
Synthesis and Biological Activities of Camphor Hydrazone and Imine Derivatives

Abstract
:1. Introduction
2. Results and Discussion
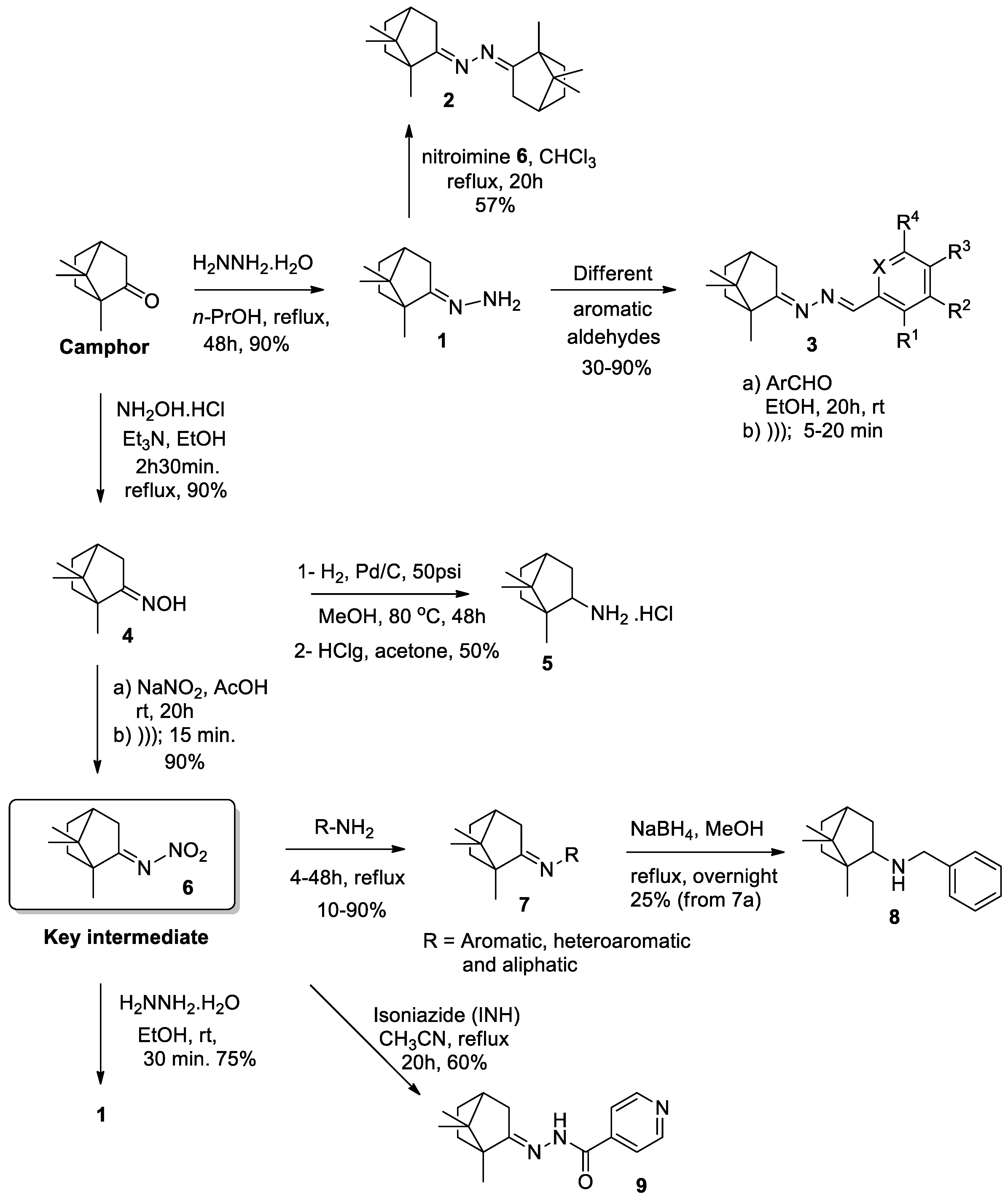
2.1. Chemistry
2.2. Crystallography
2.3. Antimycobacterial Activity against Mycobacterium tuberculosis H37Rv (ATCC 27294)
2.4. Cytotoxicity against Cancer Cell Lines
2.5. Toxicity Risks
3. Materials and Methods
3.1. Synthesis of Nitroimine (6) Using Ultrasonic Irradiation
3.2. Synthesis of (E)-(1,7,7-Trimethylbicyclo[2.2.1]heptan-2-ylidene)hydrazine (1).
3.3 Synthesis of (1E,2E)-1,2-Bis(1,7,7-trimethylbicyclo[2.2.1]heptan-2-ylidene)hydrazine (2).
3.4. General Procedures for the Synthesis of Camphor Hydrazone Derivatives (3)
3.5. General Synthetic Procedure for the Imine Derivatives 7
3.6. Synthesis of N-Benzyl-1,7,7-trimethylbicyclo[2.2.1]heptan-2-amine (8) [34].
3.7. Synthesis of N′-(1,7,7-trimethylbicyclo[2.2.1]heptan-2-ylidene)isonicotinohydrazide (9).
3.8. Biological Evaluation against Mycobacterium tuberculosis
3.9. Cytotoxicity against Cancer Cell Lines
Acknowledgments
Author Contributions
Conflicts of Interest
References and Notes
- Chen, W.; Vermaak, I.; Viljoen, A. Camphor—A Fumigant during the Black Death and a Coveted Fragrant Wood in Ancient Egypt and Babylon—A Review. Molecules 2013, 18, 5434–5454. [Google Scholar] [CrossRef] [PubMed]
- You, C.; Guo, S.; Zhang, W.; Yang, K.; Geng, Z.; Du, S.; Wang, C.; Deng, Z. Identification of repellent and insecticidal constituents from Artemisia mongolica essential oil against Lasioderma serricorne. J. Chem. 2015, 2015, 549057. [Google Scholar] [CrossRef]
- Li, Z.-J.; Yao, J.; Tao, Q.; Jiang, L.; Lu, T.-B. Enantioselective Recognition and Separation of Racemic 1-Phenylethanol by a Pair of 2D Chiral Coordination Polymers. Inorg. Chem. 2013, 52, 11694–11696. [Google Scholar] [CrossRef] [PubMed]
