Host-Guest Interactions of Plumbagin with β-Cyclodextrin, Dimethyl-β-Cyclodextrin and Hydroxypropyl-β-Cyclodextrin: Semi-Empirical Quantum Mechanical PM6 and PM7 Methods

 ,
,
Abstract
:
1. Introduction
2. Materials and Methods
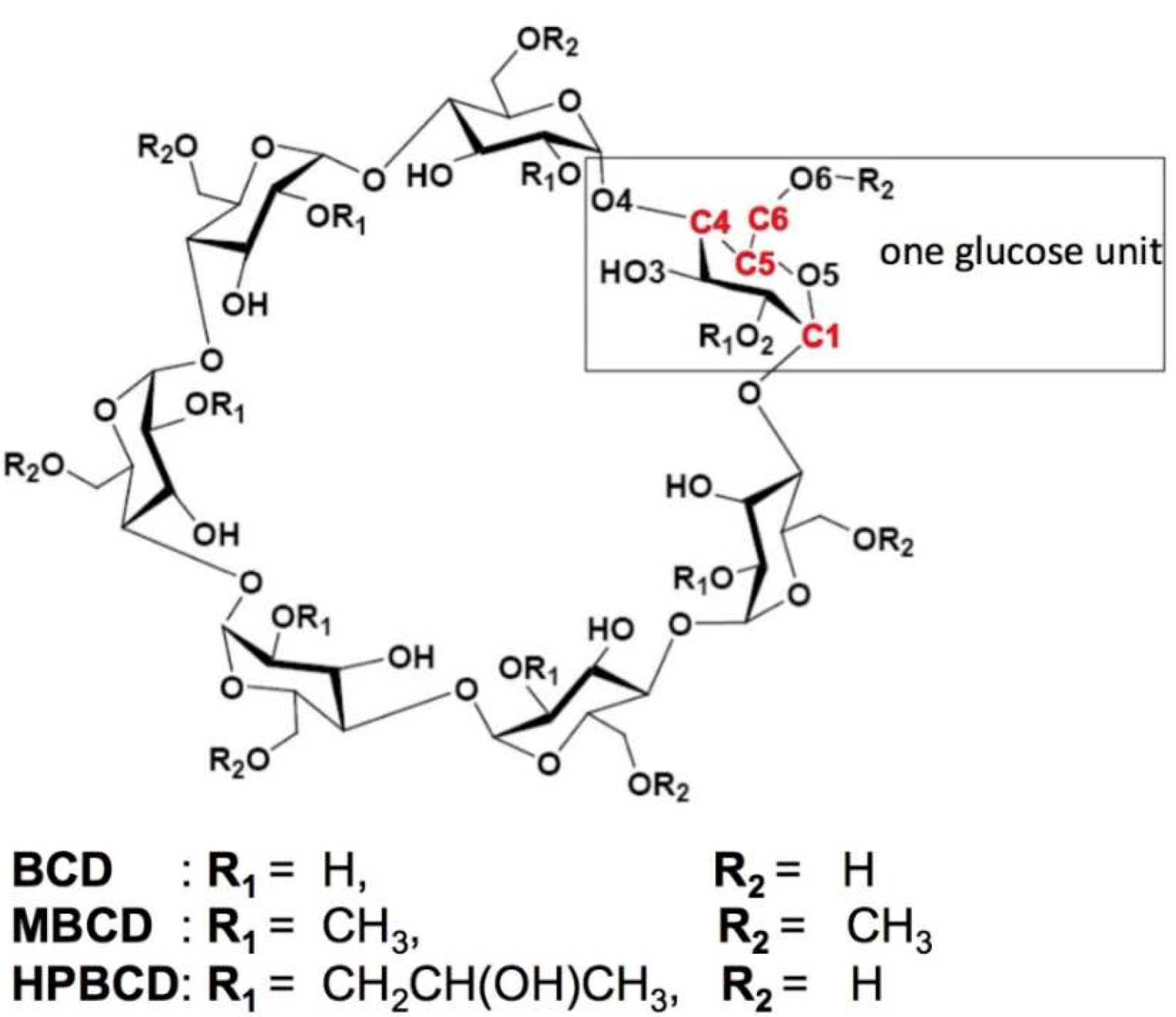
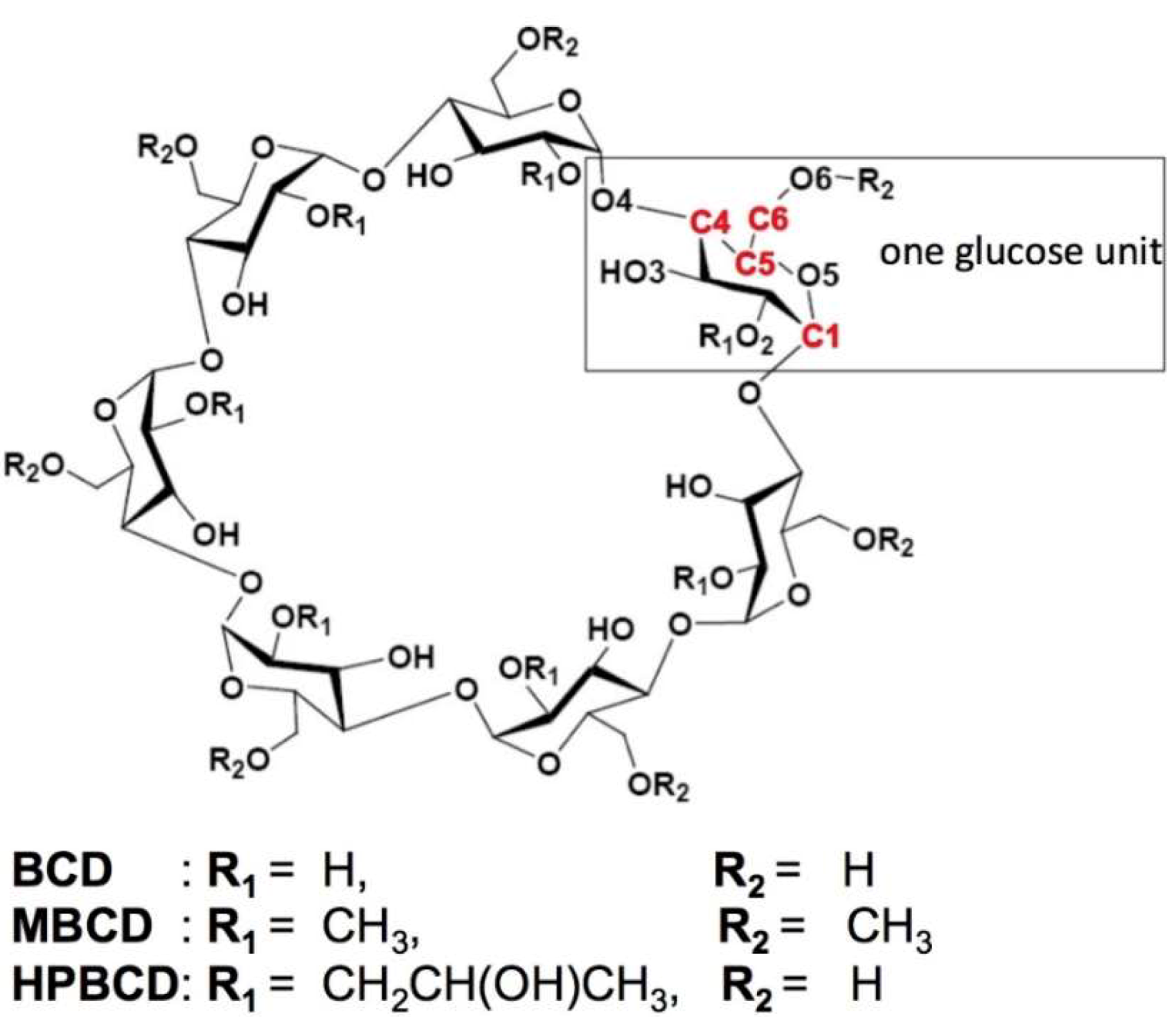
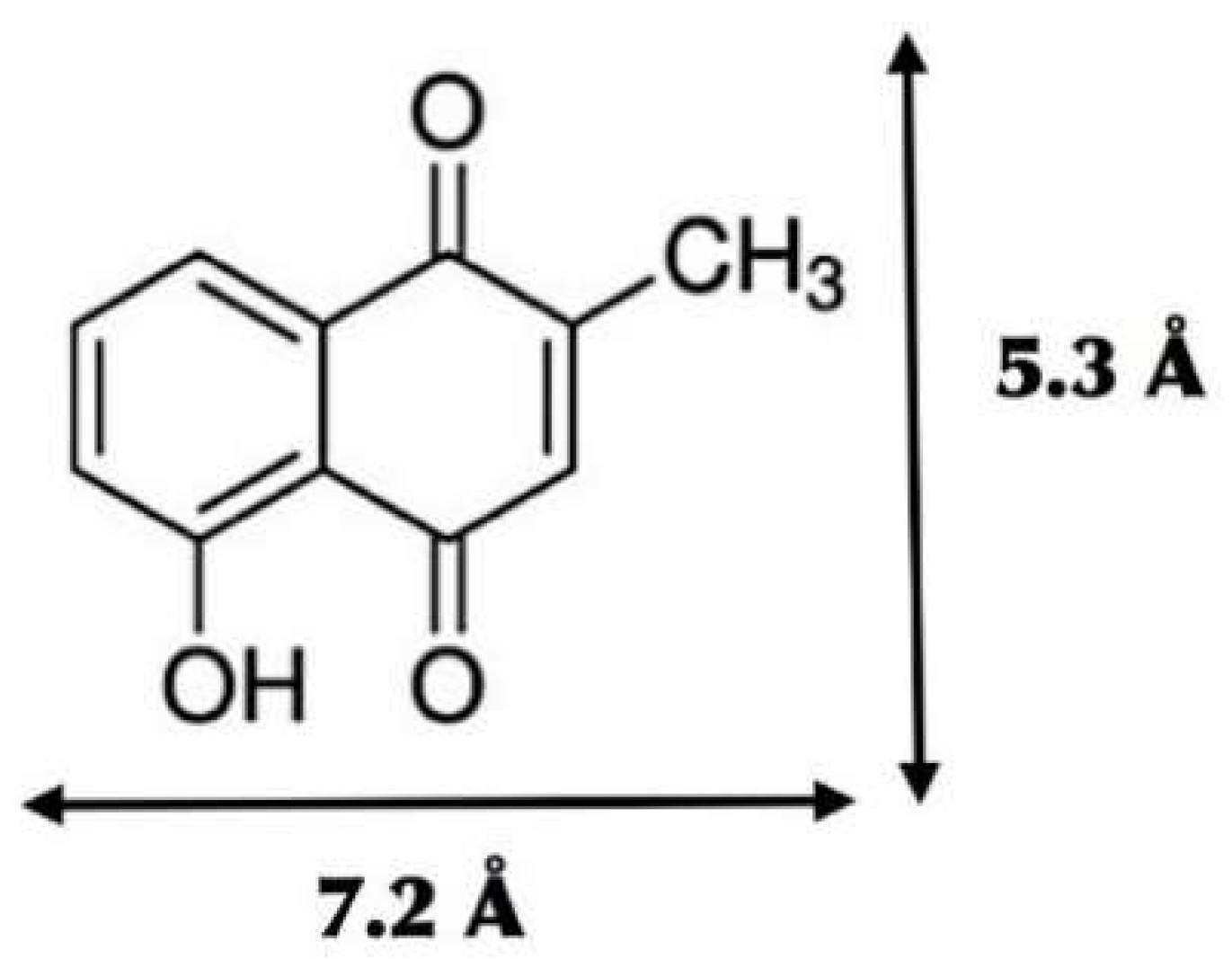
2.1. Molecular Structure Construction
2.2. Molecular Docking Calculation
2.3. Complexation Energy Calculation
3. Results and Discussion
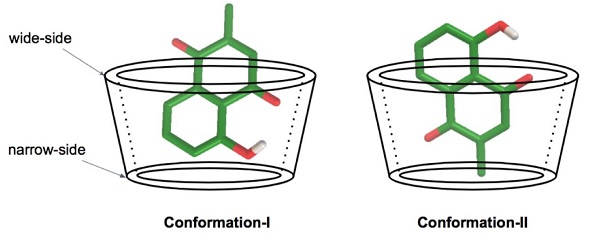
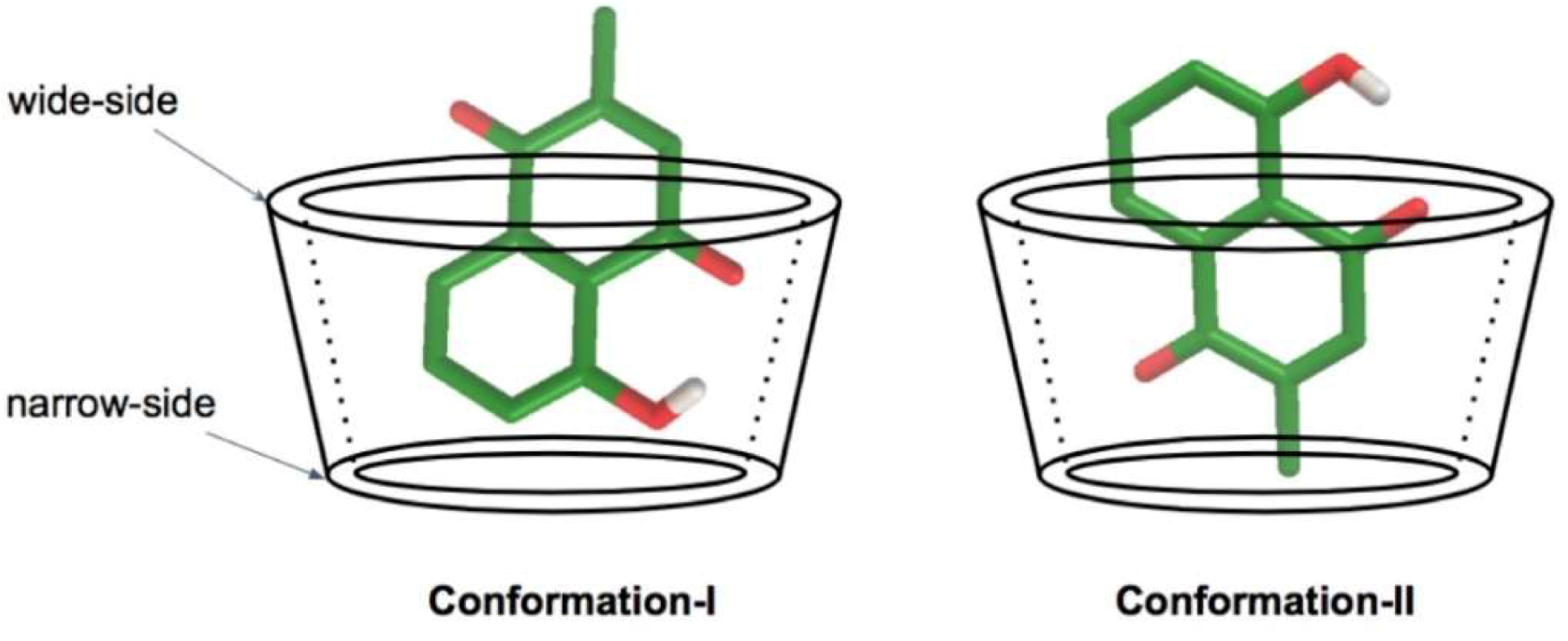
3.1. Molecular Docking Calculation
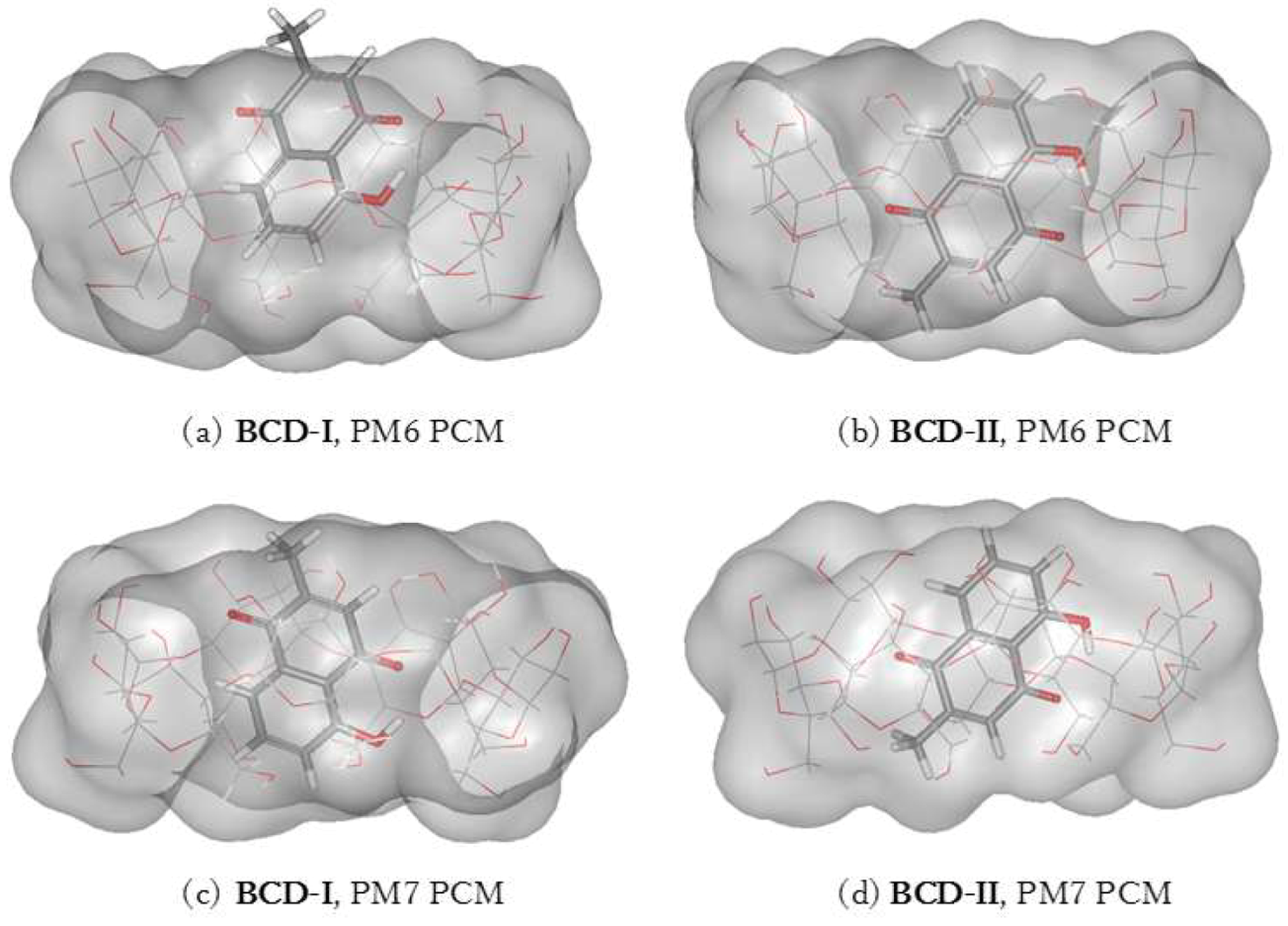
3.2. Complexation Energy Calculation
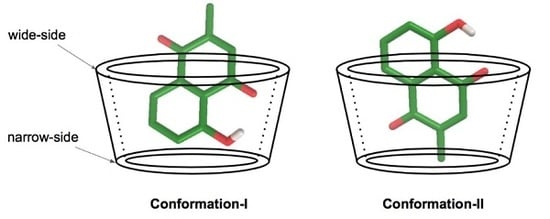
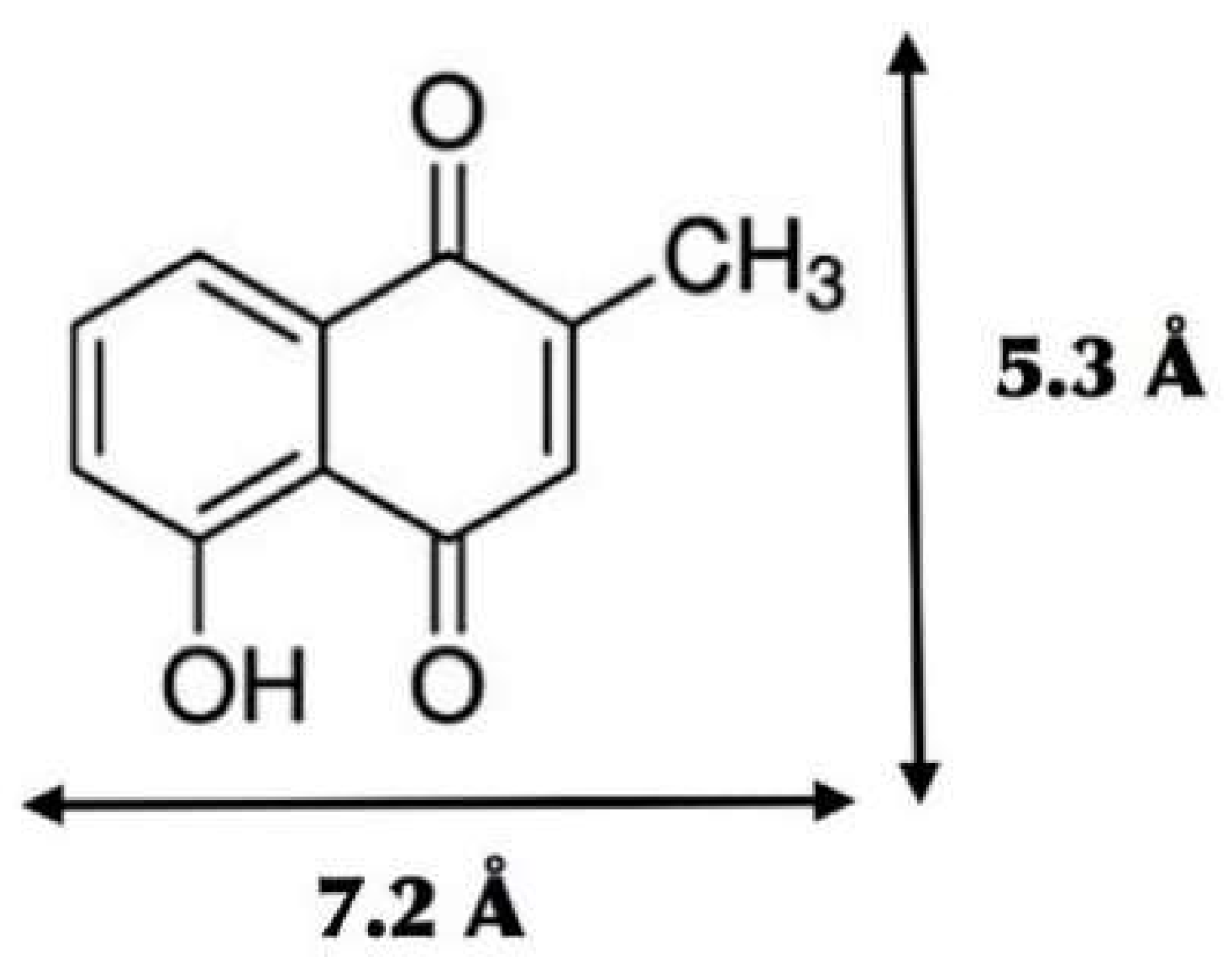
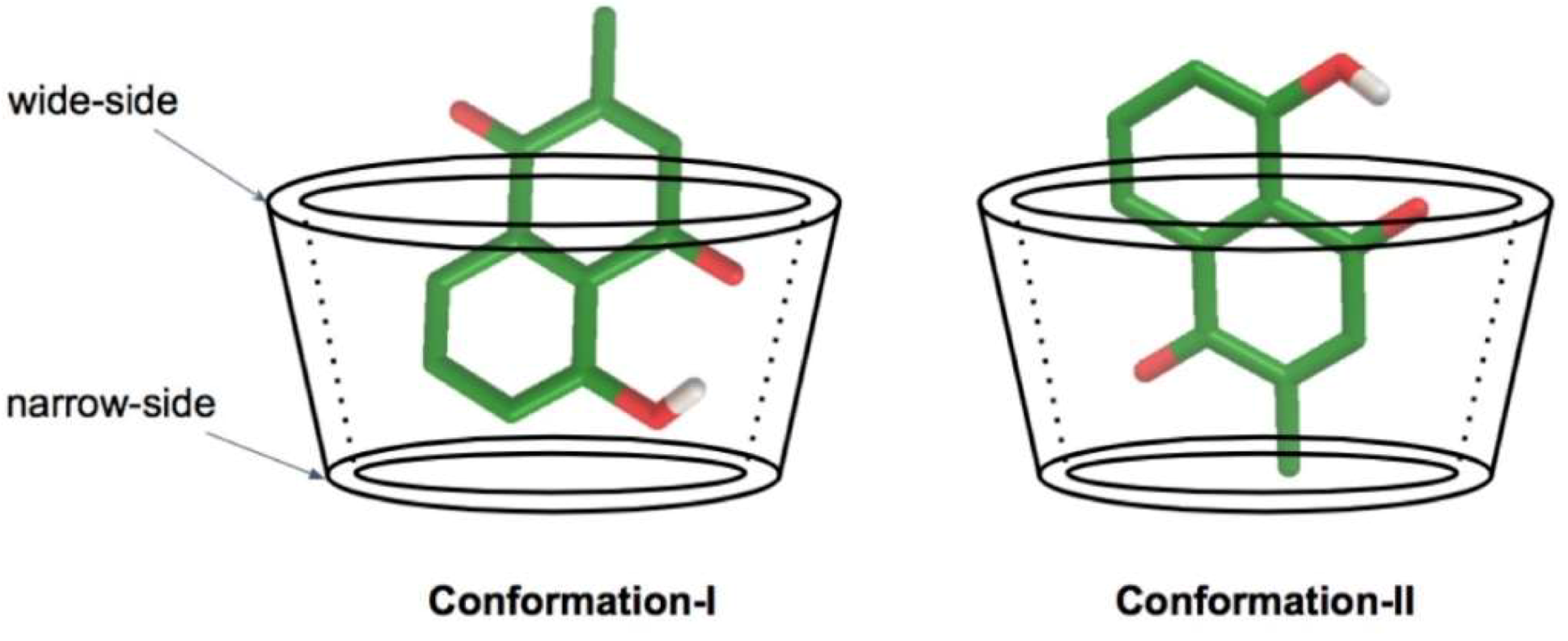
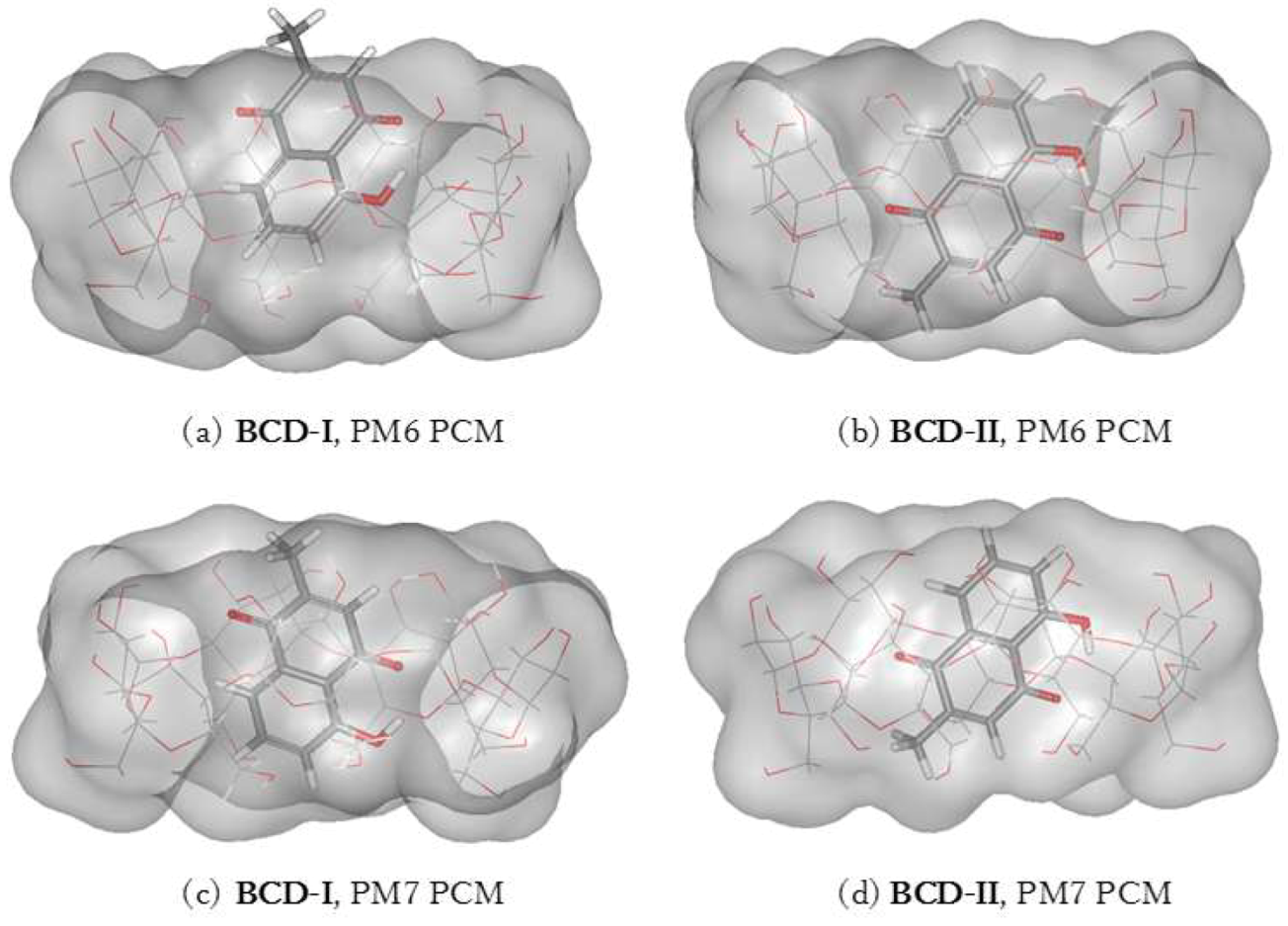
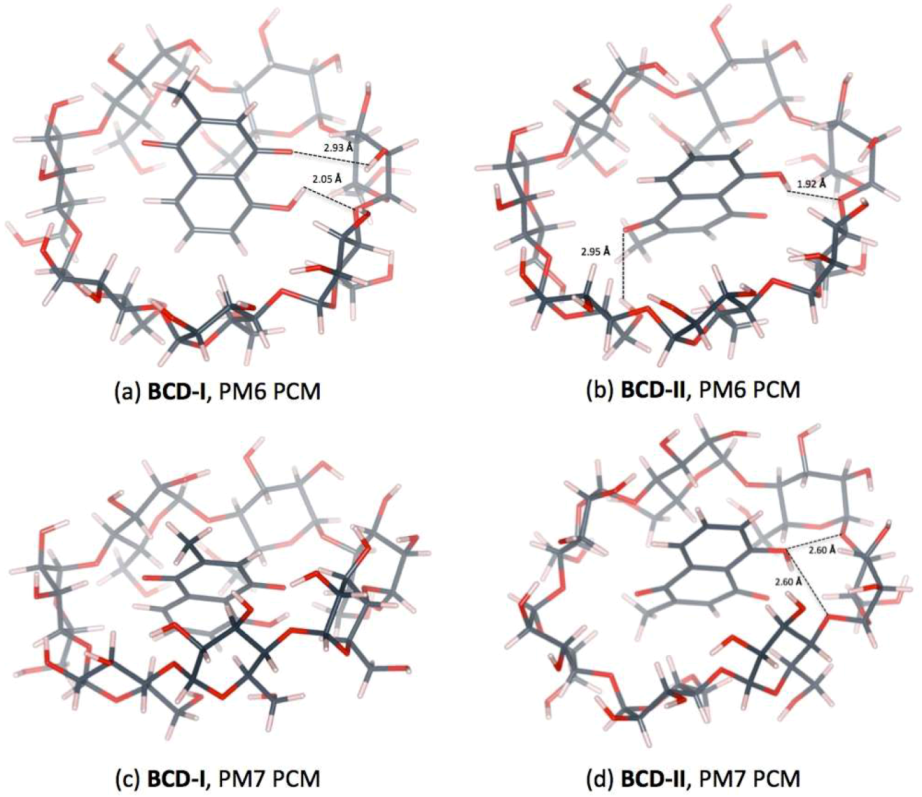
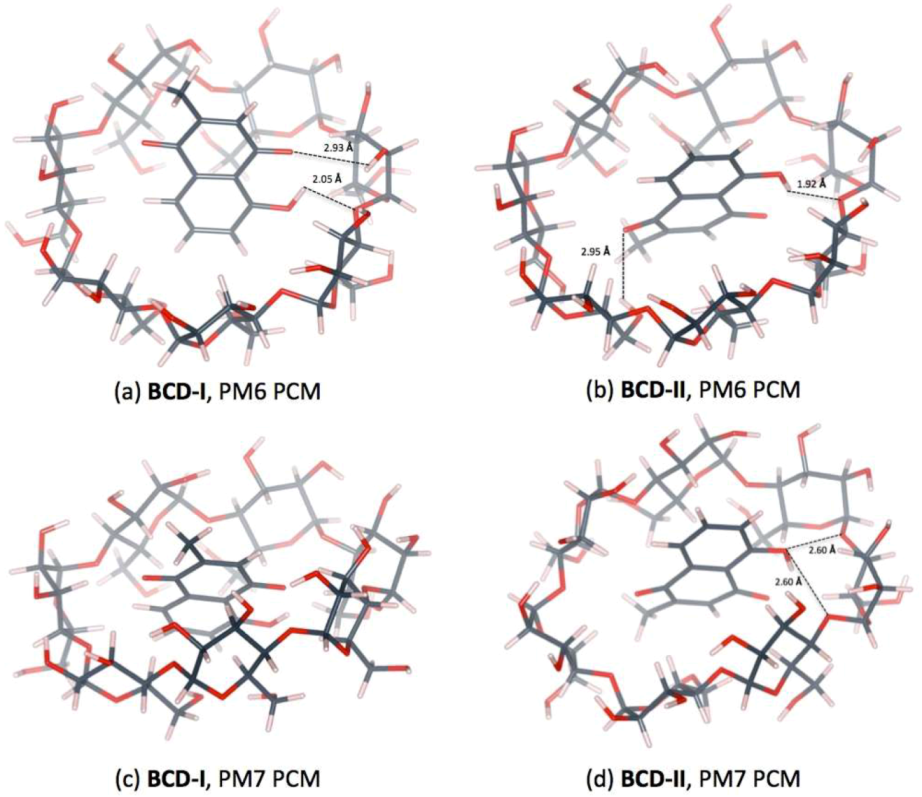
3.3. Plumbagin/β-cyclodextrin Inclusion Complex
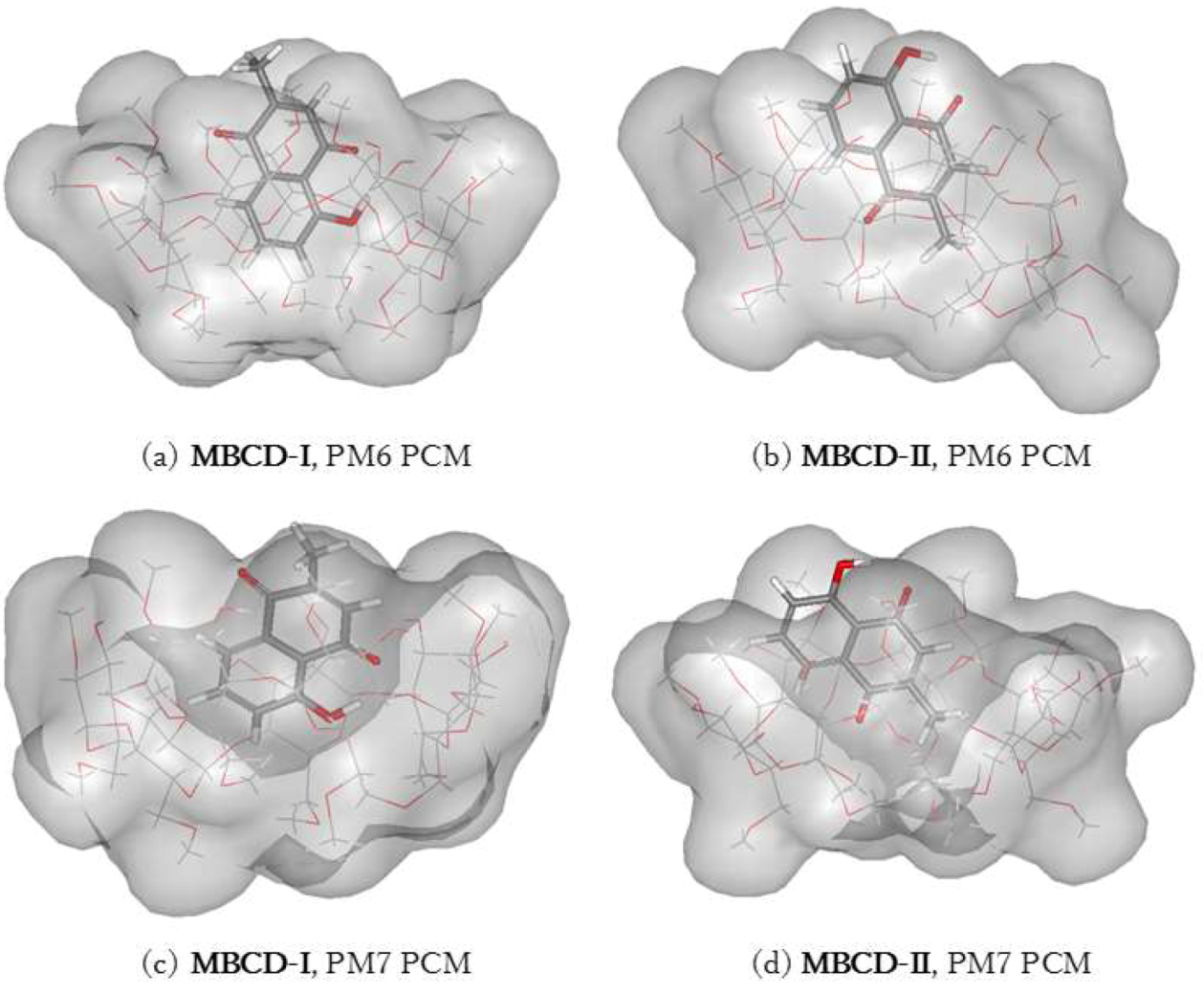
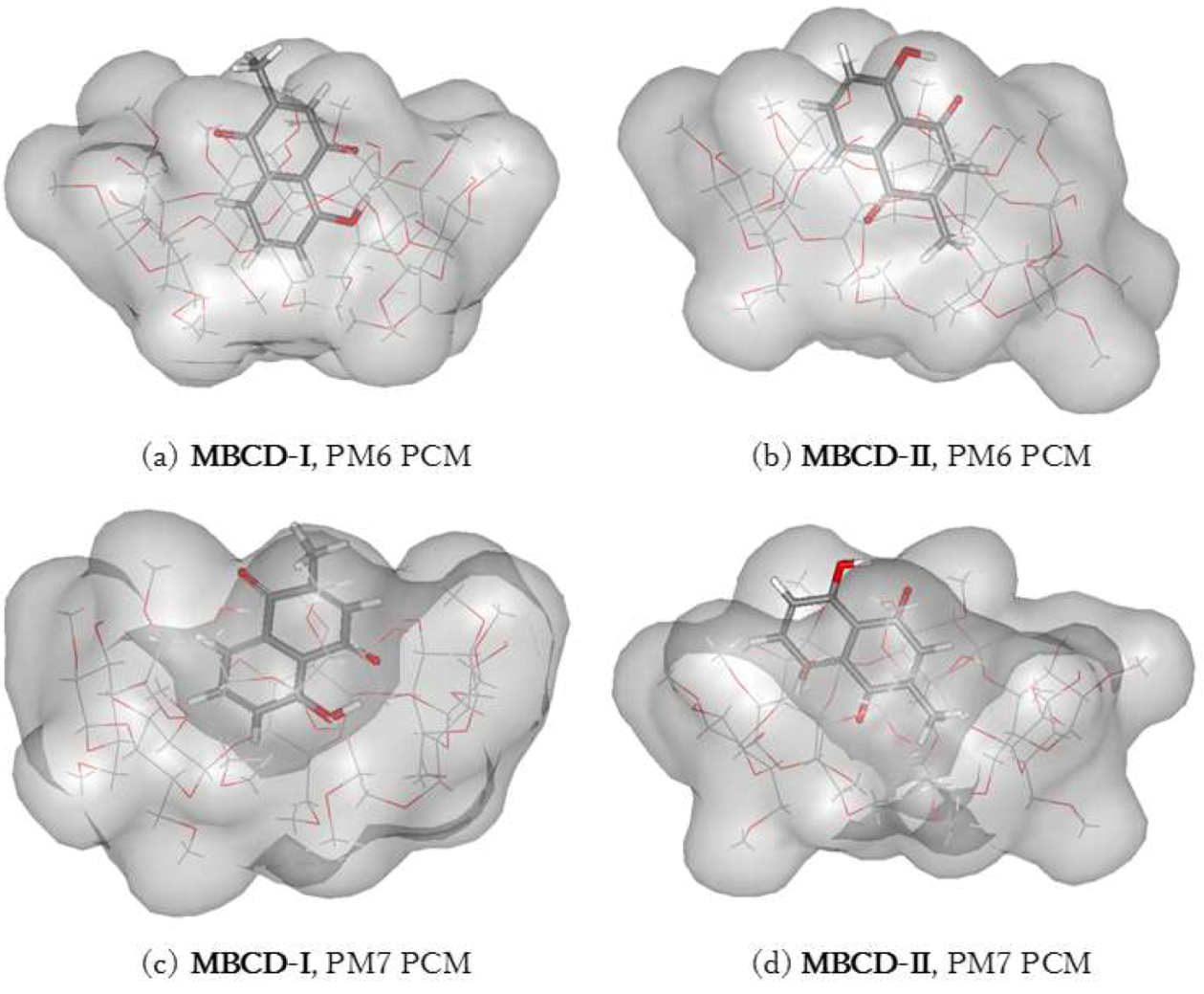
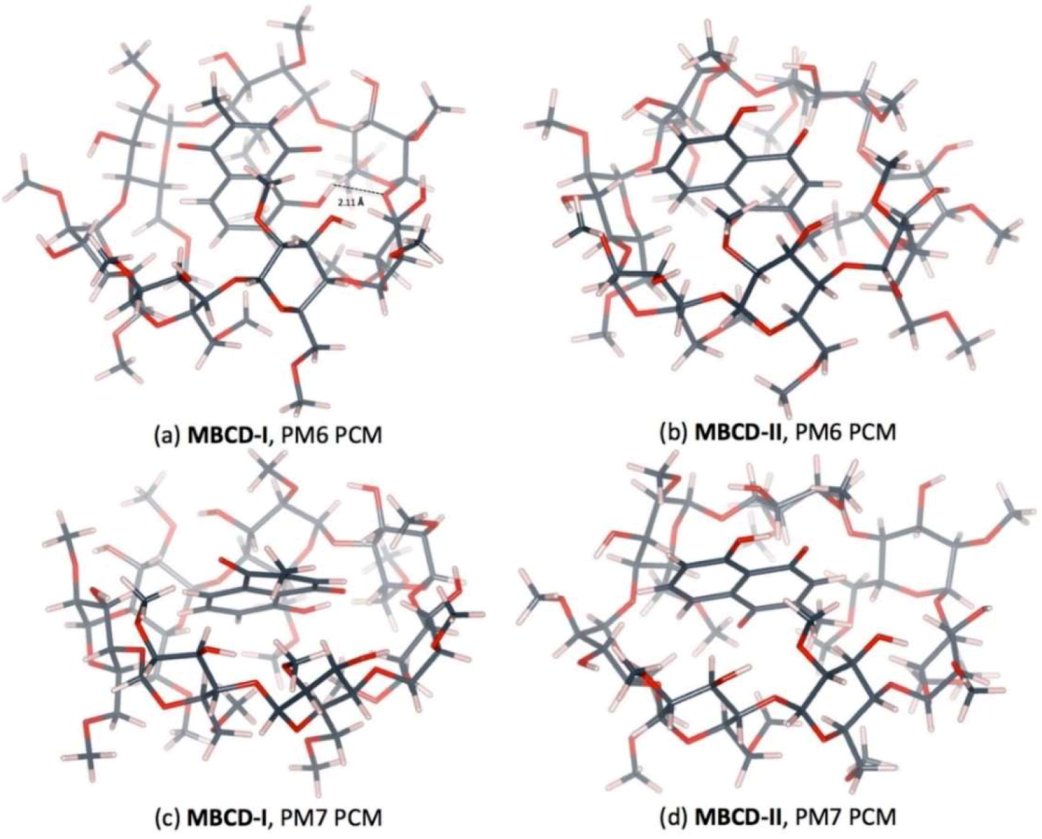
