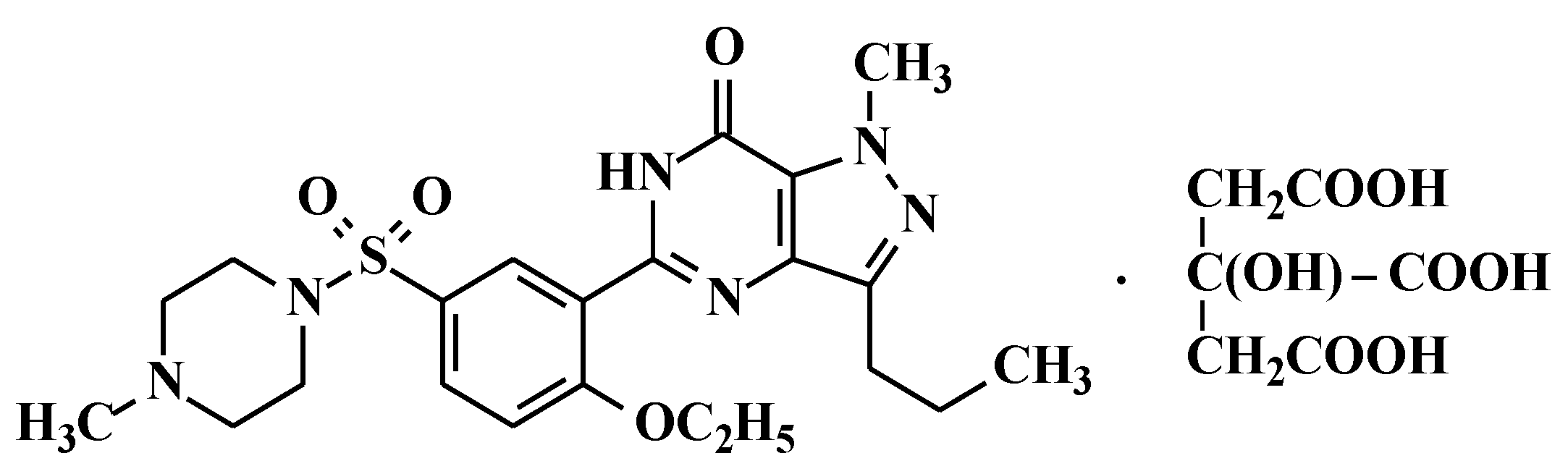
2. Materials and Methods
General Procedures: All melting points were determined with the Polmon melting point apparatus(Polmon Instruments Pvt. Ltd., Hyderabad, India). 1H-NMR and 13C-NMR spectra were recorded on a Bruker 300 & 500 spectrometer (Bruker, Fällanden, Switzerland). Chemical shifts (δ) were reported in ppmdownfield with TMS as internal standard, multiplicities are described as s: singlet, d: doublet, t: triplet, dd: double doublet, m: multiplet, brs: broad singlet. The HRMS spectra were measured on the Perkin Elmer PE SCIEX-API 2000 mass spectrometer (Waters, MA, United States). An analytical HPLC (Waters, MA, United States) was run with the Symmetry C18, 210 × 4.6 mm column at 290 nm.
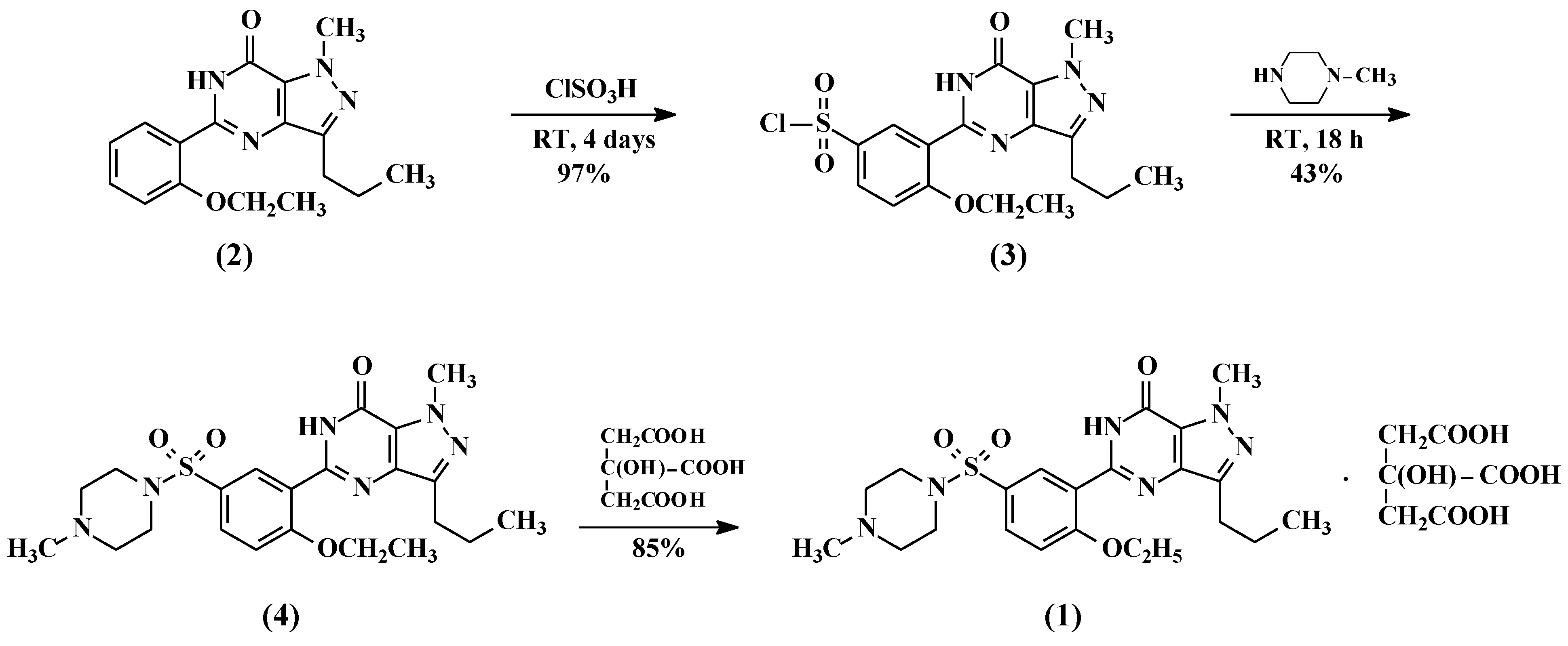
