
New Biodegradable Materials for Re-Thought Packaging from Pre-Consumer Wastes by Controlling the Storage Time as Method to Increase the Mechanical Recycling Efficiency

 ,
,  ,
,  ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Methods
2.2. Materials
2.3. Characterization
2.3.1. Rheological Properties in the Melted State
2.3.2. DSC
2.3.3. TGA
2.3.4. FTIR
2.3.5. Mechanical Characterization
3. Results
3.1. Melt Rheological Properties
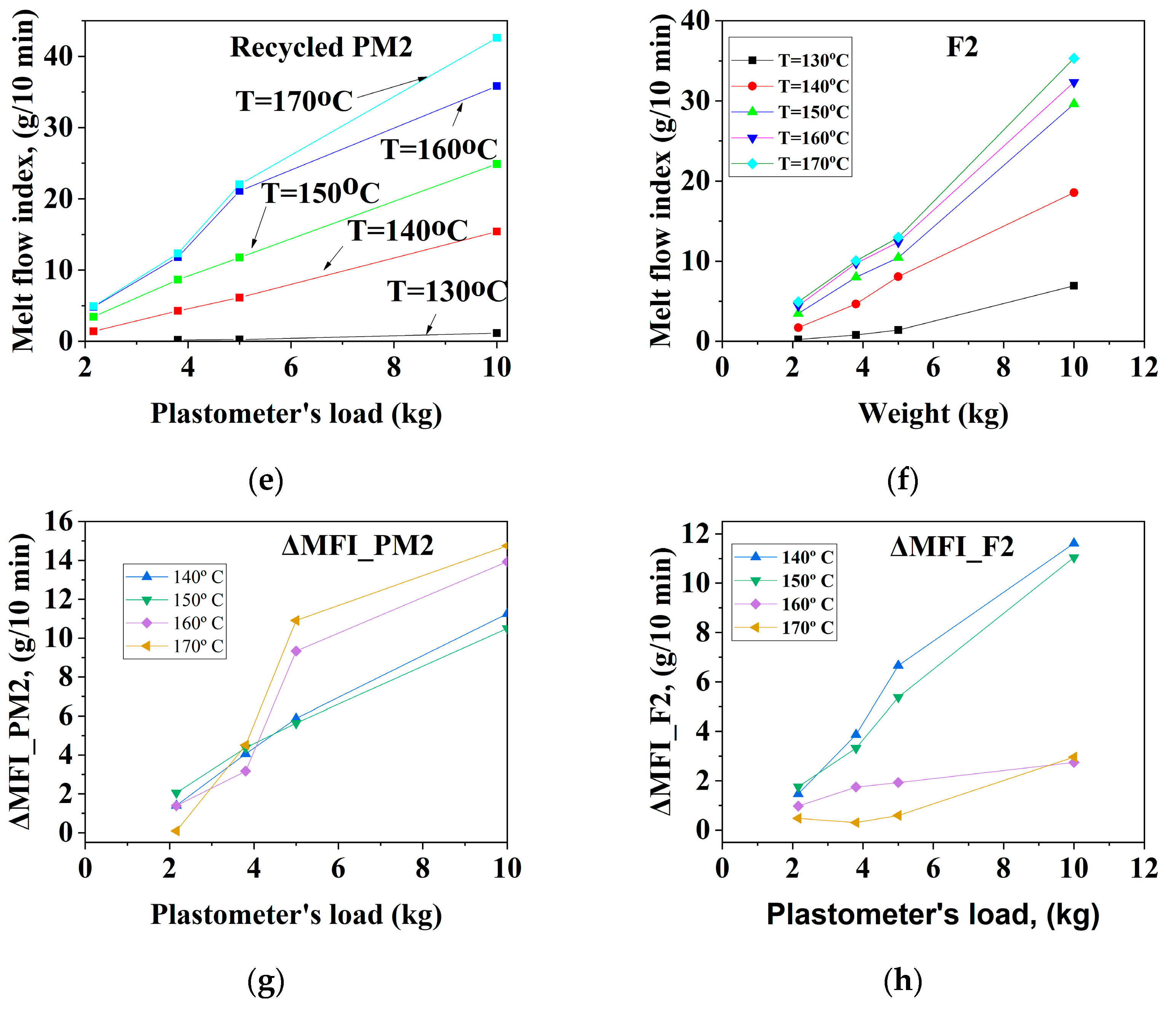
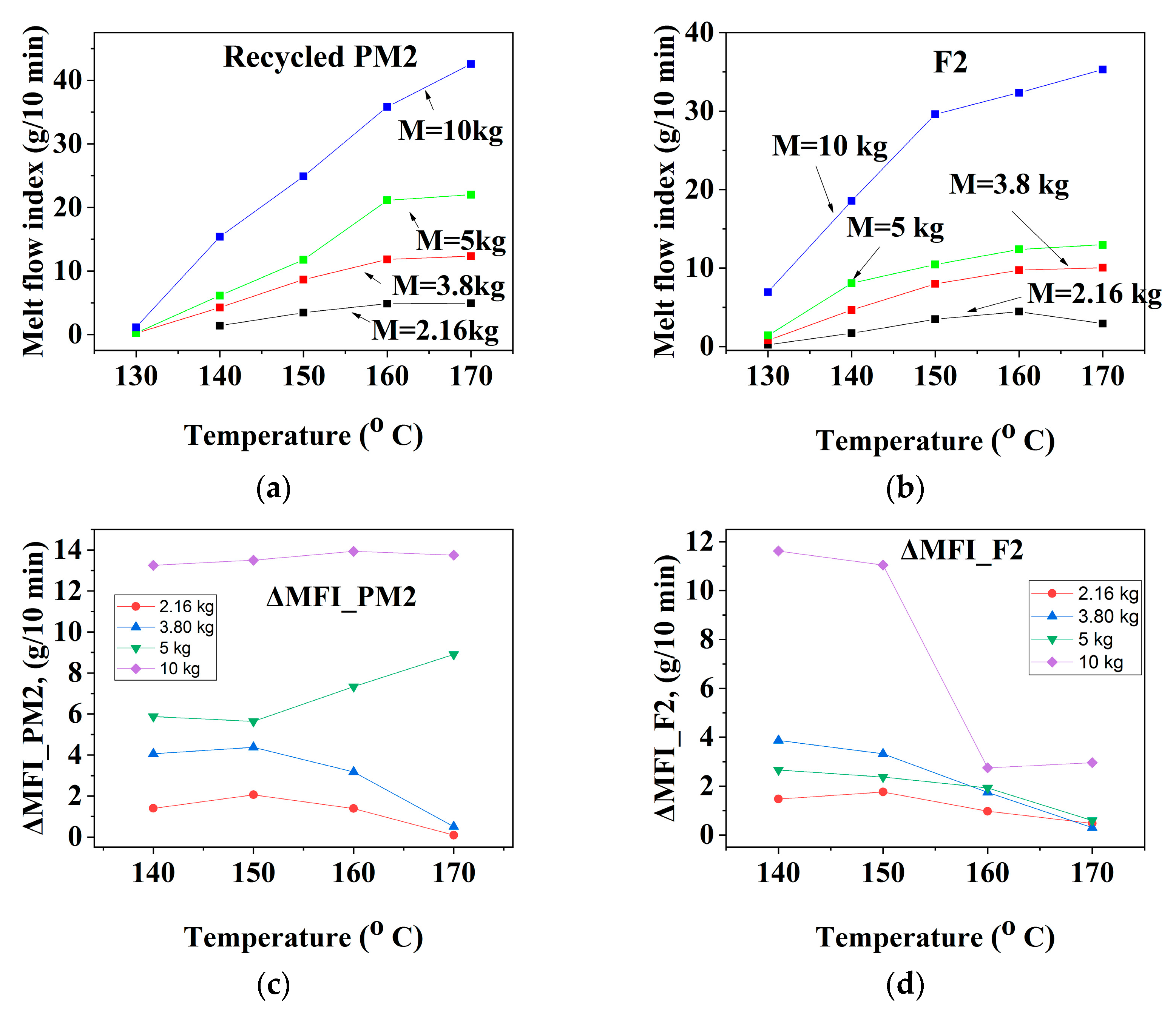
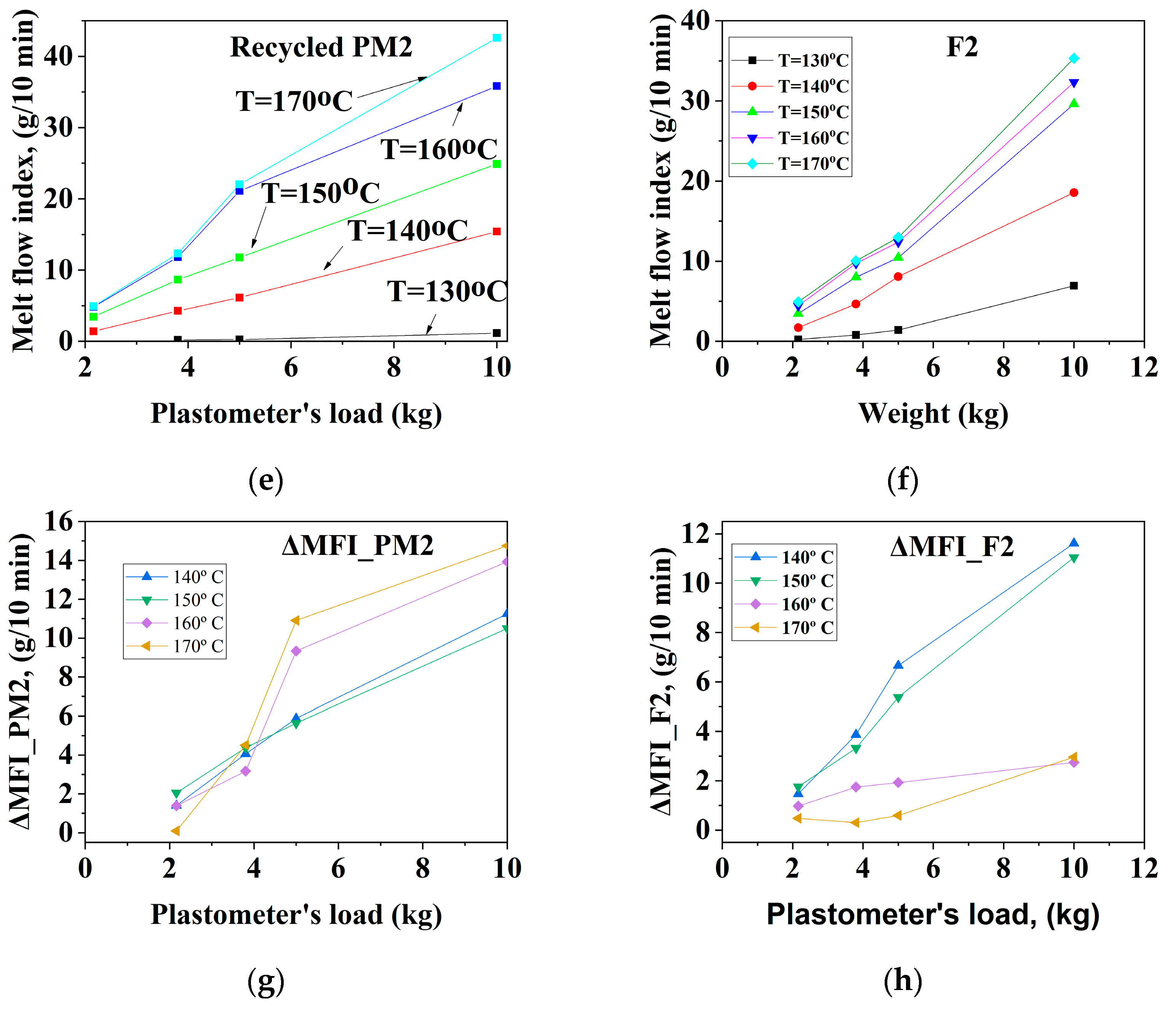