3.4. Plumbagin/Dimethyl-β-cyclodextrin Inclusion Complex
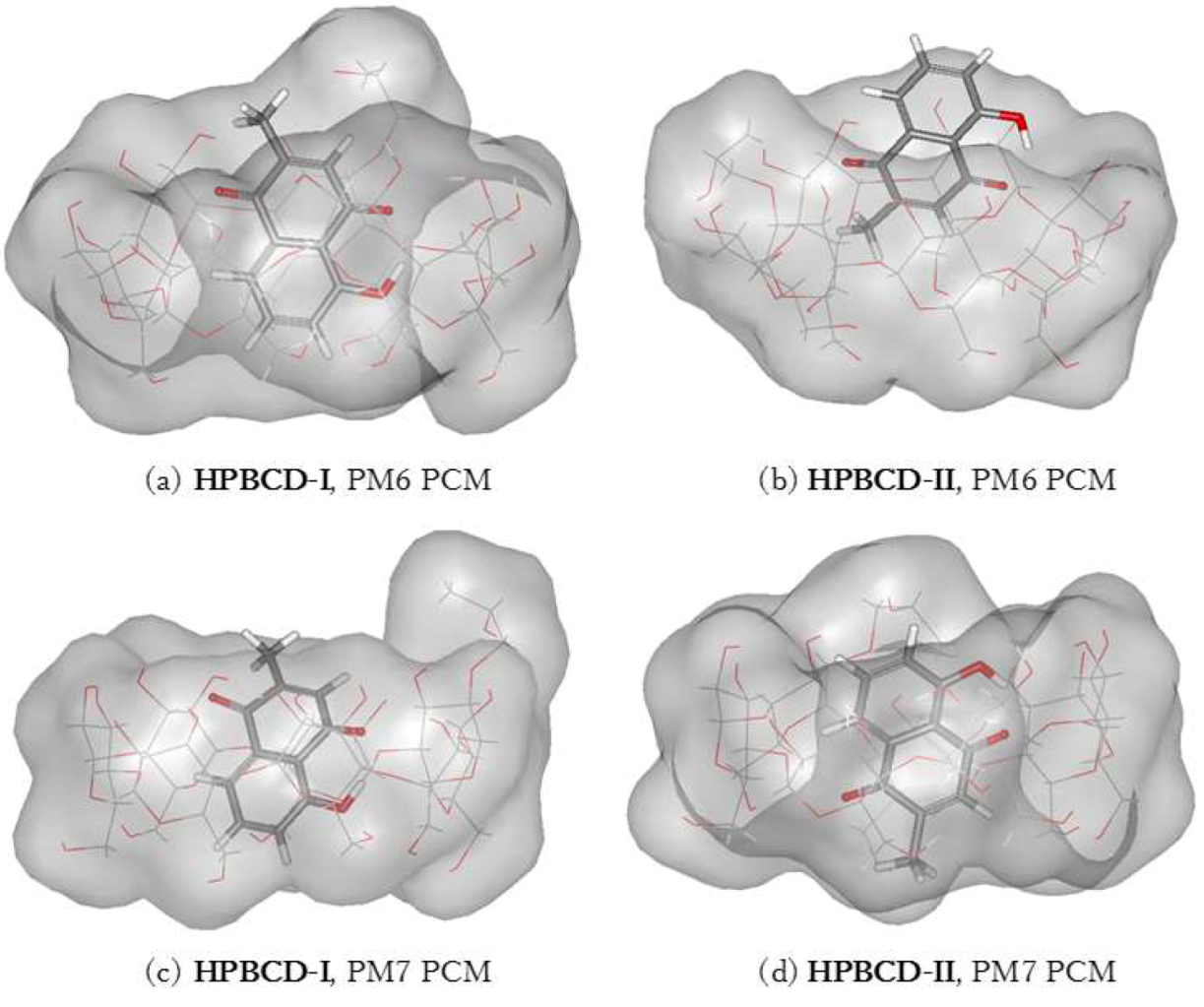
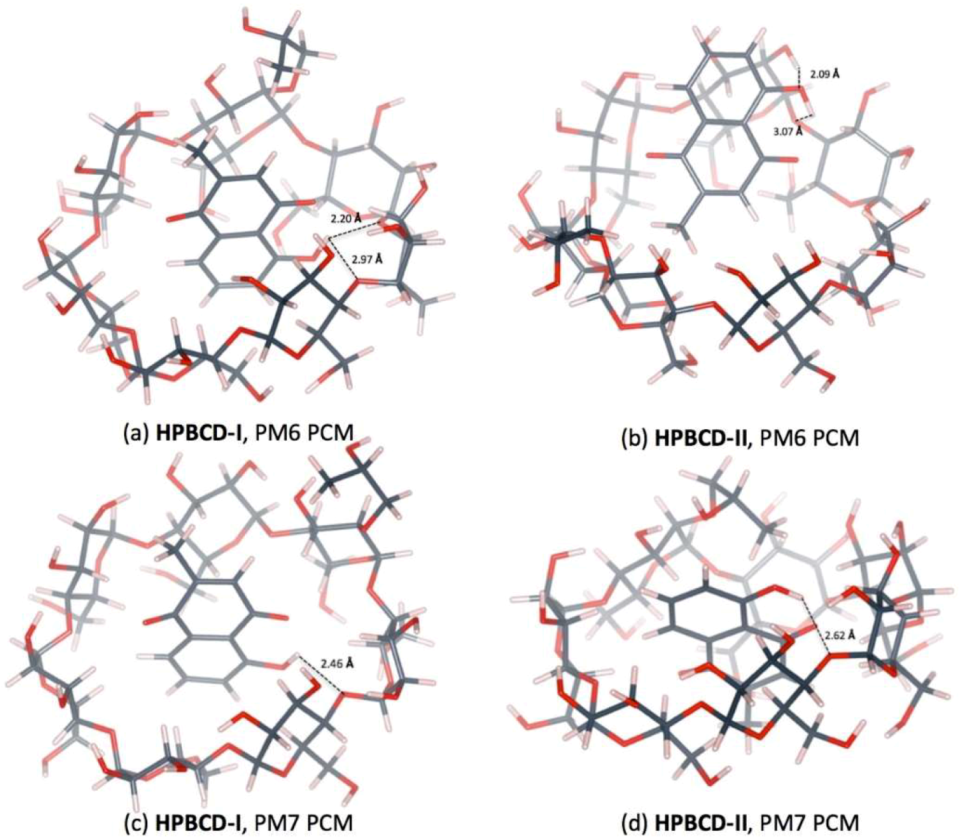
3.5. Plumbagin/Hydroxypropyl-β-cyclodextrin Inclusion Complex
4. Conclusions
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Stewart, J.J.P.; Császár, P.; Pulay, P. Fast Semiempirical Calculations. J. Comput. Chem. 1982, 2, 227–228. [Google Scholar] [CrossRef]
- Stewart, J.J.P. Optimization of Parameters for Semiempirical Methods VI: More Modifications to the NDDO Approximations and Re-Optimization of Parameters. J. Mol. Model. 2013, 19, 1–32. [Google Scholar] [CrossRef] [PubMed]
- Connors, K.A. The Stability of Cyclodextrin Complexes in Solution. Chem. Rev. 1997, 97, 1325–1358. [Google Scholar] [CrossRef] [PubMed]
- Fermeglia, M.; Ferrone, M.; Lodi, A.; Pricl, S. Host–guest Inclusion Complexes between Anticancer Drugs and β-Cyclodextrin: Computational Studies. Carbohydr. Polym. 2003, 53, 15–44. [Google Scholar] [CrossRef]
- Qi, Z.H.; Hedges, A.R. Use of Cyclodextrins for Flavors. ACS Symp. Ser. 1997, 231–243. [Google Scholar] [CrossRef]