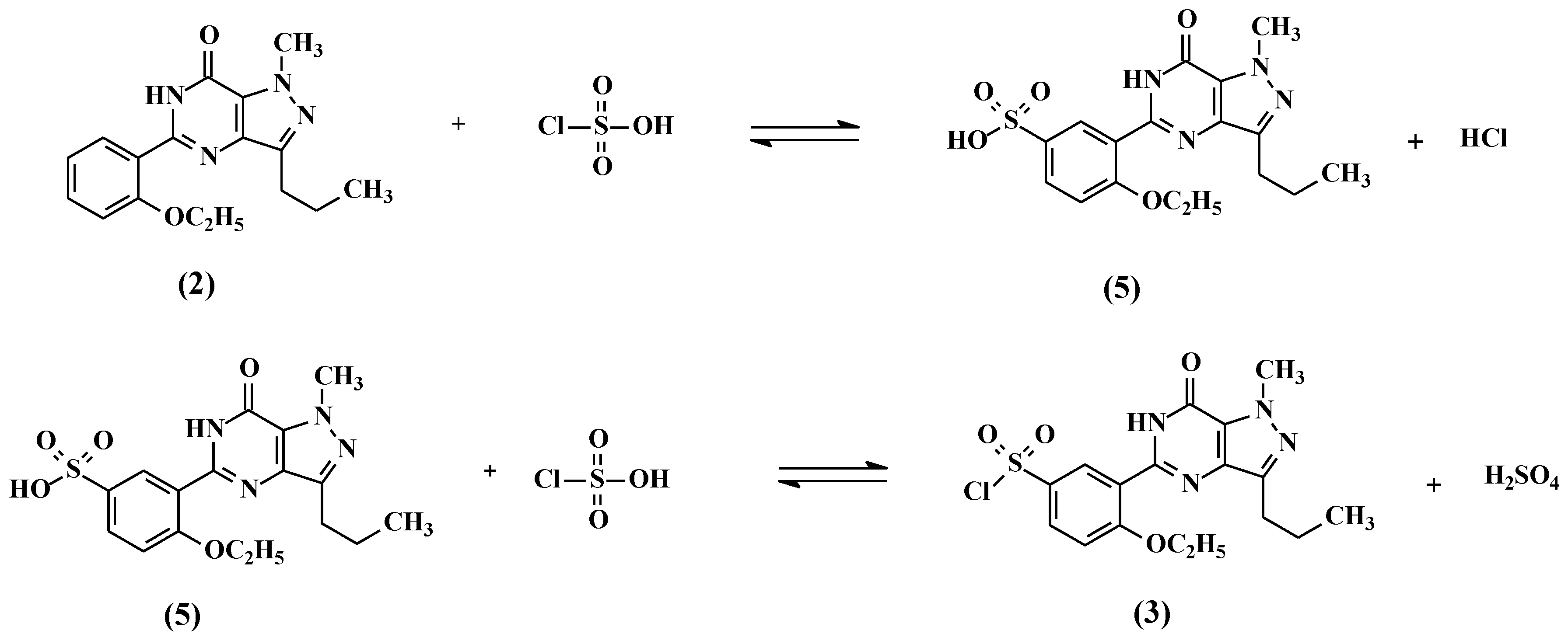
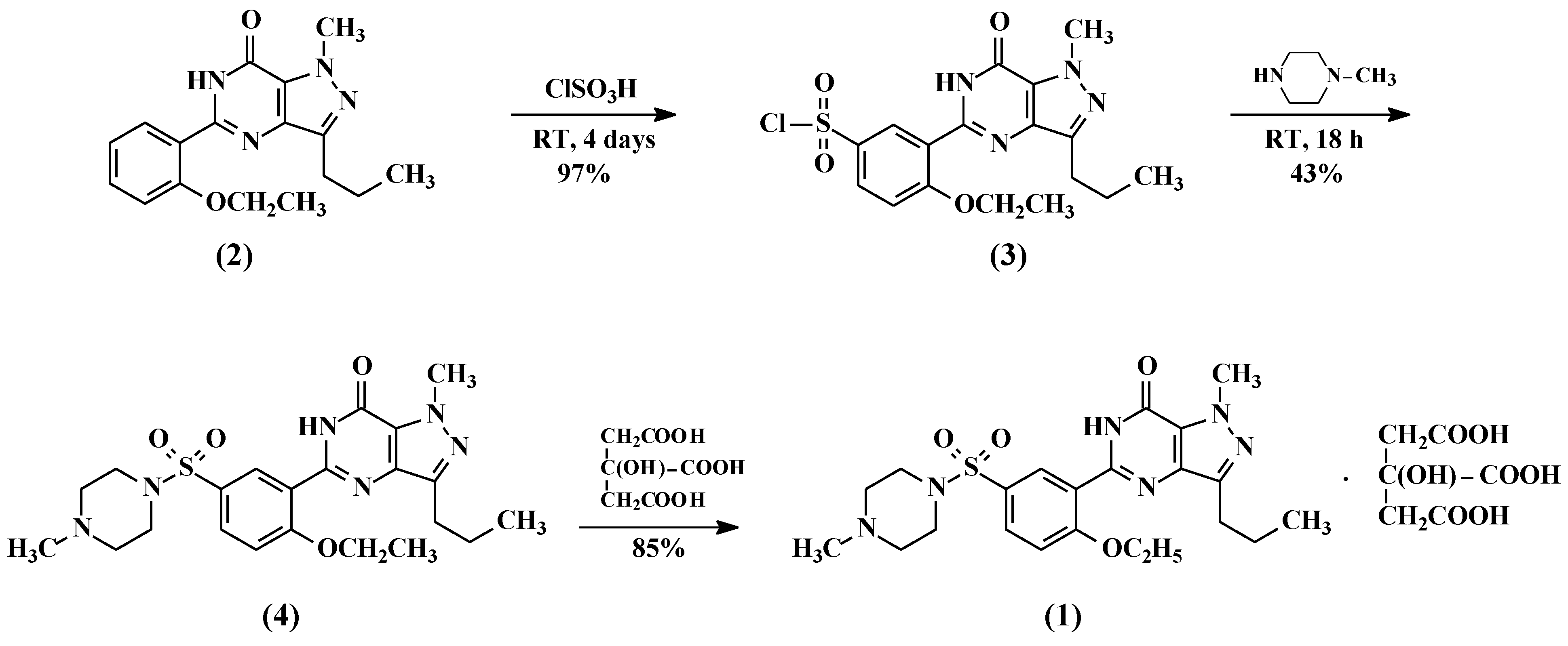
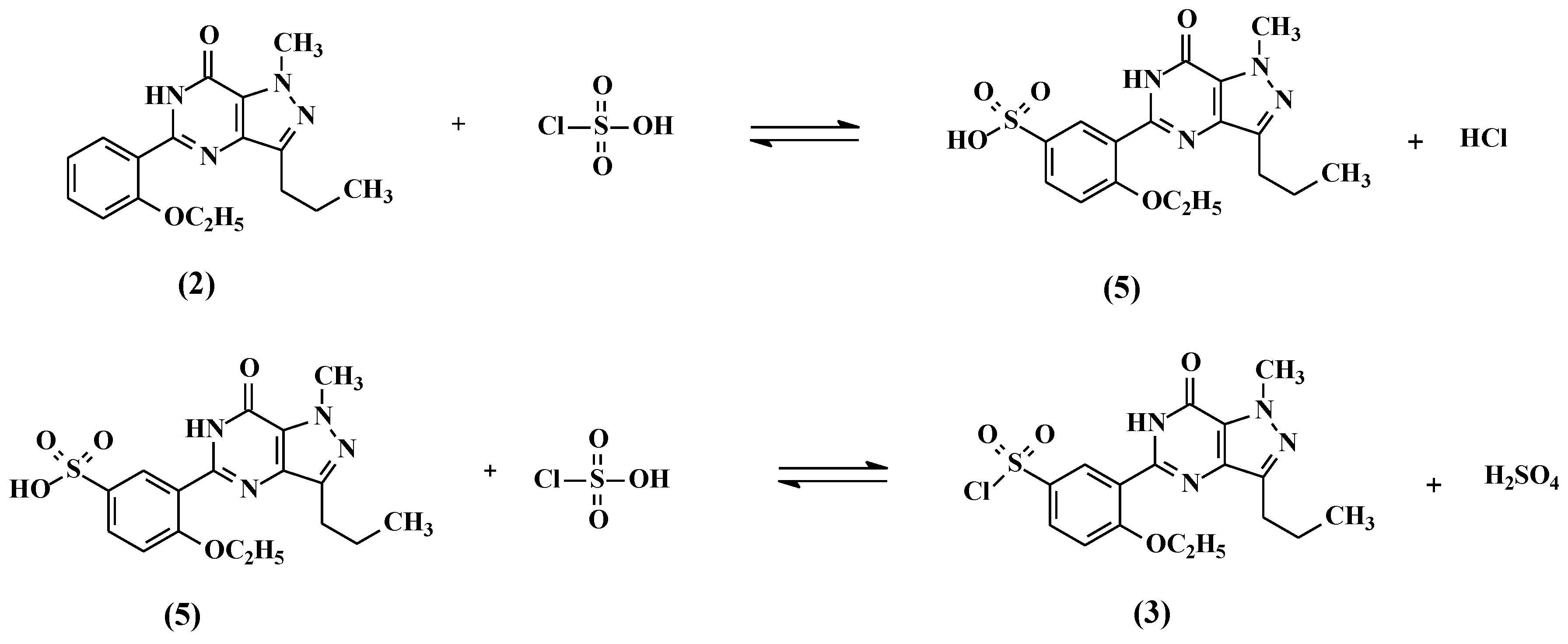
2.1. Preparation of 5-(5-Chlorosulfonyl-2-ethoxyphenyl)-1-methyl-3-n-propyl-1,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one (3)
5-(2-ethoxyphenyl)-1-methyl-3-n-propyl-1,6-dihydro-7H-pyrazolo[4,3-d]pyrimid-7-one (2, 25 g, 80.13 mmol) followed by thionyl chloride (9.53 g, 80.13 mmol) were added to chlorosulfonic acid (50 mL) portion-wise at 0–10 °C. The reaction mass temperature was raised to 20–30 °C and stirred for 4 h to complete the reaction. The reaction mass was poured onto ice (~500 g) slowly and the product was extracted with dichloromethane (250 mL). The dichloromethane layer was separated and washed with 5% w/w aqueous sodium bicarbonate (100 mL). The dichloromethane layer containing compound 3 was taken to the next step as such.