- Wenzel, T.J.; Wenzel, B.T. Diamagnetic lanthanide tris β-diketonates as organic-soluble chiral NMR shift reagents. Chirality 2008, 21, 6–10. [Google Scholar] [CrossRef] [PubMed]
- Cody, J.A.; Boeckman, R.K. Terpene derived auxiliaries. Camphor and pinene derived auxiliaries. Compr. Chirality 2012, 3, 42–105. [Google Scholar]
- Chelucci, G. Synthesis and application in asymmetric catalysis of camphor-based pyridine ligands. Chem. Soc. Rev. 2006, 35, 1230–1243. [Google Scholar] [CrossRef] [PubMed]
- Money, T.A. Chiral starting material in natural product synthesis. Nat. Prod. Rep. 1985, 2, 253–289. [Google Scholar] [CrossRef] [PubMed]
- Sokolova, A.S.; Yarovaya, O.I.; Shernyukov, A.V.; Pokrovsky, M.A.; Pokrovsky, A.G.; Lavrinenko, V.A.; Zarubaev, V.V.; Tretiak, T.S.; Anfimov, P.M.; Kiselev, O.I.; et al. New quaternary ammonium camphor derivatives and their antiviral activity, genotoxic effects and cytotoxicity. Bioorg. Med. Chem. 2013, 21, 6690–6698. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, L.F.; Bispo, M.L.; Kaiser, C.R.; Wardell, J.L.; Wardell, S.M.; Lourenço, M.C.; Bezerra, F.A.; Soares, R.P.; Rocha, M.N.; de Souza, M.V. Anti-tuberculosis evaluation and conformational study of N-acylhydrazones containing the thiophene nucleus. Arch. Pharm. Chem. Life Sci. 2014, 347, 432–448. [Google Scholar] [CrossRef] [PubMed]
- Data for compound 3k were obtained at 100(2) K with Mo-Kα radiation by means of the Rigaku Saturn724+ (2 × 2 bin mode) diffractometer. Data collection, data reduction and unit cell refinement were carried out under the control of the program CrystalClear-SM Expert 2.0. Data were collected at the NCS Crystallographic Service, based at the University of Southampton, England. Correction