
FK506-Binding Protein like (FKBPL) Has an Important Role in Heart Failure with Preserved Ejection Fraction Pathogenesis with Potential Diagnostic Utility
 , ,
, ,  , and
, and
Abstract
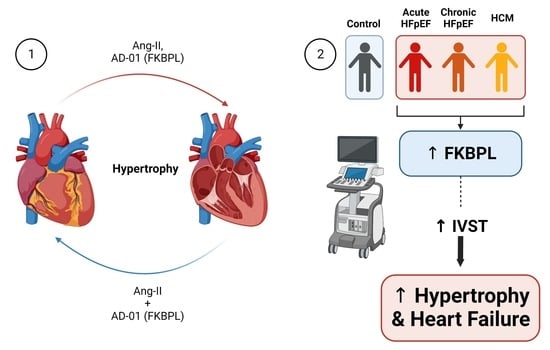
1. Introduction
2. Methods and Materials
2.1. Cell Culture and Treatments
2.2. Cell Size Analysis
2.3. Western Blot
2.4. Reverse Transcription-Polymerase Chain Reaction (RT-qPCR)
2.5. Participants and Samples
2.6. Plasma Marker Measurement
2.7. Statistical Analysis
3. Results
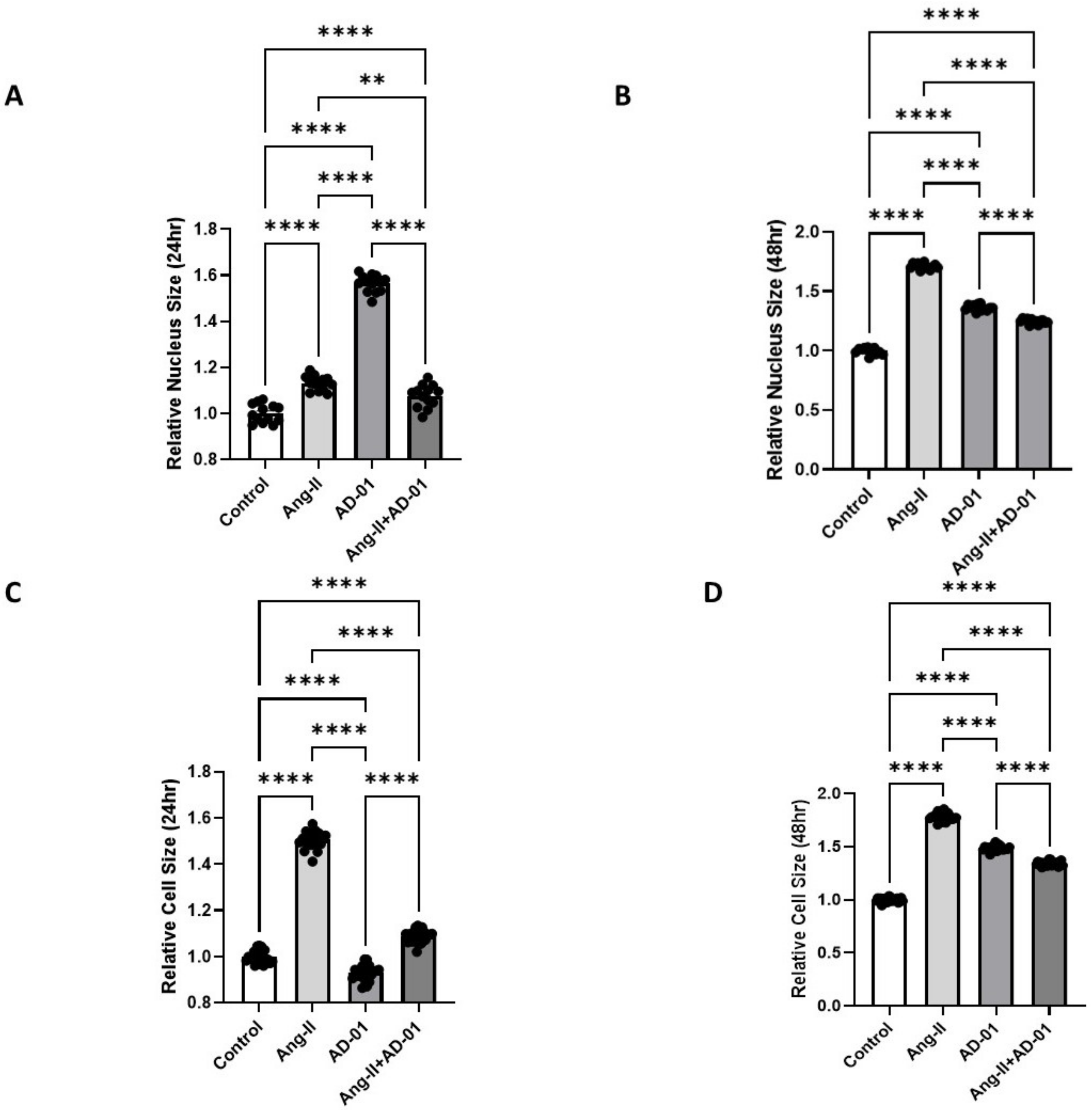
3.1. FKBPL Peptide Mimetic, AD-01, and Angiotensin-II (Ang-II) Increase Cardiomyoblast Cell and Nucleus Size; However, AD-01 in the Presence of Ang-II Abrogates Ang-II-Induced Cardiac Hypertrophy
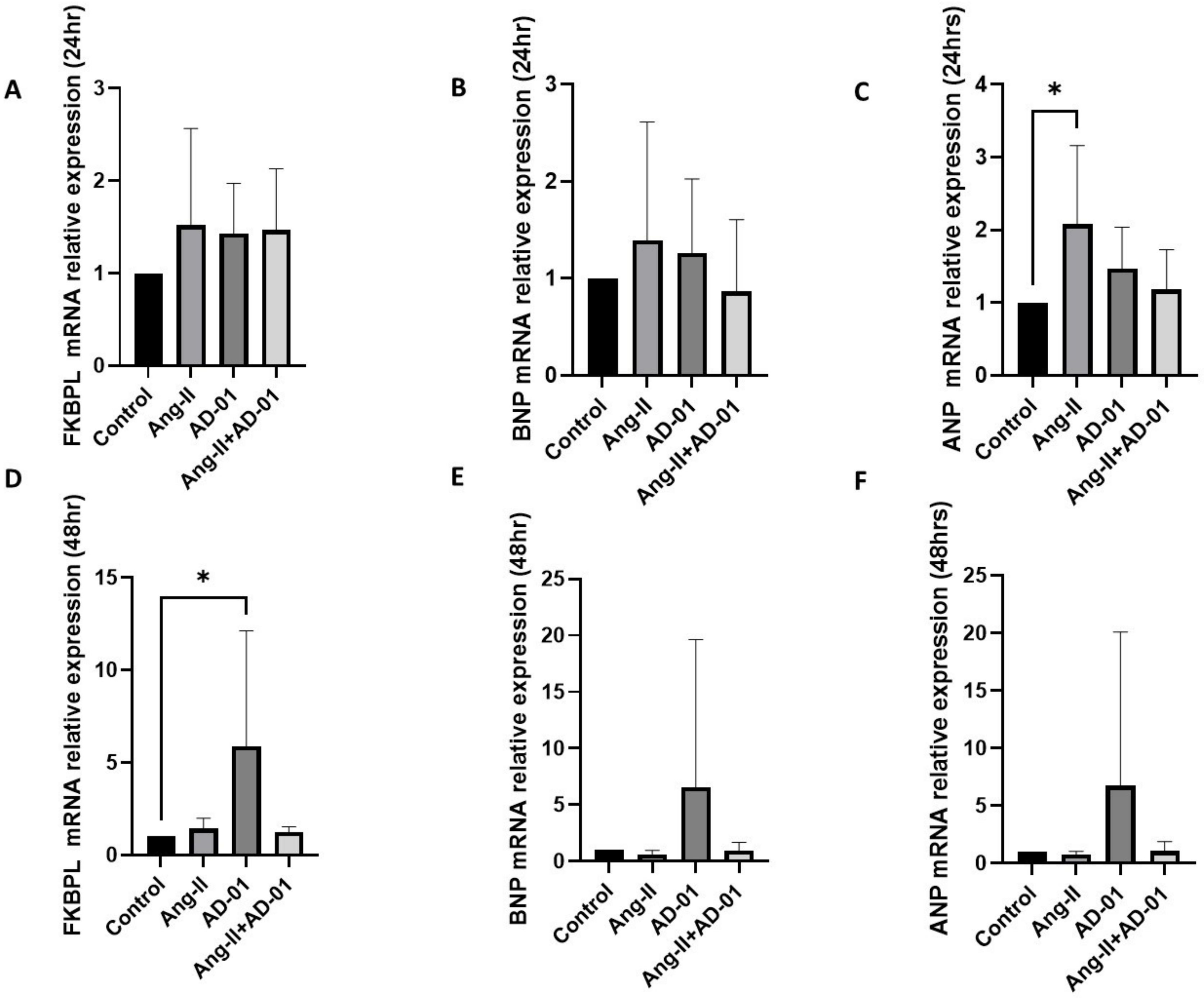
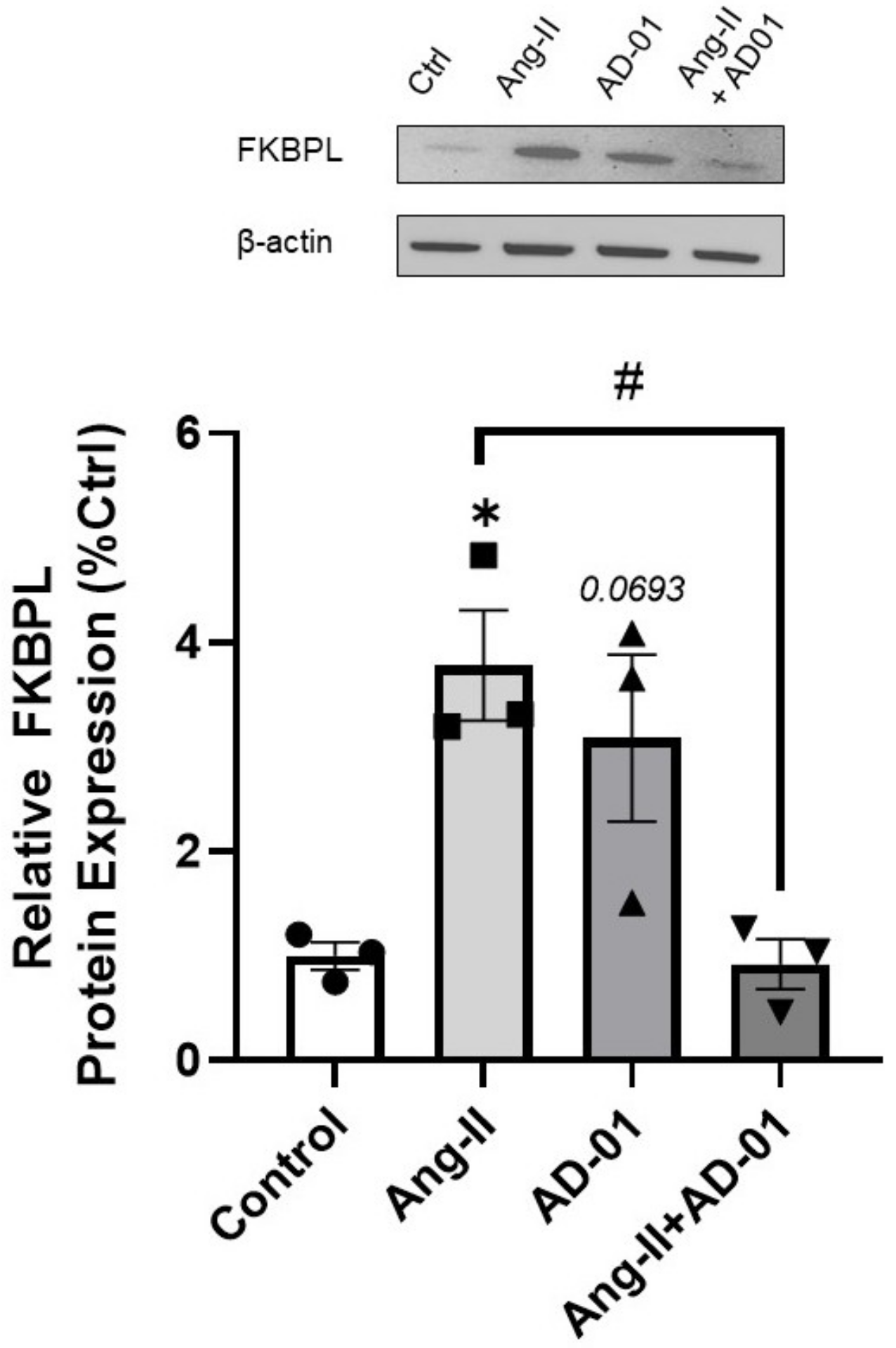
3.2. AD-01 Abrogates Ang-II-Induced Increases in FKBPL Protein Expression