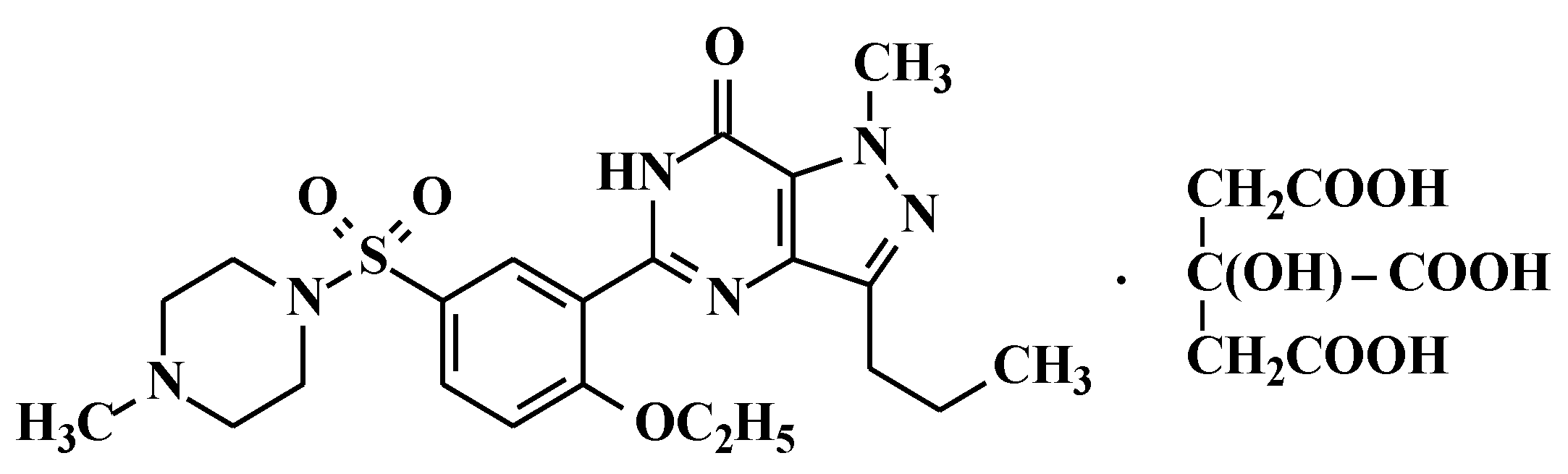
2.2. Preparation of 5-[2-Ethoxy-5-(4-methylpiperazinylsulfonyl)phenyl]-1-methyl-3-n-propyl-1,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one (Sildenafil, 4)
N-methylpiperazine (9.6 g, 96 mmol) was added to the dichloromethane layer containing sulfonyl chloride compound 3 (obtained from example 1) and stirred for 1 h at 20–25 °C. Then the reaction mass was washed with 5% w/w aqueous sodium bicarbonate (100 mL) followed by demineralised (DM) water (100 mL). The dichloromethane layer was concentrated at <50 °C and methanol was added to the residue to crystallize the product. The product was filtered and dried at 55–60 °C under vacuum to obtain 34 g of pure sildenafil (90% yield).