- Loftsson, T.; Duchêne, D. Cyclodextrins and Their Pharmaceutical Applications. Int. J. Pharm. 2007, 329, 1–11. [Google Scholar] [CrossRef] [PubMed]
- De Sousa, A.T.A.; de Melo, J.G.; Júnior, W.S.F.; Albuquerque, U.P. Medicinal Plants. In Introduction to Ethnobiology, 1st ed.; Albuquerque, U., Nóbrega Alves, R., Eds.; Springer International Publishing: New York, NY, USA, 2016; pp. 143–149. [Google Scholar] [CrossRef]
- De Paiva, S.R.; Figueiredo, M.R.; Aragão, T.V.; Coelho Kaplan, M.A. Antimicrobial Activity in Vitro of Plumbagin Isolated from Plumbago Species. Mem. Inst. Oswaldo Cruz. 2003, 98, 959–961. [Google Scholar] [CrossRef] [PubMed]
- Oommen, E.; Shenoy, B.D.; Udupa, N.; Kamath, R.; Devi, P.U. Antitumour Efficacy of Cyclodextrin-complexed and Niosome-encapsulated Plumbagin in Mice Bearing Melanoma B16F1. Pharm. Pharmacol. Commun. 1999, 5, 281–285. [Google Scholar] [CrossRef]
- D’Souza, R.; Singh, U.V.; Aithal, K.S.; Udupa, N. Antifertility Activity of Niosomal HPbCD—Plumbagin Complex. Indian J. Pharm. 1998, 60, 36–40. [Google Scholar]