for absorption was achieved by a semi-empirical method based upon the variation of equivalent reflections with Rigaku program equivalent to SADABS 2007/2. The program MERCURY was used in the preparation of the Figures. SHELXL97 and PLATON were used in the calculation of molecular geometry. The structures were solved by direct methods using SHELXS-97 and fully refined by means of the program SHELXL-97 [8]. All hydrogen atoms were placed in calculated positions.
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. 2008, A64, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Crystal data collected at 100(2) K, colourless crystal: 0.42 × 0.19 ×0.03 mm; Formula:C17H21N3O2; M = 299.37: monoclinic, P21/c: a = 14.3207(10) Å, b = 10.4920(8) Å, c = 11.0426(8) Å, β = 108.670(2) °, z = 4, V = 1571.87(19)Å3; independent reflections 2817 [R(int) = 0.04] , observed reflections 2754 [I > 2σ(I)]; parameters refined 204; number of restraints 18; R(F) 0.122 [I > 2σ(I)]; Largest diff. peak 1.54 e·Å−3 . Atomic coordinates, bond lengths, angles and thermal parameters have been deposited at the Cambridge Crystallographic Data Centre, deposition number 1405803. Full details of the crystal structure determination in cif format are available in the online version, at doi: (to be inserted Copies of these can be obtained free of charge on written application to CCDC, 12 Union Road, Cambridge, CB2 1EZ, UK (fax: +44 1223 336033); on request by e-mail to deposit@ccdc.cam.ac.uk or by access to http://www.ccdc.cam.ac.uk.
- Howie, R.A.; de Souza, M.V.; Ferreira, M.L.; Kaiser, C.R.; Wardell, J.L.; Wardell, S.M. Structures of arylaldehyde 7-chloroquinoline-4-hydrazones: Supramolecular arrangements derived from N-H···N, C-H···X (X = N, O, or π) and π···π interactions. Z. Kristallogr 2010, 225, 440–447. [Google Scholar] [CrossRef]