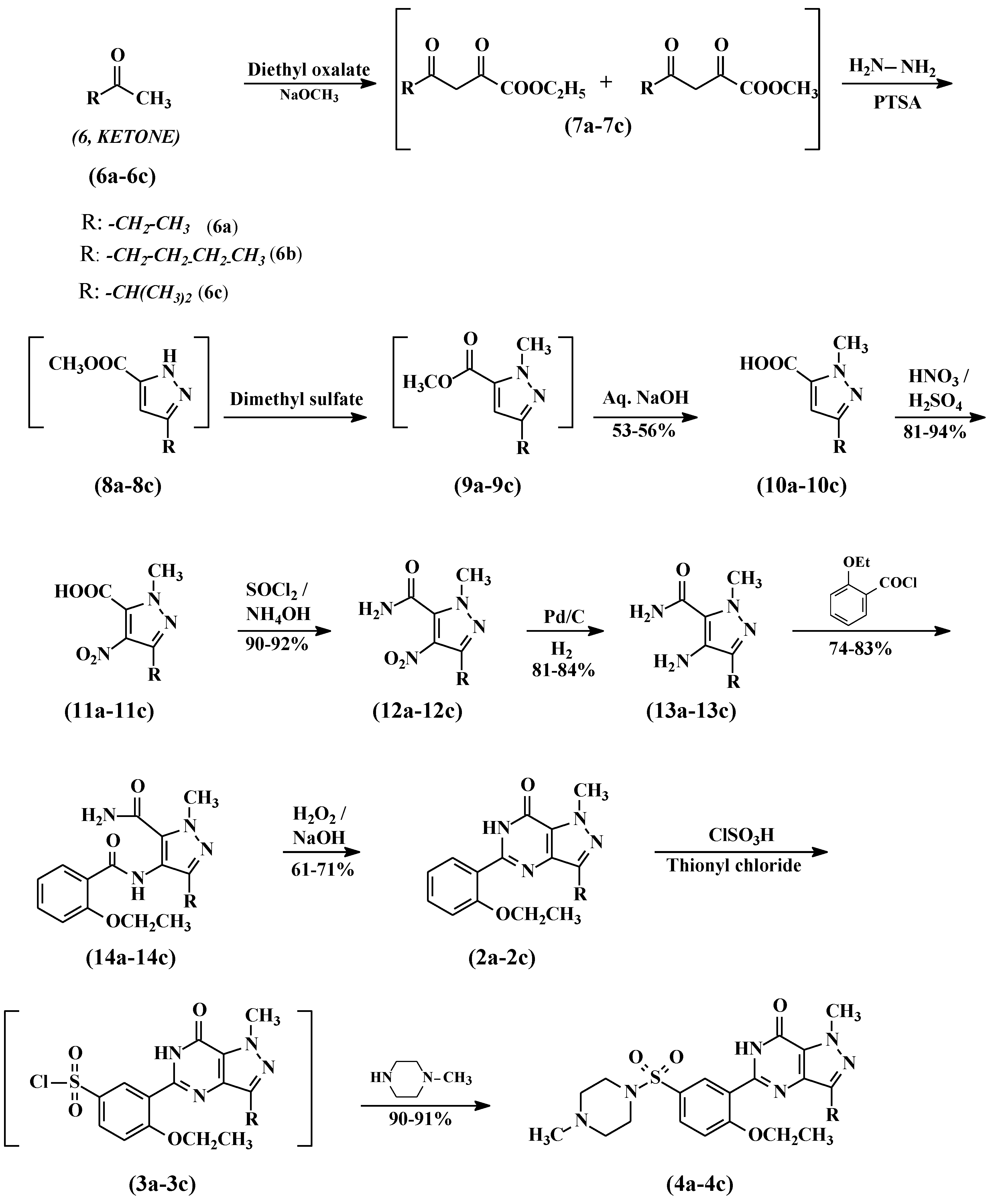
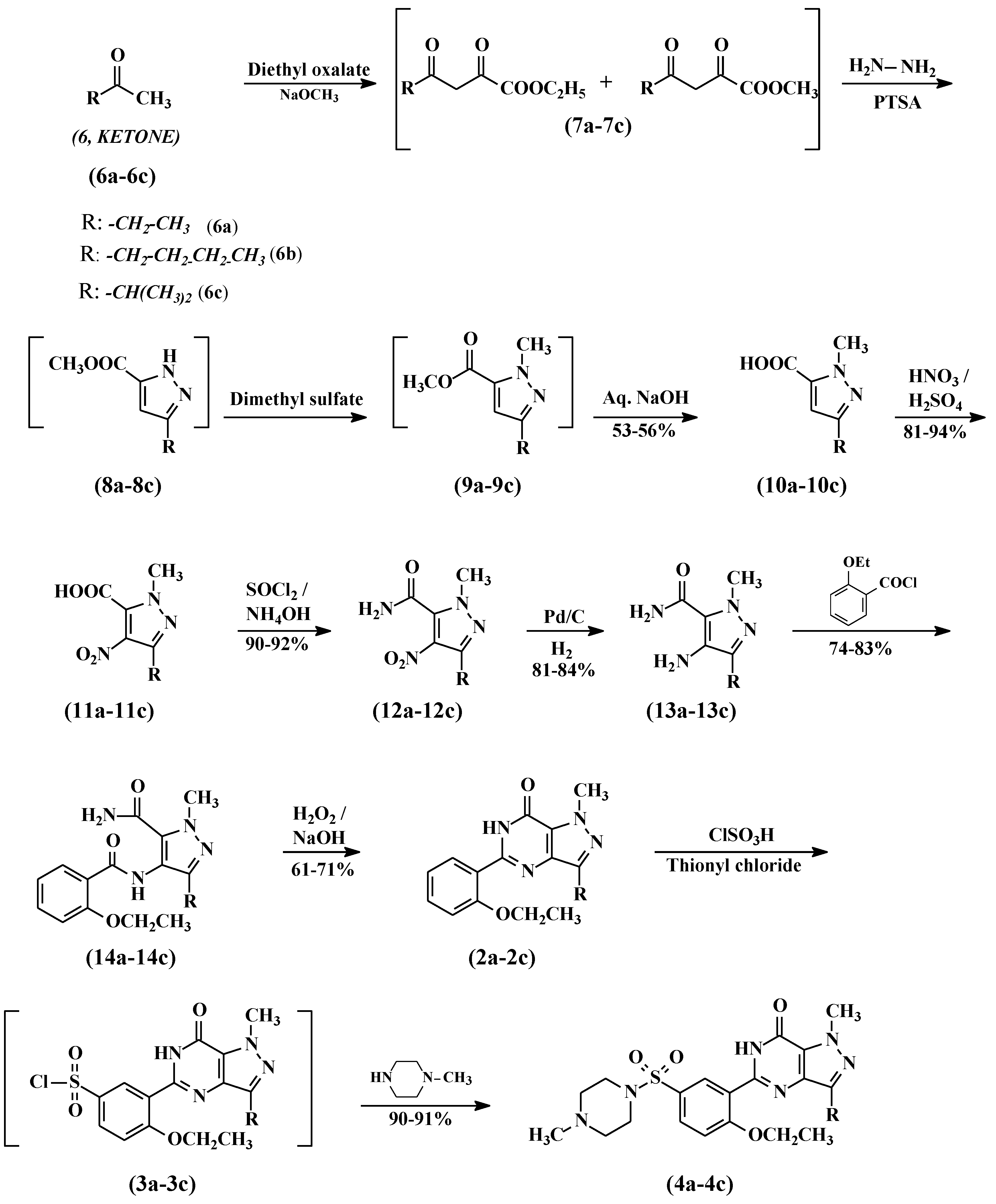
2.3. General Procedure for the Preparation of Compounds 10a–10c
To a mixture of diethyl oxalate (100 g, 685 mmol) and appropriate ketone (685 mmol), powdered sodium methoxide (37.8 g, 685 mmol) was added in portions at 20 °C. The reaction was heated to 50–55 °C and stirred for 1 h, the reaction was cooled to 20–25 °C, then diluted with ethyl acetate (200 mL) and DM water (200 mL). Further, the pH was adjusted to 1.8 with diluted hydrochloric acid and separated into two layers. The ethyl acetate layer was washed with 20% aqueous sodium chloride and dried over anhydrous sodium sulfate. The ethyl acetate layer was cooled to ~10 °C and para-toluensulfonic acid (2.96 g, 15.58 mmol) was added followed by hydrazine hydrate (30 mL, 80%) over a period of 2 h at 10–15 °C. The reaction mass was stirred for 1 h and diluted with water (200 mL). The pH of the reaction mass was adjusted to 7.2 with 30% aqueous sodium hydroxide and the layers were separated. The bottom aqueous layer was extracted with ethyl acetate (100 mL) and mixed with the first ethyl acetate layer. The combined ethyl acetate layers were concentrated at 40–45 °C under reduced pressure to obtain the title compounds 7a–7c as an oily residue.
The residue was cooled to 20–25 °C and dimethyl sulphate (86.31 g, 685 mmol) was added in 2 h at 20–25 °C. The temperature was raised to 55–60 °C and maintained for 2 h. The reaction mixture was cooled to 10–15 °C and poured into a mixture of dichloromethane (500 mL) and DM water (500 mL). The pH of the reaction was adjusted to 7.2 with 30% aqueous sodium hydroxide and the two layers were separated. The organic layer was concentrated at 40–45 °C to obtain compounds 9a–9c as an oily mass. Five N aqueous sodium hydroxide (200 mL, 1 mol) was added to the oily residue and the mixture was heated to 80 °C for 2 h. The reaction temperature was brought down to 25–30 °C and the pH was adjusted to ~1.0 with hydrochloric acid. The solid precipitate was collected by filtration, washed with water, and dried to obtain the carboxylic acid compounds 10a–10c.
Preparation of 3-ethyl-1-methyl-1H-pyrazole-5-carboxylic acid (10a). Compound 10a was prepared from 2-butanone as a pale yellow solid. Yield 53%, 1H-NMR (DMSO-d6) δ: 1.131–1.182 (t, 3H, CH3), 2.510–2.549 (m, 2H, CH2), 3.759 (s, 3H, CH3), 6.91 (s, 1H, COOH). Mass: 155.217 (M+H)+.
Preparation of 3-butyl-1-methyl-1H-pyrazole-5-carboxylic acid (10b). Compound 10b was prepared from 2-hexanone as a pale yellow solid. Yield 55%, 1H-NMR (DMSO-d6) δ: 0.86–0.91 (q, 3H, CH3), 1.27–1.34 (m, 2H, CH2), 1.50–1.57 (m, 2H, CH2), 2.49–2.54 (t, 2H, CH2), 3.99 (s, 3H, N-CH3), 6.60 (s, 1H, CH), 13.18 (bs, 1H, COOH). Mass: 183.22 (M + H)+.
Preparation of 3-isopropyl-1-methyl-1H-pyrazole-5-carboxylic acid (10c). Compound 10c was prepared from 3-methyl-2-butanone as a pale yellow solid. Yield 56%, 1H-NMR (DMSO-d6) δ: 1.17–1.20 (m, 6H, 2xCH3), 2.80–2.92 (m, 1H, CH(CH3)2), 4.0 (s, 3H, N-CH3), 6.63 (s, 1H, COOH). Mass: 169.27 (M + H)+.
2.4. General Procedure for the Preparation of Compounds 11a–11c
Sodium nitrate (380 mmol) was slowly added to sulfuric acid (192 mL) in about 30 min at 0–5 °C. The contents were heated to 25–30 °C and stirred for 1 h. Thereafter, carboxylic acid (10a–10c, 380 mmol) was added portion–wise, keeping the temperature below 40 °C. After the addition, the reaction was heated at 60 °C and maintained for 18 h. The reaction was cooled to room temperature before it was poured onto ice, then filtration and drying gave 4-nitrocarboxylic acid (11a–11c) as a white solid.