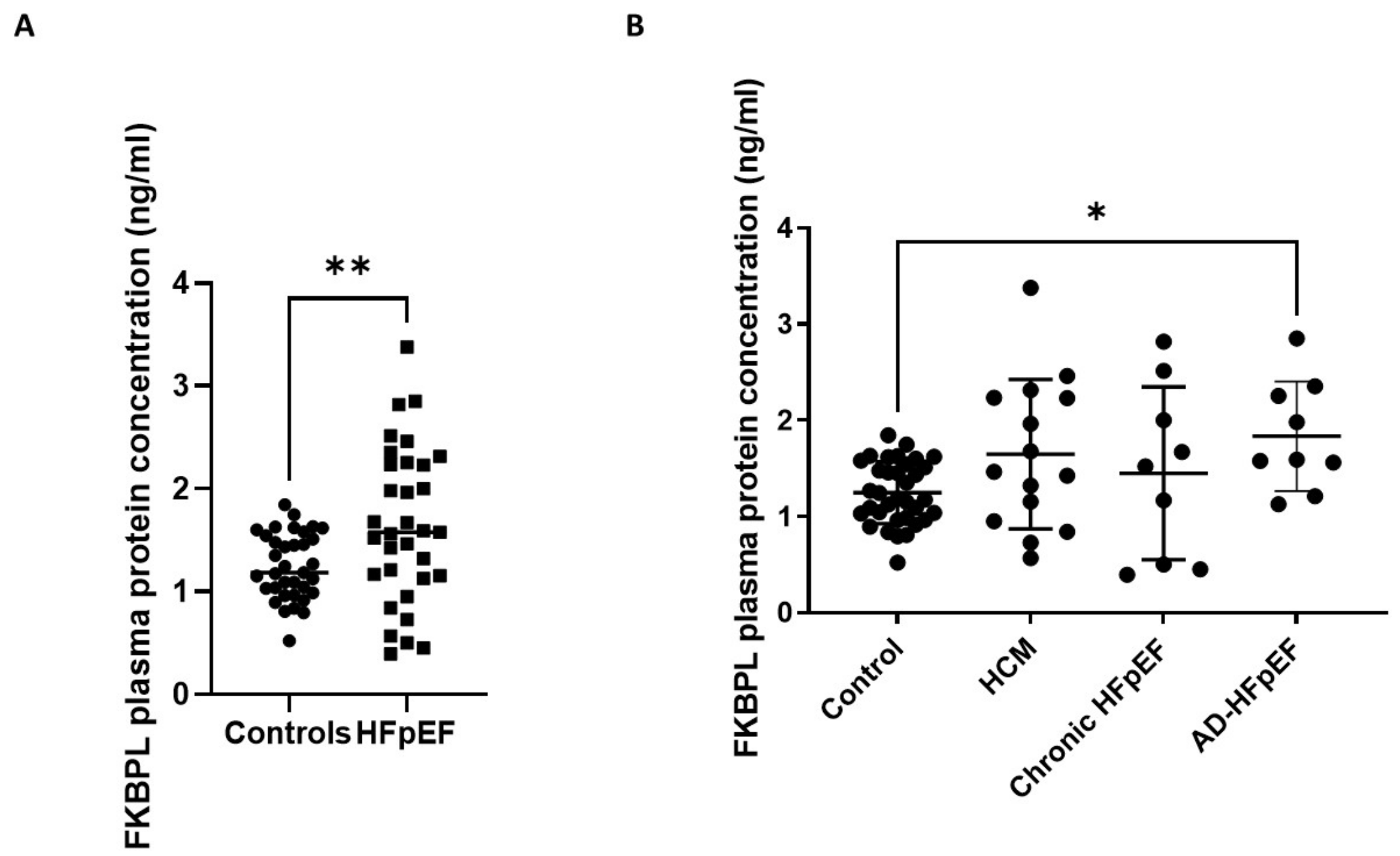
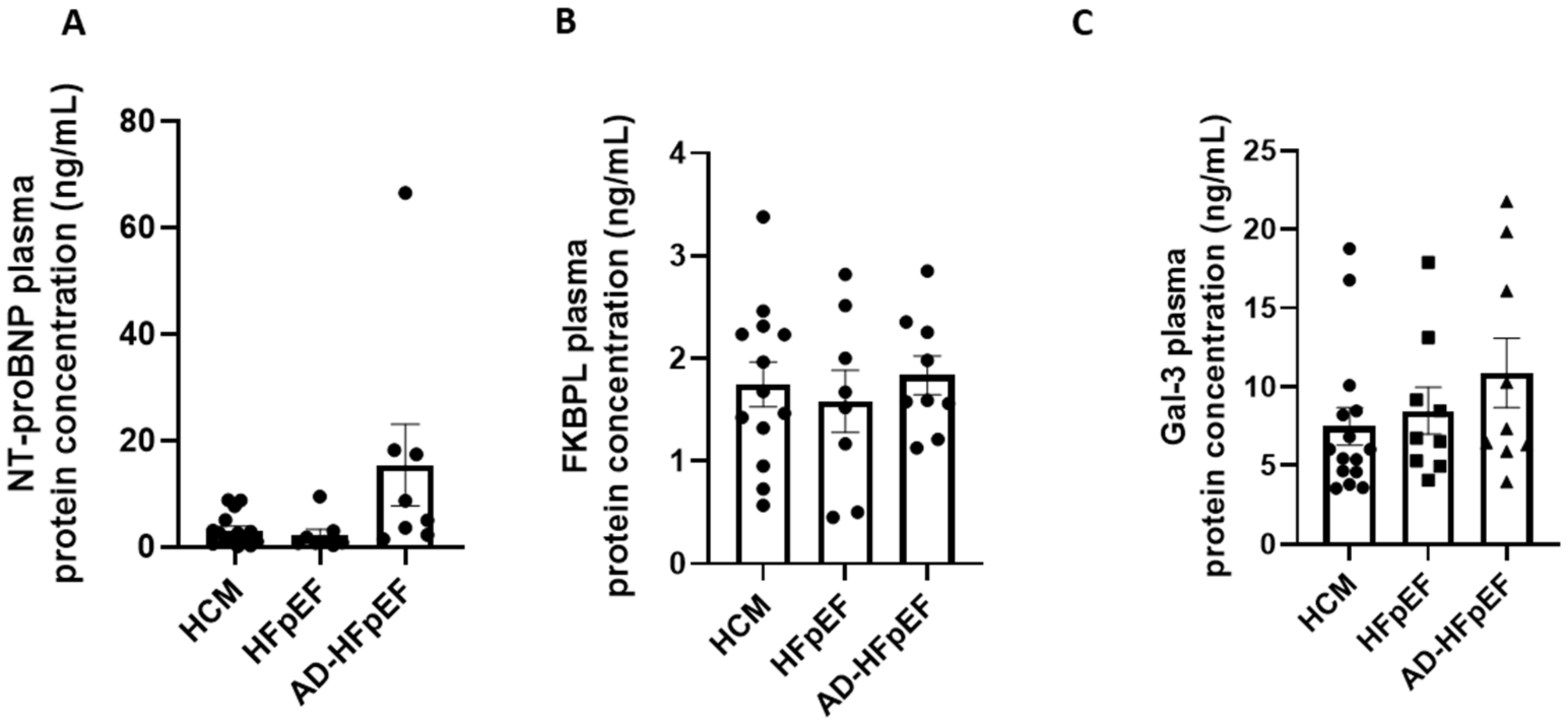
3.3. FKBPL Plasma Concentration Is Increased in Patients with HFpEF but Does Not Differ between Subgroups
3.4. FKBPL Is Positively Correlated with IVST, Indicative of Microvascular Dysfunction
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Malik, A.; Brito, D.; Vaqar, S.; Chhabra, L. Congestive Heart Failure; StatPearls: Tampa, FL, USA, 2022. [Google Scholar]
- Baman, J.R.; Ahmad, F.S. Heart Failure. JAMA 2020, 324, 1015. [Google Scholar] [CrossRef] [PubMed]
- Australian Institute of Health and Welfare. Heart, Stroke and Vascular Disease: Australian Facts; AIHW, Australian Government: Darlinghurst, Australia. Available online: https://www.aihw.gov.au/reports/heart-stroke-vascular-diseases/hsvd-facts/contents/about (accessed on 13 February 2023).