- Singh, U.V.; Udupa, N. Reduced Toxicity and Enhanced Antitumor Efficacy of Betacyclodextrin Plumbagin Inclusion Complex in Mice Bearing Ehrlich Ascites Carcinoma. Indian J. Physiol. Pharmacol. 1997, 41, 171–175. [Google Scholar] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16; Revision B.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Lichtenthaler, F.W.; Immel, S. Towards Understanding Formation and Stability of Cyclodextrin Inclusion Complexes: Computation and Visualization of their Molecular Lipophilicity Patterns[1]. Starch 1996, 48–154. [Google Scholar] [CrossRef]
- Vijayalakshmi, J.; Rajan, S.S.; Srinivasan, R. Structure of Plumbagin. Acta Crystallogr. Sect. C Cryst. Struct. Commun. 1987, 43, 2375–2377. [Google Scholar] [CrossRef]
- Steiner, T.; Koellner, G. Crystalline Beta-Cyclodextrin Hydrate at Various Humidities: Fast, Continuous, and Reversible Dehydration Studied by X-ray Diffraction. J. Am. Chem. Soc. 1994, 116, 5122–5128. [Google Scholar] [CrossRef]
- Aree, T.; Hoier, H.; Schulz, B.; Reck, G.; Saenger, W. Novel Type of Thermostable Channel Clathrate Hydrate Formed by Heptakis (2,6-di-O-methyl)-β-cyclodextrin-15H2O—A Paradigm of the Hydrophobic Effect. Angew. Chem. Int. Ed. 2000, 39, 897–899. [Google Scholar] [CrossRef]