Preparation of 3-ethyl-1-methyl-4-nitro-1H-pyrazole-5-carboxylic acid (11a). Compound 11a was prepared from 10a as a white solid. Yield 82%, 1H-NMR (DMSO-d6) δ: 1.17–1.26 (t, 3H, CH3), 2.807–2.857 (m, 2H, CH2), 3.894 (s, 3H, N-CH3). Mass: 200.27 (M + H)+.
Preparation of 3-butyl-methyl-4-nitro-1H-pyrazole-5-carboxylic acid (11b). Compound 11b was prepared from 10b as a white solid. Yield 94%, 1H-NMR (DMSO-d6) δ: 0.874–0.966 (m, 3H, CH3), 1.328–1.397 (m, 2H, CH2), 1.546–1.646 (m, 2H, CH2), 2.766–2.817 (t, 2H, CH2), 3.912 (s, 3H, N-CH3). Mass: 228.22 (M + H)+.
Preparation of 3-isopropyl-1-methyl-4-nitro-1H-pyrazole-5-carboxylic acid (11c). Compound 11c was prepared from 10c as a white solid. Yield 81%, 1H-NMR (DMSO-d6) δ: 1.23–1.29 (m, 6H, 2CH3), 3.32–3.41 (m, 1H, CH(CH3)2), 3.90 (s, 3H, N-CH3). Mass: 214.19 (M + H)+.
2.5. General Procedure for the Preparation of Compounds 12a–12c
4-Nitrocarboxylic acid (11a–11c, 352 mmol) was added to thionyl chloride (200 mL) and the resulting mixture was heated under reflux for 3 h. Thereafter, excess thionyl chloride was removed by evaporation under vacuum. The oily residue was dissolved in acetone (750 mL) and the pH was adjusted to 10 with aqueous ammonia. Acetone was distilled off and the mass was diluted by adding DM water (750 mL). The resulting slurry was filtered and washed with DM water to provide 4-nitrocarboxamide (12a–12c) as a pale yellow solid.
Preparation of 3-ethyl-1-methyl-4-nitro-1-H-pyrazole-5-carboxamide (12a). Compound 12a was prepared from compound 11a as a pale yellow solid. Yield 92%, 1H-NMR (DMSO-d6) δ: 1.045–1.083 (t, 3H, CH3), 2.359–2.456 (q, 2H, CH2), 3.92 (s, 3H, N-CH3), 7.483–7.695 (brs, 2H, NH2). Mass: 199.18 (M + H)+.
Preparation of 3-butyl-1-methyl-4-nitro-1H-pyrazole-5-carboxamide (12b). Compound 12b was prepared from compound 11b as a pale yellow solid. Yield 90%, 1H-NMR (DMSO-d6) δ: 0.88–0.93 (t, 3H, CH3), 1.27–1.39 (m, 2H, CH2), 1.55–1.65 (m, 2H, CH2), 2.80–2.85 (t, 2H, CH2), 3.78 (s, 3H, N-CH3), 8.23, 8.42 (2brs, 2H, CONH2). Mass: 227.3 (M + H)+.
Preparation of 3-isopropyl-1-methyl-4-nitro-1H-pyrazoIe-5-carboxamide (12c). Compound 12c was prepared from compound 11c as a pale yellow solid. Yield 90%, 1H-NMR (DMSO-d6) δ: 1.23–1.26 (m, 6H, 2xCH3), 3.34–3.51 (m, 1H, CH(CH3)2), 3.79 (s, 3H, N-CH3), 8.23, 8.43 (2brs, 2H, CONH2). Mass: 213.21 (M + H)+.
2.6. General Procedure for the Preparation of Compounds 13a–13c
A solution of 4-nitro carboxamide (12a–12c, 94 mmol) in methanol (400 mL) and 5% Pd/C (5 g, 50% wt) was charged into a one-litre hydrogenator at 25–30 °C. The reaction mixture was hydrogenated with 4 kg/cm2 pressure at 25–30 °C for ~5 h. After completion of the reaction, the hydrogen pressure was released and carefully filtered to remove the catalyst under the nitrogen atmosphere. The filtrate was concentrated under reduced pressure at 40–45 °C and the product was crystallized by adding ethyl acetate (250 mL). The product was filtered and dried to obtain the 4-aminocarboxamide compound (13a–13c).
Preparation of 3-ethyl-1-methyl-4-amino-1-H-pyrazole-5-carboxamide (13a). Compound 13a was prepared from compound 12a. Yield 84%, 1H-NMR (DMSO-d6) δ: 1.055–1.108 (t, 3H, CH3), 2.359–2.456 (q, 2H, CH2), 3.994 (s, 3H, N-CH3), 7.483–7.695 (brs, 2H, NH2). Mass: 169.20 (M + H)+.
Preparation of 3-butyl-1-methyl-4-amino-1-H-pyrazole-5-carboxamide (13b). Compound 13b was prepared from compound 12b. Yield 82%, 1H-NMR (DMSO-d6) δ: 0.86–0.93 (m, 3H, CH3), 1.27–1.35 (m, 2H, CH2), 1.48–1.53 (m, 2H, CH2), 2.42–2.50 (m, 2H, CH2), 3.85 (s, 3H, N-CH3), 4.09 (brs, 2H, NH2), 7.48 (brs, 2H, CONH2). Mass: 197.25 (M + H)+.
Preparation of 3-isopropyl-1-methyl-4-amino-1-H-pyrazole-5-carboxamide (13c). Compound 13c was prepared from compound 12c. Yield 81%, 1H-NMR (DMSO-d6) δ: 1.10–1.17 (m, 6H, 2CH3), 2.92–3.02 (m, 1H, CH(CH3)2), 3.94 (s, 3H, N-CH3) 4.08 (brs, 2H, NH2), 7.50 (brs, 2H, CONH2). Mass: 183.1 (M + H)+.
2.7. General Procedure for the Preparation of Compounds 14a–14c
To a solution of 4-amino carboxamide (13a–13c, 50 mmol) and triethylamine (10.12 g, 100 mmol) in dichloromethane (250 mL), 2-ethoxybenzoyl chloride (10 g, 54.5 mmol) was added at 0–5 °C. The resulting mixture was allowed to warm to room temperature and it was stirred for a further 2 h. The reaction mixture was washed with DM water twice and the organic layer was concentrated under reduced pressure. Then n-hexane (250 mL) was added to the residue and stirred for 1 h to precipitate the product. The product was collected by filtration and dried to obtain pyrazole carboxamide, compounds (14a–14c).