- McDonagh, T.A.; Metra, M.; Adamo, M.; Gardner, R.S.; Baumbach, A.; Böhm, M.; Burri, H.; Butler, J.; Čelutkienė, J.; Chioncel, O.; et al. 2021 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure. Developed by the Task Force for the diagno-sis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC) With the special contribution of the Heart Failure Association (HFA) of the ESC. Eur. Heart J. 2021, 42, 4901. [Google Scholar] [CrossRef] [PubMed]
- Inamdar, A.A.; Inamdar, A.C. Heart Failure: Diagnosis, Management and Utilization. J. Clin. Med. 2016, 5, 62. [Google Scholar] [CrossRef] [PubMed]
- Lewis, R.A.; Durrington, C.; Condliffe, R.; Kiely, D.G. BNP/NT-proBNP in pulmonary arterial hypertension: Time for point-of-care testing? Eur. Respir. Rev. 2020, 29, 200009. [Google Scholar] [CrossRef]
- Chen, H.; Chhor, M.; Rayner, B.S.; McGrath, K.; McClements, L. Evaluation of the diagnostic accuracy of current biomarkers in heart failure with preserved ejection fraction: A systematic review and meta-analysis. Arch. Cardiovasc. Dis. 2021, 114, 793–804. [Google Scholar] [CrossRef]
- Andrejic, O.M.; Vucic, R.M.; Pavlovic, M.; McClements, L.; Stokanovic, D.; Jevtovic–Stoimenov, T.; Nikolic, V.N. Association between Galectin-3 levels within central and peripheral venous blood, and adverse left ventricular remodelling after first acute myocardial infarction. Sci. Rep. 2019, 9, 13145. [Google Scholar] [CrossRef]
- Annett, S.; Spence, S.; Garciarena, C.; Campbell, C.; Dennehy, M.; Drakeford, C.; Lai, J.; Dowling, J.; Moore, G.; Yakkundi, A.; et al. The immunophilin protein FKBPL and its peptide derivatives are novel regulators of vascular integrity and inflammation via NF-κB signaling. bioRxiv 2021. [Google Scholar] [CrossRef]
- Yakkundi, A.; Bennett, R.; Hernández-Negrete, I.; Delalande, J.-M.; Hanna, M.; Lyubomska, O.; Arthur, K.; Short, A.; McKeen, H.; Nelson, L.; et al. FKBPL Is a Critical Antiangiogenic Regulator of Developmental and Pathological Angiogenesis. Arter. Thromb. Vasc. Biol. 2015, 35, 845–854. [Google Scholar] [CrossRef]
- Alqudah, A.; Eastwood, K.-A.; Jerotic, D.; Todd, N.; Hoch, D.; McNally, R.; Obradovic, D.; Dugalic, S.; Hunter, A.J.; Holmes, V.A.; et al. FKBPL and SIRT-1 Are Downregulated by Diabetes in Pregnancy Impacting on Angiogenesis and Endothelial Function. Front. Endocrinol. 2021, 12, 459. [Google Scholar] [CrossRef]
- Januszewski, A.S.; Watson, C.J.; O’Neill, V.; McDonald, K.; Ledwidge, M.; Robson, T.; Jenkins, A.J.; Keech, A.C.; McClements, L. FKBPL is associated with metabolic parameters and is a novel determinant of cardiovascular disease. Sci. Rep. 2020, 10, 1–7. [Google Scholar] [CrossRef]
- Annett, S.; Moore, G.; Short, A.; Marshall, A.; McCrudden, C.; Yakkundi, A.; Das, S.; McCluggage, W.G.; Nelson, L.; Harley, I.; et al. FKBPL-based peptide, ALM201, targets angiogenesis and cancer stem cells in ovarian cancer. Br. J. Cancer 2019, 122, 361–371. [Google Scholar] [CrossRef]
- Chen, H.; Tesic, M.; Nikolic, V.N.; Pavlovic, M.; Vucic, R.M.; Spasic, A.; Jovanovic, H.; Jovanovic, I.; Town, S.E.L.; Padula, M.P.; et al. Systemic Biomarkers and Unique Pathways in Different Phenotypes of Heart Failure with Preserved Ejection Fraction. Biomolecules 2022, 12, 1419. [Google Scholar] [CrossRef]
- Laemmli, U.K. Cleavage of Structural Proteins during the Assembly of the Head of Bacteriophage T4. Nature 1970, 227, 680–685. [Google Scholar] [CrossRef]