- De Souza, M.V.; Nogueira, T.C.; Wardell, S.M.; Wardell, J.L. Crystal structures of (E)-2-(2-benzylidenehydrazinyl)quinoxalines: Persistent N-H···N intermolecular hydrogen bonds but variable π···π interactions. Z. Kristallogr. 2014, 229, 587–594. [Google Scholar] [CrossRef]
- Lindgren, E.B.; Yoneda, J.D.; Leal, K.Z.; Nogueira, A.F.; Vasconcelos, T.R.; Wardell, J.L.; Wardell, S.M. Structures of hydrazones, (E)-2-(1,3-benzothiazolyl)-NH-N=CHAr, [Ar =4-(pyridin-2-yl)phenyl, pyrrol-2-yl, thien-2-yl and furan-2-yl]: Difference in conformations and intermolecular hydrogen bonding. J. Mol. Struct. 2013, 1036, 19–27. [Google Scholar] [CrossRef]
- Carvalho, S.A.; Harrison, W.T.; Fraga, C.A.; da Silva, E.F.; Wardell, J.L.; Wardell, S.M. 5-Phenyl-2-(benzalhydrazonyl)-1,3,4-thiadiazoles, potential trypanocidal agents: Consistent dimer formation via N-H · · · N intermolecular hydrogen bonds. Z. Kristallogr. 2009, 224, 598–606. [Google Scholar] [CrossRef]
- Steed, J.W.; Gale, J.P. Crystal engineering. In Supramolecular Chemistry: From Molecules to Nanomaterials; Tiekink, E.R.T., Ed.; John Wiley & Sons Ltd.: Chichester, UK, 2012; Volume 6, pp. 2791–2828. [Google Scholar]
- Canetti, J.; Rist, E.; Grosset, R. Measurement of sensitivity of the tuberculosis bacillus to antibacillary drugs by the method of proportions. Methodology, resistance criteria, results and interpretation. Rev. Tuberc. Pneumol. 1963, 27, 212–215. [Google Scholar]
- Franzblau, S.G.; Witzig, R.S.; McLaughlin, J.C.; Torres, P.; Madico, G.; Hernandez, A.; Degnan, M.T.; Cook, M.B.; Quenzer, V.K.; Ferguson, R.M.; et al. Rapid, low-technology MIC determination with clinical Mycobacterium tuberculosis isolates by using the microplate Alamar Blue assay. J. Clin. Microbiol. 1998, 36, 362–366. [Google Scholar] [PubMed]