Preparation of 3-ethyl-4-(2-ethoxybenzamido)-1-methyl-1H-pyrazole-5-carboxamide (14a). Compound 14a was prepared from compound 13a. Yield 74%, 1H-NMR (DMSO-d6) δ: 1.11–1.16 (t, 3H, CH2CH3), 1.37–1.41 (t, 3H, OCH2CH3), 2.45–2.51 (q, 2H, CH2CH3), 3.92 (s, 3H, N-CH3), 4.17–4.23 (q, 2H, OCH2CH3), 7.03–7.08 (t, 1H, Ar), 7.16–7.19 (d, 1H, Ar), 7.47–7.53 (m, 1H, Ar), 7.64–7.67 (m, 1H, Ar), 7.37 & 7.76 (2brs,2H, CONH2), 9.49 (s, 1H, -CONH). Mass: 317.35 (M + H)+.
Preparation of 3-butyl-4-(2-ethoxybenzamido)-1-methyl-1H-pyrazole-5-carboxamide (14b). Compound 14b was prepared from compound 13b. Yield 83%, 1H-NMR (DMSO-d6) δ: 0.83–0.88 (t, 3H, CH2CH3), 1.27–1.41 (t, 5H, CH2 & CH3), 1.52–1.57 (m, 2H, CH2), 2.44–2.51 (m, 2H, OCH2), 3.92 (s, 3H, N-CH3), 4.17–4.24 (q, 2H, OCH2), 7.03–7.08 (m, 1H, Ar), 7.17–7.19 (dd, 1H, Ar), 7.47–7.53 (m, 1H, Ar), 7.63 & 7.66 (m, 1H, Ar), 7.36 & 7.76 (2brs, 2H, CONH2), 9.48 (s, 1H, CONH). Mass: 345.31 (M + H)+.
Preparation of 3-isopropyl-4-(2-ethoxybenzamido)-1-methyl-1H-pyrazole-5-carboxamide (14c). Compound 14c was prepared from compound 13c. Yield 81%, 1H-NMR (DMSO-d6) δ: 1.14–1.22 (m, 6H, CH3), 1.37–1.41 (t, 3H, CH3), 2.88–2.97 (m, 1H, CH(CH3)2), 3.92 (s, 3H, N-CH3), 4.17–4.24 (q, 2H, OCH2CH3), 7.03–7.08 (m, 1H, Ar), 7.17–7.19 (dd, 1H, Ar), 7.47–7.53 (m, 1H, Ar), 7.63–7.66 (m, 1H, Ar), 7.33 & 7.74 (2brs, 2H, CONH2), 9.45 (s, 1H, CONH). Mass: 331.42 (M + H)+.
2.8. General Procedure for the Preparation of Compounds 2a–2c
Pyrazole carboxamide compound (14a–14c, 42.42 mmol) was added portion-wise to a solution of sodium hydroxide (3.4 g, 85 mmol) and 30% hydrogen peroxide solution (10 mL) in DM water (200 mL). Ethanol (50 mL) was added and the resulting mixture was heated under reflux for 8 h. Thereafter, the reaction mixture was cooled and evaporated under vacuum. The resulting solid was treated with 2 N hydrochloric acid (100 mL) at 25–30 °C and the mixture was extracted with dichloromethane (2 × 200 mL). The combined organic extracts were washed successively with saturated aqueous sodium carbonate (100 mL) and saturated sodium chloride solution (100 mL). The dichloromethane layer was concentrated and n-hexane was added to the residue to crystallize the product. The product was collected by filtration and dried to obtain the cyclized compounds 2a–2c.
Preparation of 3-ethyl-5-(2-ethoxyphenyl)-1-methyl-7H-pyrazolo[4,3-d]pyrimidin-7-one (2a). Compound 2a was prepared from compound 14a as a white solid. Yield 61%, 1H-NMR (DMSO-d6) δ: 1.26–1.35 (m, 6H, 2CH3), 2.78–2.86 (q, 2H, CH2), 4.09–4.14 (q, 2H, OCH2), 4.15 (s, 3H, N-CH3), 7.04–7.09 (t, 1H, Ar), 7.14–7.16 (dd, 1H, Ar), 7.46–7.51 (m, 1H, Ar), 7.65–7.68 (t, 1H, Ar), 11.97 (s, 1H, NH). Mass: 299.1 (M + H)+.
Preparation of 3-butyl-5-(2-ethoxyphenyl)-1-methyl-7H-pyrazolo[4,3-d]pyrimidin-7-one (2b). Compound 2b was prepared from compound 14b as a white solid. Yield 65%, 1H-NMR (DMSO-d6) δ: 0.88–0.93 (t, 3H, CH2CH3), 1.31–1.39 (m, 5H, CH2 & CH3), 1.68–1.73 (m, 2H, CH2), 2.77–2.82 (t, 2H, CH2), 4.09–4.12 (m, 2H, OCH2), 4.15 (s, 3H, N-CH3), 7.04–7.09 (t, 1H, Ar), 7.14–7.16 (dd, 1H, Ar), 7.45–7.51 (m, 1H, Ar), 7.64, 7.66 (m, 1H, Ar), 11.95(s, 1H, -NH), 9.48 (s, 1H, -CONH). Mass: 327.2 (M + H)+.
Preparation of 3-isopropyl-5-(2-ethoxyphenyl)-1-methyl-7H-pyrazolo[4,3-d]pyrimidin-7H-one (2c): Compound 2c was prepared from compound 14c as a white solid. Yield 71%, 1H-NMR (DMSO-d6) δ: 1.31–1.36 (m, 9H, 3xCH3), 3.34–3.42 (m, 1H, CH(CH3)2), 4.09–4.15 (q, 2H, OCH2), 4.16 (s, 3H, N-CH3), 7.04–7.09 (t, 1H, Ar), 7.14–7.16 (dd, 1H, Ar), 7.45–7.50 (m, 1H, Ar), 7.65–7.68 (m, 1H, Ar), 11.95 (s, 1H, NH). Mass: 313.1 (M + H)+.