- Ponikowski, P.; Voors, A.; Anker, S.; Bueno, H.; Cleland, J.; Coats, A.; Falk, V.; González-Juanatey, J.R.; Harjola, V.-P.; Jankowska, E.; et al. 2016 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure: The Task Force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC)Developed with the special contribution of the Heart Failure Association (HFA) of the ESC. Eur. Heart J. 2016, 37, 2129–2200. [Google Scholar]
- Van Heerebeek, L.; Hamdani, N.; Handoko, M.L.; Falcao-Pires, I.; Musters, R.J.; Kupreishvili, K.; Ijsselmuiden, A.J.; Schalkwijk, C.G.; Bronzwaer, J.G.; Diamant, M.; et al. Diastolic stiffness of the failing diabetic heart: Importance of fibrosis, advanced glycation end products, and myocyte resting tension. Circulation 2008, 117, 43–51. [Google Scholar] [CrossRef]
- Watkins, S.J.; Borthwick, G.M.; Arthur, H.M. The H9C2 cell line and primary neonatal cardiomyocyte cells show similar hypertrophic responses in vitro. Vitr. Cell. Dev. Biol. Anim. 2011, 47, 125–131. [Google Scholar] [CrossRef]
- Peter, A.K.; Bjerke, M.A.; Leinwand, L.A. Biology of the cardiac myocyte in heart disease. Mol. Biol. Cell 2016, 27, 2149–2160. [Google Scholar] [CrossRef]
- Nakano, S.; Muramatsu, T.; Nishimura, S.; Senbonmatsu, T. Cardiomyocyte and Heart Failure. In Current Basic and Pathological Approaches to the Function of Muscle Cells and Tissues—From Molecules to Humans; IntechOpen: London, UK, 2012. [Google Scholar] [CrossRef]
- Orsborne, C.; Chaggar, P.S.; Shaw, S.M.; Williams, S.G. The renin-angiotensin-aldosterone system in heart failure for the non-specialist: The past, the present and the future. Postgrad. Med. J. 2016, 93, 29–37. [Google Scholar] [CrossRef]
- Xu, L.; Pagano, J.; Chow, K.; Oudit, G.Y.; Haykowsky, M.J.; Mikami, Y.; Howarth, A.G.; White, J.A.; Howlett, J.G.; Dyck, J.R.; et al. Cardiac remodelling predicts outcome in patients with chronic heart failure. ESC Heart Fail. 2021, 8, 5352–5362. [Google Scholar] [CrossRef]
- Todd, N.; McNally, R.; Qudhah, A.; Jerotic, D.; Suvakov, S.; Obradovic, D.; Hoch, D.; Hombrebueno, J.R.; Campos, G.L.; Watson, C.J.; et al. Role of A Novel Angiogenesis FKBPL-CD44 Pathway in Preeclampsia Risk Stratification and Mesenchymal Stem Cell Treatment. J. Clin. Endocrinol. Metab. 2020, 106, 26–41. [Google Scholar] [CrossRef]
- Yakkundi, A.; McCallum, L.; O’Kane, A.; Dyer, H.; Worthington, J.; McKeen, H.D.; McClements, L.; Elliott, C.; McCarthy, H.; Hirst, D.G.; et al. The Anti-Migratory Effects of FKBPL and Its Peptide Derivative, AD-01: Regulation of CD44 and the Cytoskeletal Pathway. PLoS ONE 2013, 8, e55075. [Google Scholar] [CrossRef] [PubMed]
- Zeng, H.; Chen, J.-X. Microvascular Rarefaction and Heart Failure With Preserved Ejection Fraction. Front. Cardiovasc. Med. 2019, 6, 15. [Google Scholar] [CrossRef] [PubMed]
- De Boer, R.A.; Edelmann, F.; Cohen-Solal, A.; Mamas, M.A.; Maisel, A.; Pieske, B. Galectin-3 in heart failure with preserved ejection fraction. Eur. J. Heart Fail. 2013, 15, 1095–1101. [Google Scholar] [CrossRef] [PubMed]
- Zhong, X.; Qian, X.; Chen, G.; Song, X. The role of galectin-3 in heart failure and cardiovascular disease. Clin. Exp. Pharmacol. Physiol. 2018, 46, 197–203. [Google Scholar] [CrossRef]
- Zhao, W.; Chen, Y.; Yang, W.; Han, Y.; Wang, Z.; Huang, F.; Qiu, Z.; Yang, K.; Jin, W. Effects of Cardiomyocyte-Specific Deletion of STAT3–A Murine Model of Heart Failure With Preserved Ejection Fraction. Front. Cardiovasc. Med. 2020, 7, 613123. [Google Scholar] [CrossRef]