- Reis, R.S.; Neves, I., Jr.; Lourenço, S.L.; Fonseca, L.S.; Lourenço, M.C. Comparison of flow cytometric and Alamar Blue tests with the proportional method for testing susceptibility of Mycobacterium tuberculosis to rifampin and isoniazid. J. Clin. Microbiol. 2004, 42, 2247–2248. [Google Scholar] [CrossRef] [PubMed]
- Vanitha, J.D.; Paramasivan, C.N. Evaluation of microplate Alamar blue assay for drug susceptibility testing of Mycobacterium avium complex isolates. Diagn. Microbiol. Infect. Dis. 2004, 49, 179–182. [Google Scholar] [CrossRef] [PubMed]
- Organic Chemistry Portal. Available online: http://www.organic-chemistry.org/prog/peo/ (accessed on 30 March 2015).
- Page, P.C.; Murrell, V.L.; Limousin, C.; Laffan, D.D.; Bethell, D.; Slawin, A.M.; Smith, T.A. The First Stable Enantiomerically Pure Chiral N-H Oxaziridines: Synthesis and Reactivity. J. Org. Chem. 2000, 65, 4204–4207. [Google Scholar] [CrossRef] [PubMed]
- Barton, D.H.; McGhie, J.F.; Batten, P.L. On the oxidation of hydrazones by lead tetra-acetate. J. Chem. Soc. C 1970, 1033–1042. [Google Scholar] [CrossRef]
- Nanjundaswamy, H.M.; Pasha, M.A. Rapid, Chemoselective and Facile Synthesis of Azines by Hydrazine/I2. Synth. Commun. 2007, 37, 3417–3420. [Google Scholar] [CrossRef]
- Zhou, Y.; Dong, J.; Zhang, F.; Gong, Y. Synthesis of C1-Symmetric Chiral Secondary Diamines and Their Applications in the Asymmetric Copper(II)-Catalyzed Henry (Nitroaldol) Reactions. J. Org. Chem. 2011, 76, 588–600. [Google Scholar] [CrossRef] [PubMed]
- Mostowicz, D.; BeJzecki, C. Absolute configuration at chiral nitrogen in oxaziridines. J. Org. Chem. 1977, 42, 3917–3921. [Google Scholar] [CrossRef]