- Harata, K.; Rao, C.T.; Pitha, J.; Fukunaga, K.; Uekama, K. Crystal Structure of 2-O-[(S)-2-Hydroxypropyl]cyclomaltoheptaose. Carbohydr. Res. 1991, 222, 37–45. [Google Scholar] [CrossRef]
- Barone, V.; Cossi, M.; Tomasi, J. A New Definition of Cavities for the Computation of Solvation Free Energies by the Polarizable Continuum Model. J. Chem. Phys. 1997, 107, 3210–3221. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated Docking with Selective Receptor Flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.M.; Goodsell, D.S.; Halliday, R.S.; Huey, R.; Hart, W.E.; Belew, R.K.; Olson, A.J. Automated Docking Using a Lamarckian Genetic Algorithm and an Empirical Binding Free Energy Function. J. Comput. Chem. 1998, 19, 1639–1662. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Guest/Host | Cluster | Conformation | Frequency (%) | ΔG (kcal/mol) | |
|---|---|---|---|---|---|
| Lowest | Average | ||||
| plumbagin/BCD | 1 | I | 100 | −6.21 | −6.19 |
| plumbagin/MBCD | 1 | I | 85 | −5.14 | −5.13 |
| 2 | II | 15 | −5.03 | −5.02 | |
| plumbagin/HPBCD | 1 | II | 48 | −5.76 | −5.75 |
| 2 | I | 2 | −5.74 | −5.73 | |
| 3 | I | 50 | −5.72 | −5.71 | |
| Guest/Host | Cluster | Conformation | Frequency (%) | ΔG (kcal/mol) | |
|---|---|---|---|---|---|
| Lowest | Average | ||||
| plumbagin/BCD | 1 | I | 61 | −5.34 | −5.26 |
| 2 | I | 2 | −5.24 | 5.23 | |
| 3 | II | 24 | −5.22 | −5.21 | |
| 4 | II | 13 | −5.20 | −5.18 | |
| plumbagin/MBCD | 1 | I | 100 | −5.12 | −5.12 |
| plumbagin/HPBCD | 1 | II | 100 | −5.89 | −5.87 |
| PM6 | PM7 | |||
|---|---|---|---|---|
| E (kcal/mol) | ∆E (kcal/mol) | E (kcal/mol) | ∆E (kcal/mol) | |
| Isolated molecule | ||||
| Plumbagin | −84.56 | −87.09 | ||
| BCD | −1614.25 | −1648.53 | ||
| MBCD | −1543.64 | −1573.87 | ||
| HPBCD | −1659.94 | −1701.90 | ||
| Inclusion Complex | ||||
| BCD-I | −1704.99 | −6.18 | −1768.09 | −32.47 |
| BCD-II | −1704.97 | −6.15 | −1765.72 | −30.10 |
| BCD-I–BCD-II | −0.03 | −2.37 | ||
| MBCD-I | −1636.23 | −8.03 | −1702.37 | −41.41 |
| MBCD-II | −1640.97 | −12.78 | −1699.12 | −38.17 |
| MBCD-I–MBCD-II | 4.75 | −3.24 | ||