- Cecchi, F.; Olivotto, I.; Gistri, R.; Lorenzoni, R.; Chiriatti, G.; Camici, P.G. Coronary Microvascular Dysfunction and Prognosis in Hypertrophic Cardiomyopathy. N. Engl. J. Med. 2003, 349, 1027–1035. [Google Scholar] [CrossRef]
- Tesic, M.; Beleslin, B.; Giga, V.; Jovanovic, I.; Marinkovic, J.; Trifunovic, D.; Petrovic, O.; Dobric, M.; Aleksandric, S.; Juricic, S.; et al. Prognostic Value of Transthoracic Doppler Echocardiography Coronary Flow Velocity Reserve in Patients With Asymmetric Hypertrophic Cardiomyopathy. J. Am. Heart Assoc. 2021, 10, e021936. [Google Scholar] [CrossRef]
- Bravo, P.E.; Pinheiro, A.; Higuchi, T.; Rischpler, C.; Merrill, J.; Santaularia-Tomas, M.; Abraham, M.R.; Wahl, R.L.; Abraham, T.P.; Bengel, F.M. PET/CT Assessment of Symptomatic Individuals with Obstructive and Nonobstructive Hypertrophic Cardiomyopathy. J. Nucl. Med. 2012, 53, 407–414. [Google Scholar] [CrossRef]
- Ismail, T.F.; Hsu, L.-Y.; Greve, A.M.; Gonçalves, C.; Jabbour, A.; Gulati, A.; Hewins, B.; Mistry, N.; Wage, R.; Roughton, M.; et al. Coronary microvascular ischemia in hypertrophic cardiomyopathy—A pixel-wise quantitative cardiovascular magnetic resonance perfusion study. J. Cardiovasc. Magn. Reson. 2014, 16, 49. [Google Scholar] [CrossRef]
- Yang, Y.; Li, Z.; Guo, X.; Zhou, Y.; Chang, Y.; Yang, H.; Yu, S.; Ouyang, N.; Chen, S.; Sun, G.; et al. Interventricular Septum Thickness for the Prediction of Coronary Heart Disease and Myocardial Infarction in Hypertension Population: A Prospective Study. J. Clin. Med. 2022, 11, 7152. [Google Scholar] [CrossRef]
- Kansal, S.; Roitman, D.; Sheffield, L.T. Interventricular septal thickness and left ventricular hypertrophy. An echocardiographic study. Circulation 1979, 60, 1058–1065. [Google Scholar] [CrossRef]
- Ledwidge, M.; Gallagher, J.; Conlon, C.; Tallon, E.; O’Connell, E.; Dawkins, I.; Watson, C.; O’Hanlon, R.; Bermingham, M.; Patle, A.; et al. Natriuretic peptide-based screening and collaborative care for heart failure: The STOP-HF randomized trial. JAMA 2013, 310, 66–74. [Google Scholar] [CrossRef]
- Tonry, C.; McDonald, K.; Ledwidge, M.; Hernandez, B.; Glezeva, N.; Rooney, C.; Morrissey, B.; Pennington, S.R.; Baugh, J.A.; Watson, C.J. Multiplexed measurement of candidate blood protein biomarkers of heart failure. ESC Heart Fail. 2021, 8, 2248–2258. [Google Scholar] [CrossRef]
- Watson, C.J.; Gallagher, J.; Wilkinson, M.; Russell-Hallinan, A.; Tea, I.; James, S.; O’Reilly, J.; O’Connell, E.; Zhou, S.; Ledwidge, M.; et al. Biomarker profiling for risk of future heart failure (HFpEF) development. J. Transl. Med. 2021, 19, 61. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristics | Controls (n = 40) | Acute HFpEF (n = 9) | Chronic HFpEF (n = 9) | HCM (n = 15) |
|---|---|---|---|---|
| Age (years) | 72.43 ± 6.4 | 73.4 ± 13.3 | 64.6 ± 10.6 | 50.7 ± 13.6 |
| Female (no. [%]) | 13 (37.1) | 4 (44.4) | 3 (33.3) | 3 (20) |
| BMI (kg/m2) | 27.6 ± 5.3 | 32 ± 4.4 | 28 ± 2.5 | 25.9 ± 4.1 |
| EF (%) | n/a | 57.6 ± 10.9 | 57.4 ± 8.0 | 64.5 ± 3.8 |
| NYHA Class | n/a | I/II/III | I/II | I/II |
| Diabetes n (%) | 20 (54) | 5 (56) | 2 (22) | 0 (0) |
| NT-proBNP (ng/mL) | n/a | 13.8 ± 20.9 | 2.3 ± 3.0 | 3.2 ± 3.0 |
| FKBPL (ng/mL) | 1.26 ± 0.3 | 1.8 ± 0.6 | 1.5 ± 0.9 | 1.6 ± 0.8 |
| Gal-3 (ng/mL) | n/a | 10.9 ± 6.6 | 8.5 ± 4.5 | 7.5 ± 4.6 |
