 30 mL MeOH was added to the flask and the mixture was sonicated for a further 30 min. The solution was allowed to cool to room temperature (22 °C) prior to making up to volume with MeOH. The mixture was then stirred for one hour using a digital hot plate stirrer (Lasec®, Port Elizabeth, South Africa). A 5 mL aliquot of the resultant mixture was filtered through a 0.45 µm Millipore® Millex-HV Hydrophilic PVDF filter membrane (Millipore® Co., Bedford, MA, USA) and analyzed using the validated HPLC method after dilution to obtain a 25 µg/mL solution in ACN.
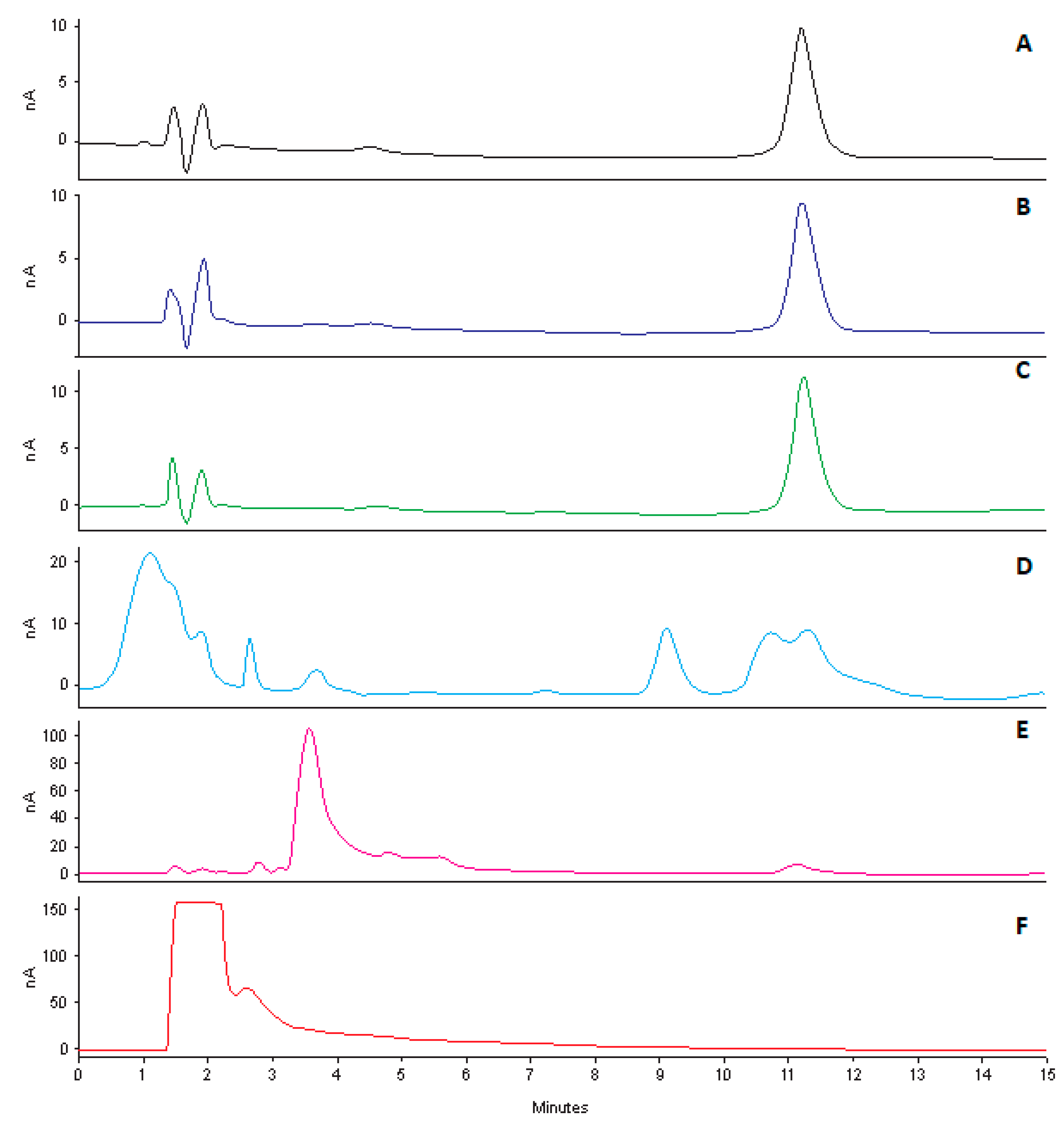
2.8. Forced Degradation Studies
Stress studies were conducted by exposing CLA to acidic, alkaline, hydrogen peroxide and light conditions [
39]. Stock solutions (100 µg/mL) were prepared as described in
Section 2.3. These solutions were then exposed to different stress conditions viz., 0.1 M HCL, 0.1 M NaOH, 4% H
2O
2 at 80 °C and 500 W/m
2 for 12 h, prior to analysis using the validated analytical method. A tolerance level of 10% degradation is considered optimal for the purposes of validating the analytical method intended for assay [
39]. Consequently, a tolerance of 10% was used to determine if CLA had degraded as a result of exposure to stress conditions. These studies were also performed to determine interference, if any, of degradation products with the chromatography.
4. Conclusions
A simple, selective and sensitive high-performance liquid chromatographic method with electrochemical detection for the quantitation of clarithromycin in bulk samples and oral dosage forms has been developed. The method complies with ICH validation parameters. In addition, an attempted investigation into the applicability of USP method scaling across different L-designation proved feasible. Method scaling allows for translation of an analytical method while achieving an equivalent separation without the need for re-validation. In this study, a revalidation was undertaken and confirmed the applicability of method scaling across analytical columns of different L-designation, to the HPLC-ECD analytical determination of CLA. This, in turn, saves costs and is environmentally sustainable as less solvent is used and reduced analytical run times can be achieved. The scaled method resulted in an increase in throughput with shorter run times that resulted in less solvent consumption in the laboratory. HPLC methods can thus be scaled on condition that they meet USP system suitability requirements, maintaining separation quality, thereby eliminating the need for method re-validation. Although USP method scaling is only permitted for columns within the same L-designation, its applicability should be investigated across analytical columns of different L-designation to further streamline regulatory requirements. This is, to the best of our knowledge, the first evaluation of an HPLC-ECD method using a modified scaling approach.












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}