- Francesca, C.; Leriverend, C.; Metzner, P. Carbon versus sulfur addition of nucleophiles to sulfines: The case of amines. Tetrahedron Lett. 1993, 34, 6741–6742. [Google Scholar]
- Markovic, S.; Markovic, V.; Joksovic, M.D.; Todorovic, N.; Joksovic, L.; Divjakovic, V.; Trifunovic, S. Debromination of endo-(+)-3-bromocamphor with primary amines. J. Braz. Chem. Soc. 2013, 24, 1099–1108. [Google Scholar]
- Saccardi, P.; Romagnoli, E. Some new compounds of camphor with amines. Arch. Farmacol. Sper. Sci. Affin 1927, 43, 197–204. [Google Scholar]
- Love, B.E.; Ren, J. Synthesis of Sterically Hindered Imines. J. Org. Chem. 1993, 58, 5556–5557. [Google Scholar] [CrossRef]
- Suquet, M.; Callet, G.; Raveux, R. Antifibrillant compounds derived from phenyl (2-hydroxy-phenyl)amine and from exo-2-[(2-hydroxyphenyl) amino]-bornane. Bull. Soc. Chim. France 1962, 444–455. [Google Scholar]
- Botteghi, C.; Schionato, A.; Chelucci, G.; Brunner, H.; Kuerzinger, A.; Obermann, U. Asymmetric catalysis. XLVI. Enantioselective hydrosilylation of ketones with [Rh(COD)Cl]2 and optically active nitrogen ligands. J. Organomet. Chem. 1989, 370, 17–31. [Google Scholar] [CrossRef]
- Li, S.J.; Mi, A.; Yang, G.; Jiang, Y. Facile preparation of some highly hindered chiral 1,2-diphenyl-2-(N-monoalkyl)amino alcohols and N-benzylbornamine. Synth. Commun. 1992, 22, 1497–1503. [Google Scholar] [CrossRef]
- Bhat, M.A.; Al-Omar, M.A. Synthesis, characterization, and in vitro anti-Mycobacterium tuberculosis activity of terpene Schiff bases. Med. Chem. Res. 2013, 22, 4522–4528. [Google Scholar] [CrossRef]
- Mosmann, T.J. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Meth. 1983, 65, 55–63. [Google Scholar] [CrossRef]
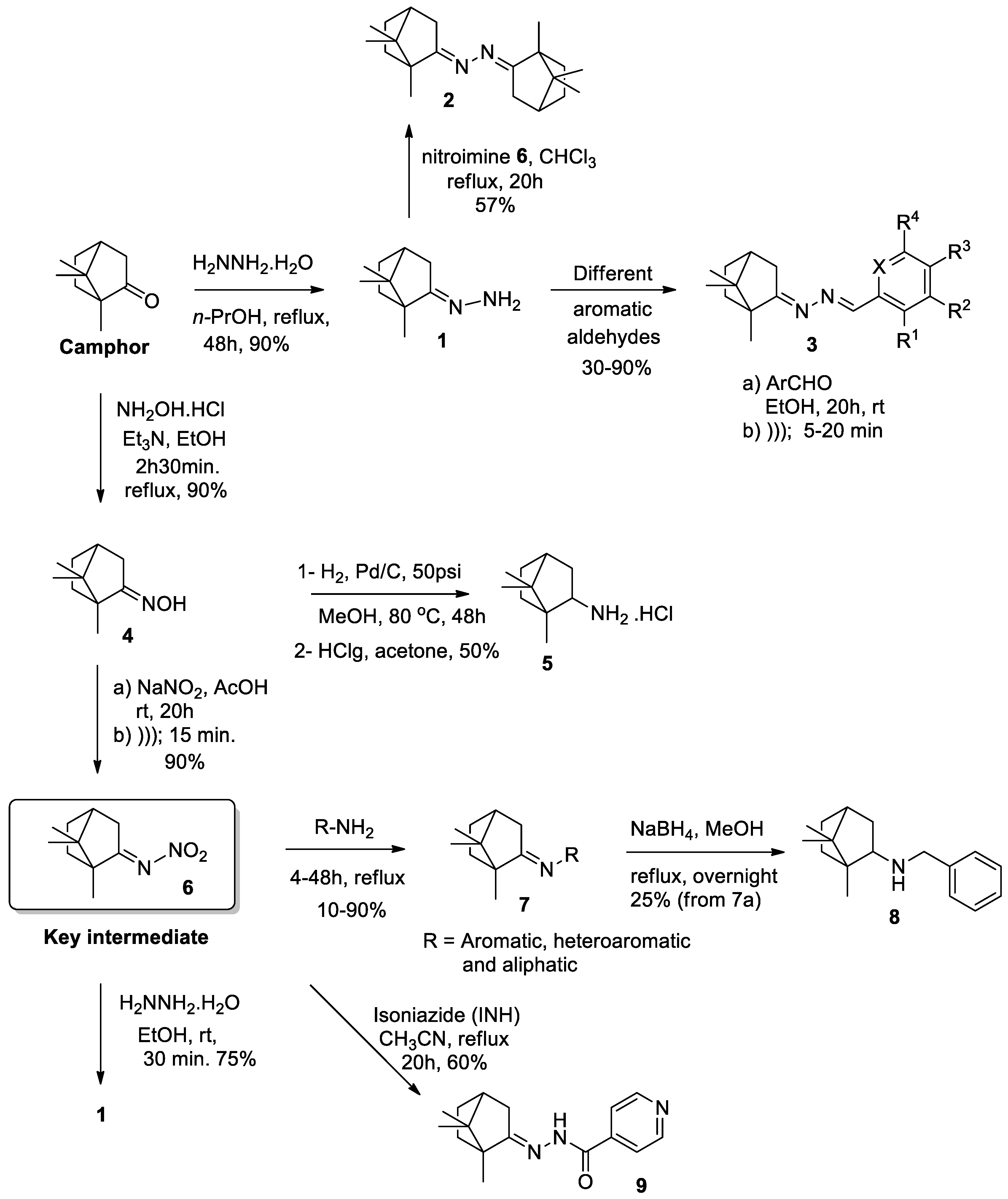
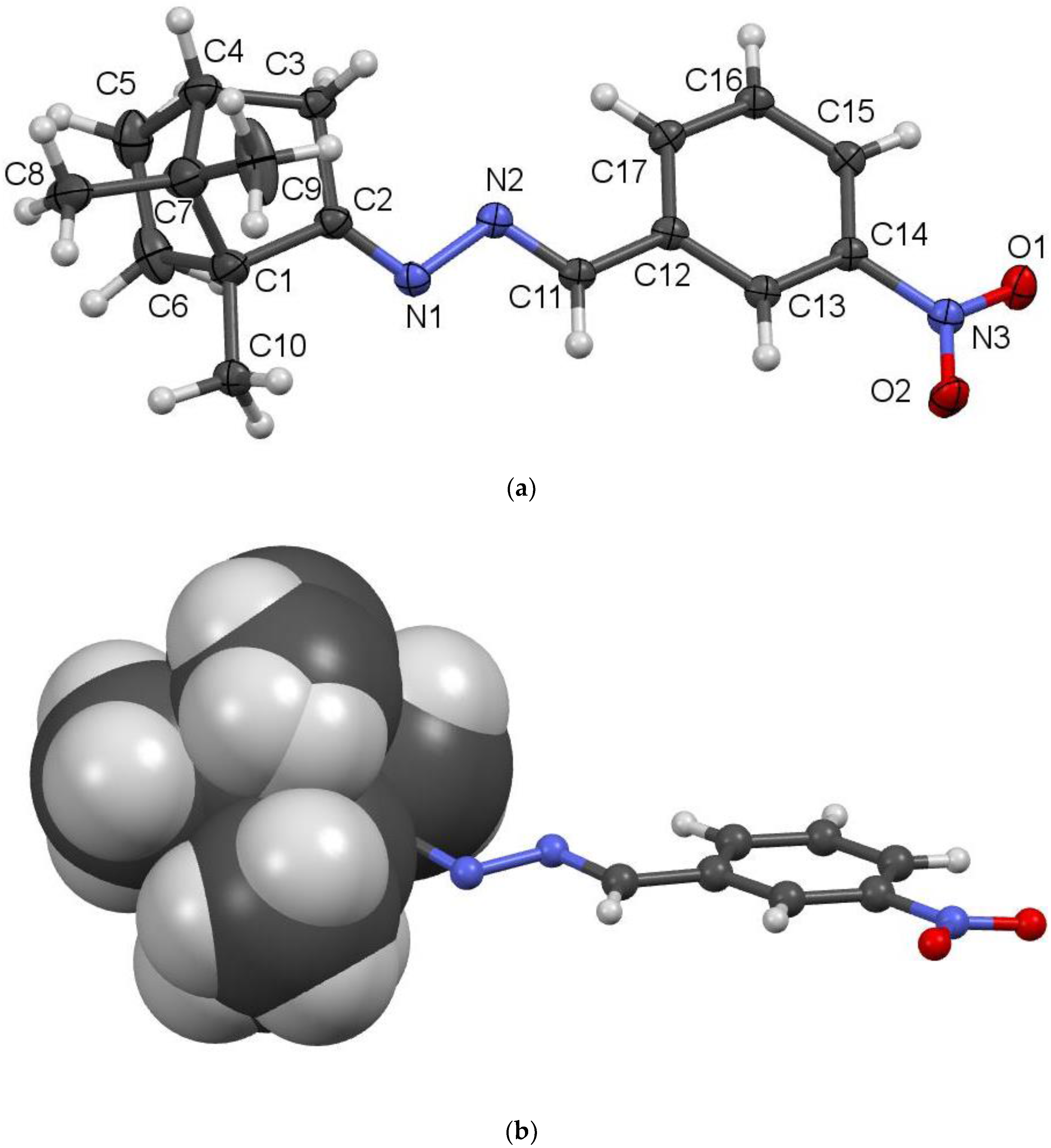

{kind=link}
{kind=link}