| Echocardiography measurement | ||||
| EDD (mm) | n/a | 55.0 ± 11.6 | 52.8 ± 6.9 | 47.5 ± 5.5 |
| ESD (mm) | n/a | 37 ± 9.6 | 35.3 ± 8.2 | 28.9 ± 4.2 |
| IVST (mm) | n/a | 12.3 ± 2.9 | 12.4 ± 2.4 | 17.9 ± 2.3 |
| PWT (mm) | n/a | 11.7 ± 2.1 | 12.1 ± 1.5 | 9.3 ± 1.7 |
| Medications | ||||
| Aspirin (no. [%]) | n/a | 7 (78) | 4 (44) | 1 (7) |
| Purinergic receptor antagonists (no. [%]) | n/a | 5 (56) | 3 (33) | 0 |
| Statins (no. [%]) | n/a | 6 (67) | 3 (33) | 2 (13) |
| Isosorbide mononitrate (no. [%]) | n/a | 3 (33) | 1 (11) | 0 |
| Beta-blockers (no. [%]) | n/a | 9 (100) | 6 (67) | 14 (93) |
| ACE-inhibitors (no. [%]) | n/a | 7 (78) | 5 (56) | 4 (27) |
| Diuretics (no. [%]) | n/a | 4 (44) | 4 (44) | 4 (27) |
| Calcium channel blockers (no. [%]) | n/a | 3 (33) | 2 (22) | 1 (7) |
| Warfarin (no. [%]) | n/a | 1 (11) | 1 (11) | 0 |
| Amiodarone (no. [%]) | n/a | 0 | 0 | 1 (7) |
| PPIs (no. [%]) | n/a | 4 (44) | 3 (33) | 0 |
| Trimetazidine (no. [%]) | n/a | 1 (11) | 1 (11) | 0 |
| Molsidomine (no. [%]) | n/a | 1 (11) | 1 (11) | 0 |
| Spironolactone (no. [%]) | n/a | 0 | 3 (33) | 0 |
| Allopurinol (no. [%]) | n/a | 0 | 1 (11) | 0 |
| Aminophylline (no. [%]) | n/a | 0 | 2 (22) | 0 |
| FKBPL | EDD | ESD | IVST | PWT | ||
|---|---|---|---|---|---|---|
| FKBPL | Pearson Correlation | 1 | −0.281 | −0.361 * | 0.621 *** | −0.401 * |
| Sig. (2-tailed) | 0.119 | 0.042 | 0.000 | 0.021 | ||
| N | 33 | 32 | 32 | 33 | 33 | |
| FKBPL | NT-proBNP | Gal-3 | ||
|---|---|---|---|---|
| FKBPL | Pearson Correlation | 1 | 0.063 | −0.042 |
| Sig. (2-tailed) | 0.731 | 0.815 | ||
| N | 33 | 32 | 33 | |
| NT-proBNP | Pearson Correlation | 0.063 | 1 | 0.464 ** |
| Sig. (2-tailed) | 0.731 | 0.007 | ||
| N | 32 | 32 | 32 | |
| Gal-3 | Pearson Correlation | −0.042 | 0.464 ** | 1 |
| Sig. (2-tailed) | 0.815 | 0.007 | ||
| N | 33 | 32 | 33 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chhor, M.; Chen, H.; Jerotić, D.; Tešić, M.; Nikolić, V.N.; Pavlović, M.; Vučić, R.M.; Rayner, B.; Watson, C.J.; Ledwidge, M.; et al. FK506-Binding Protein like (FKBPL) Has an Important Role in Heart Failure with Preserved Ejection Fraction Pathogenesis with Potential Diagnostic Utility. Biomolecules 2023, 13, 395. https://doi.org/10.3390/biom13020395
Chhor M, Chen H, Jerotić D, Tešić M, Nikolić VN, Pavlović M, Vučić RM, Rayner B, Watson CJ, Ledwidge M, et al. FK506-Binding Protein like (FKBPL) Has an Important Role in Heart Failure with Preserved Ejection Fraction Pathogenesis with Potential Diagnostic Utility. Biomolecules. 2023; 13(2):395. https://doi.org/10.3390/biom13020395
Chicago/Turabian StyleChhor, Michael, Hao Chen, Djurdja Jerotić, Milorad Tešić, Valentina N. Nikolić, Milan Pavlović, Rada M. Vučić, Benjamin Rayner, Chris J. Watson, Mark Ledwidge, and et al. 2023. "FK506-Binding Protein like (FKBPL) Has an Important Role in Heart Failure with Preserved Ejection Fraction Pathogenesis with Potential Diagnostic Utility" Biomolecules 13, no. 2: 395. https://doi.org/10.3390/biom13020395
APA StyleChhor, M., Chen, H., Jerotić, D., Tešić, M., Nikolić, V. N., Pavlović, M., Vučić, R. M., Rayner, B., Watson, C. J., Ledwidge, M., McDonald, K., Robson, T., McGrath, K. C., & McClements, L. (2023). FK506-Binding Protein like (FKBPL) Has an Important Role in Heart Failure with Preserved Ejection Fraction Pathogenesis with Potential Diagnostic Utility. Biomolecules, 13(2), 395. https://doi.org/10.3390/biom13020395