| HPBCD-I | −1753.57 | −9.08 | −1820.19 | −31.21 |
| HPBCD-II | −1750.20 | −5.70 | −1829.73 | −40.75 |
| HPBCD-I–HPBCD-II | −3.38 | 9.54 | ||
| Distance (Å) | |||
|---|---|---|---|
| PM6 | BCD-I | O4(BCD)…H(OH-PL) | 2.05 |
| O(CO-PL)…H(O3H-BCD) | 2.93 | ||
| PM6 | BCD-II | O4(BCD)…H(OH-PL) | 1.92 |
| O(CO-PL)…H(O6H-BCD) | 2.95 | ||
| PM6 | MBCD-I | O4(MBCD)…H(OH-PL) | 2.11 |
| PM6 | HPBCD-I | O4(HPBCD)n…H(OH-PL) | 2.20 |
| O4(HPBCD)n+1…H(OH-PL) | 2.97 | ||
| PM6 | HPBCD-II | O4(HPBCD)…H(OH-PL) | 3.07 |
| O(OH-PL)…H(O2H-HPBCD) | 2.09 | ||
| PM7 | BCD-II | O4(BCD)n…H(OH-PL) | 2.60 |
| O4(BCD)n+1…H(OH-PL) | 2.60 | ||
| PM7 | HPBCD-I | O4(HPBCD …H(OH-PL) | 2.46 |
| PM7 | HPBCD-II | O4(HPBCD)…H(OH-PL) | 2.62 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Srihakulung, O.; Maezono, R.; Toochinda, P.; Kongprawechnon, W.; Intarapanich, A.; Lawtrakul, A.L. Host-Guest Interactions of Plumbagin with β-Cyclodextrin, Dimethyl-β-Cyclodextrin and Hydroxypropyl-β-Cyclodextrin: Semi-Empirical Quantum Mechanical PM6 and PM7 Methods. Sci. Pharm. 2018, 86, 20. https://doi.org/10.3390/scipharm86020020