| Final Compound (3) | R1 | R2 | R3 | R4 | X | MIC (µg/mL) | Time Classic Method (h) | Yield (%) | Time Ultrasound Method a (min) |
|---|---|---|---|---|---|---|---|---|---|
| a | H | H | H | H | CH | Res | 20 | 64 | 5 |
| b | H | H | F | H | CH | Res | 20 | 80 | 5 |
| c | H | H | Cl | H | CH | Res | 20 | 71 | 5 |
| d | H | H | Br | H | CH | 50 | 20 | 57 | 5 |
| e | H | H | OH | H | CH | Res | 20 | 70 | 60 |
| f | H | H | OCH3 | H | CH | 100 | 20 | 75 | 5 |
| g | H | H | NO2 | H | CH | ins | 20 | 36 | 5 |
| h | H | Cl | H | H | CH | 100 | 20 | 70 | 5 |
| i | H | CN | H | H | CH | Res | 20 | 85 | 5 |
| j | H | OCH3 | H | H | CH | Res | 20 | 61 | 5 |
| k | H | NO2 | H | H | CH | ins | 20 | 50 | 5 |
| l | F | H | H | H | CH | Res | 20 | 68 | 5 |
| m | Cl | H | H | H | CH | 100 | 20 | 51 | 5 |
| n | Br | H | H | H | CH | 50 | 20 | 55 | 5 |
| o | OH | H | H | H | CH | 50 | 20 | 64 | 5 |
| p | NO2 | H | H | H | CH | 25 | 20 | 77 | 5 |
| q | Cl | Cl | H | H | CH | Res | 20 | 78 | 5 |
| r | OH | OH | H | H | CH | 100 | 20 | 73 | 7 |
| s | OH | H | OH | H | CH | 100 | 20 | 30 | 10 |
| t | H | OH | OH | H | CH | 100 | 20 | 38 | 5 |
| u | OH | H | H | OH | CH | Res | 20 | 64 | 5 |
| v | OH | H | OCH3 | H | CH | 50 | 20 | 78 | 18 |
| w | OH | H | H | NO2 | CH | Res | 20 | 54 | 5 |
| x | H | H | H | H | N | 100 | 20 | 38 | 5 |
| C2-N1 | 1.272(5) | O1-N3 | 1.218(5) |
| N1-N2 | 1.420(4) | O2-N3 | 1.226(5) |
| N2-C11 | 1.268(5) | C11-C12 | 1.467(5) |
| N2-N1-C2 | 112.2(3) | N1-N2-C11 | 112.0(3) |
| N2-C11-C12 | 121.9(4) | O1-N3-O2 | 123.5(4) |
| C2-N1-N2-C11 | 173.4(3) | N1-N2-C11-C12 | 175.6(3) |
| Final Compound (7) | R | MIC (µg/mL) | Condition, Time | Yield (%) |
|---|---|---|---|---|
| a | –CH2Ph | Res | CHCl3, reflux, 4 h | 55 |
| b | –CH2(CH2)2CH3 | Res | CHCl3, reflux, 4 h | 70 |
| c |  | Res | CHCl3, reflux, 3 h | 65 |