2.9. General Procedure for the Preparation of Compounds 3a–3c
Cyclized compounds (2a–2c, 80.13 mmol) followed by thionyl chloride (9.53 g, 80.13 mmol) were added to chlorosulfonic acid (50 mL) portion-wise at 0–10 °C. The reaction temperature was raised to 20–30 °C and stirred for 4 h. The reaction mass was poured onto ice (500 g) and extracted with dichloromethane (250 mL). The dichloromethane layer was separated and washed with 5% w/w aqueous sodium bicarbonate (100 mL). The dichloromethane layer containing compound 3 was taken to next step as such.
Preparation of 5-(5-chlorosulfonyl-2-ethoxyphenyl)-1-methyl-3-ethyl-1,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one (3a). Compound 3a was prepared from compound 2a. Dichloromethane solution containing compound 3a was used as such in the next step.
Preparation of 5-(5-chlorosulfonyl-2-ethoxyphenyl)-1-methyl-3-butyl-1,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one (3b). Compound 3b was prepared from compound 2b. Dichloromethane solution containing compound 3b was used as such in the next step.
Preparation of 5-(5-chlorosulfonyl-2-ethoxyphenyl)-1-methyl-3-isopropyl-1,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one (3c). Compound 3c was prepared from compound 2c. Dichloromethane solution containing compound 3c was used as such in the next step.
2.10. General Procedure for the Preparation of Compounds 4a–4c
N-methylpiperazine (9.6 g, 96.15mmol) was added to the dichloromethane layer, containing sulfonyl chloride (3a–3c, obtained from example 9) at 20–25 °C. The reaction was stirred for 4 h and then washed with 5% w/w aqueous sodium bicarbonate (100 mL) followed by DM water (100 mL). The dichloromethane layer was concentrated at <40 °C under reduced pressure to obtain a foamy material. Methanol was added to crystallize the product and it was dried to result in compounds 4a–4c.
Preparation of 5-[2-ethoxy-5-(4-methylpiperazinylsulfonyl)phenyl]-1-methyl-3-ethyl-1,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one (sildenafil ethyl analogue, 4a). Sildenafil ethyl analogue (4a) was prepared from compound 3a. Yield: 91%, Melting point: 203–206 °C, HRMS for C21H28N6O4S (M + H)+ calc.: 460.1892, found: 460.1912, 1H-NMR (DMSO-d6) δ: 1.26–1.35 (m, 6H, 2CH3), 2.14 (s, 3H, CH3), 2.36 (s, 4H, 2xCH2 of piperazine), 2.79–2.86 (m, 2H, CH2, of piperazine), 2.90 (s, 4H, 2xCH2, of piperazine), 4.16 (s, 3H, N-CH3), 4.20–4.25 (q, 2H, OCH2), 7.36–7.39 (d, J = 9 Hz, 1H, Ar), 7.81–7.85 (t, J = 9.3 Hz, 2H, Ar),12.21 (brs, 1H, NH). 13C-NMR (DMSO-d6) δ:13.1, 14.2, 18.6, 37.8, 38.7, 39.2, 39.8, 40.3, 45.3, 45.7, 53.5, 64.8, 113.2, 123.7, 124.5, 126.2, 129.9, 131.4, 137.4, 146.1, 148.1, 153.7, 159.9. IR (KBr pellet): 3104, 2976, 2946, 2802, 1690, 1598, 1456, 1348, 1287, 1171 cm−1.
Preparation of 5-[2-ethoxy-5-(4-methylpiperazinylsulfonyl)phenyl]-1-methyl-3-n-butyl-1,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one (sildenafil butyl analogue, 4b). Sildenafil butyl analogue (4b) was prepared from compound 3b. Yield: 90%, Melting point: 189–192 °C, HRMS for C23H32N6O4S (M + H)+ calc.: 488.2205, found: 488.2310, 1H-NMR (DMSO-d6) δ: 0.88–0.93 (t, J = 9,6 Hz, 3H, CH3), 1.31–1.39 (m, 5H, CH2& CH3), 1.70 (m, 2H, CH2 of piperazine), 2.14 (s, 3H, CH3), 2.36 (s, 4H, 2CH2, of piperazine), 2.50–2.80 (m, 2H, -CH2), 2.90 (s, 4H, 2CH2 of piperazine), 4.16 (s, 3H, N-CH3), 4.20–4.23 (q, 2H, -OCH2), 7.36–7.39 (d, J = 9 Hz, 1H, Ar), 7.81–7.85 (t, J = 12 Hz, 2H, Ar),12.19 (brs, 1H, -NH).13C-NMR (DMSO-d6) δ:13.6, 14.2, 21.8, 24.7, 30.5, 37.8, 38.9, 39.2, 39.5, 39.8, 40.1, 40.3, 45.3, 45.7, 53.5, 64.8, 113.2, 123.7, 124.4, 126.2, 129.9, 131.5, 137.7, 145.1, 148.1, 153.7, 159.9. IR (KBr pellet): 3107, 2959, 2937, 2796, 1689, 1599, 1488, 1349, 1288, 1171 cm−1.
Preparation of 5-[2-ethoxy-5-(4-methylpiperazinylsulfonyl)phenyl]-1-methyl-3-isopropyl-1,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one (sildenafil isopropyl analogue, 4c). Sildenafil isopropyl analogue (4c) was prepared from compound 3c. Yield: 90%, Melting point: 212–215 °C, HRMS for C22H30N6O4S (M + H)+ calc.: 474.2049, found: 474.2105, 1H-NMR (DMSO-d6) δ: 1.31–1.36 (m, 9H, -3xCH3), 2.14 (s, 3H, -CH3), 2.36 (s, 4H, 2CH2 of piperazine), 2.91 (s, 4H, 2CH2, of piperazine), 4.16 (s, 3H, -NCH3), 4.18–4.23 (q, 2H, -OCH2), 7.36–7.39 (d, 1H, Ar), 7.82–7.85 (d, 2H, Ar), 12.20 (brs, 1H, -NH). 13C-NMR (DMSO-d6) δ: 14.2, 21.8, 26.0, 37.8, 38.7, 38.9, 39.2, 39.8, 40.1, 40.3, 45.3, 45.7, 53.5, 64.8, 113.2, 123.7, 124.6, 126.3, 130.0, 131.4, 137.0, 147.8, 149.8, 149.9, 153.8, 159.9. IR (KBr pellet): 3147, 3078, 2980, 2937, 2797, 1689, 1602, 1457, 1349, 1289, 1168 cm−1.


{kind=link}
{kind=link}
{kind=link}
{kind=link}