Srihakulung O, Maezono R, Toochinda P, Kongprawechnon W, Intarapanich A, Lawtrakul AL. Host-Guest Interactions of Plumbagin with β-Cyclodextrin, Dimethyl-β-Cyclodextrin and Hydroxypropyl-β-Cyclodextrin: Semi-Empirical Quantum Mechanical PM6 and PM7 Methods. Scientia Pharmaceutica. 2018; 86(2):20. https://doi.org/10.3390/scipharm86020020
Chicago/Turabian StyleSrihakulung, Ornin, Ryo Maezono, Pisanu Toochinda, Waree Kongprawechnon, Apichart Intarapanich, and And Luckhana Lawtrakul. 2018. "Host-Guest Interactions of Plumbagin with β-Cyclodextrin, Dimethyl-β-Cyclodextrin and Hydroxypropyl-β-Cyclodextrin: Semi-Empirical Quantum Mechanical PM6 and PM7 Methods" Scientia Pharmaceutica 86, no. 2: 20. https://doi.org/10.3390/scipharm86020020
APA StyleSrihakulung, O., Maezono, R., Toochinda, P., Kongprawechnon, W., Intarapanich, A., & Lawtrakul, A. L. (2018). Host-Guest Interactions of Plumbagin with β-Cyclodextrin, Dimethyl-β-Cyclodextrin and Hydroxypropyl-β-Cyclodextrin: Semi-Empirical Quantum Mechanical PM6 and PM7 Methods. Scientia Pharmaceutica, 86(2), 20. https://doi.org/10.3390/scipharm86020020