| d |  | Res | CHCl3, reflux, 3 h | 73 |
| e |  | Res | CH3CN, reflux, 48 h | 79 |
| f |  | 100 | CH3CN, reflux, 30 h | 52 |
| g |  | 100 | CH3CN, reflux, 43 h | 73 |
| h |  | Res | CH3CN, reflux, 17 h | 81 |
| i |  | Res | CH3CN, reflux, 48 h | 90 |
| j |  | 3.12 | CH3CN, reflux, 20 h | 53 |
| k |  | Res | CH3CN, reflux, 48 h | 46 |
| l |  | Res | CH3CN, reflux, 64 h | 39 |
| m |  | Res | CH3CN, reflux, 24 h | 10 |
| n |  | Res | CH3CN, reflux, 72 h | 15 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Da Silva, E.T.; Da Silva Araújo, A.; Moraes, A.M.; De Souza, L.A.; Silva Lourenço, M.C.; De Souza, M.V.N.; Wardell, J.L.; Wardell, S.M.S.V. Synthesis and Biological Activities of Camphor Hydrazone and Imine Derivatives. Sci. Pharm. 2016, 84, 467-483. https://doi.org/10.3390/scipharm84030467
Da Silva ET, Da Silva Araújo A, Moraes AM, De Souza LA, Silva Lourenço MC, De Souza MVN, Wardell JL, Wardell SMSV. Synthesis and Biological Activities of Camphor Hydrazone and Imine Derivatives. Scientia Pharmaceutica. 2016; 84(3):467-483. https://doi.org/10.3390/scipharm84030467
Chicago/Turabian StyleDa Silva, Emerson T., Adriele Da Silva Araújo, Adriana M. Moraes, Leidiane A. De Souza, Maria Cristina Silva Lourenço, Marcus V. N. De Souza, James L. Wardell, and Solange M. S. V. Wardell. 2016. "Synthesis and Biological Activities of Camphor Hydrazone and Imine Derivatives" Scientia Pharmaceutica 84, no. 3: 467-483. https://doi.org/10.3390/scipharm84030467
APA StyleDa Silva, E. T., Da Silva Araújo, A., Moraes, A. M., De Souza, L. A., Silva Lourenço, M. C., De Souza, M. V. N., Wardell, J. L., & Wardell, S. M. S. V. (2016). Synthesis and Biological Activities of Camphor Hydrazone and Imine Derivatives. Scientia Pharmaceutica, 84(3), 467-483. https://doi.org/10.3390/scipharm84030467