3.1.1. Mechanically Recycled Compounds from the PET-CW Resulted from Manufacture of Films Flexographically Printed with Black (PM2) and Primary Compound (F2) Used in this Production
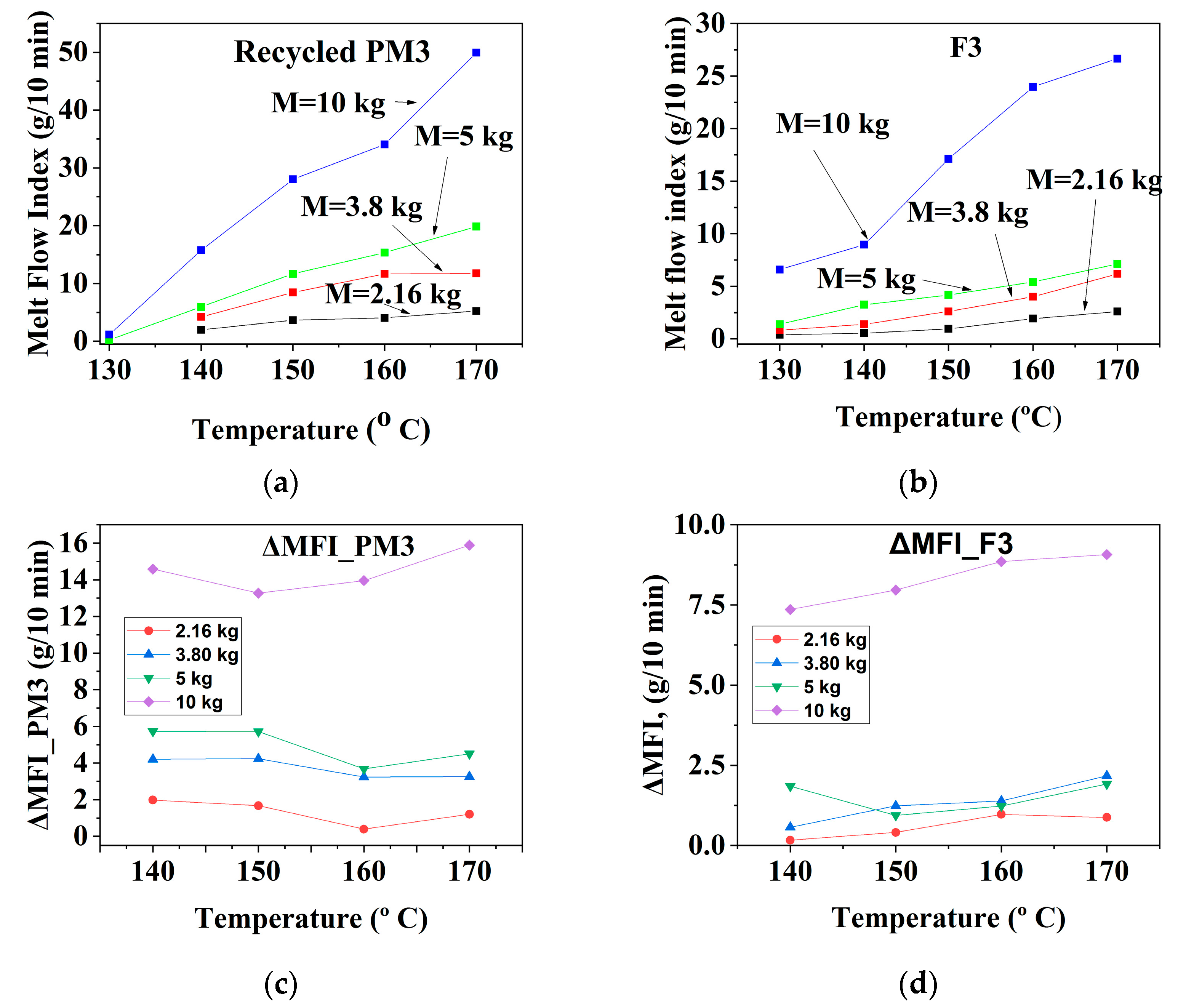
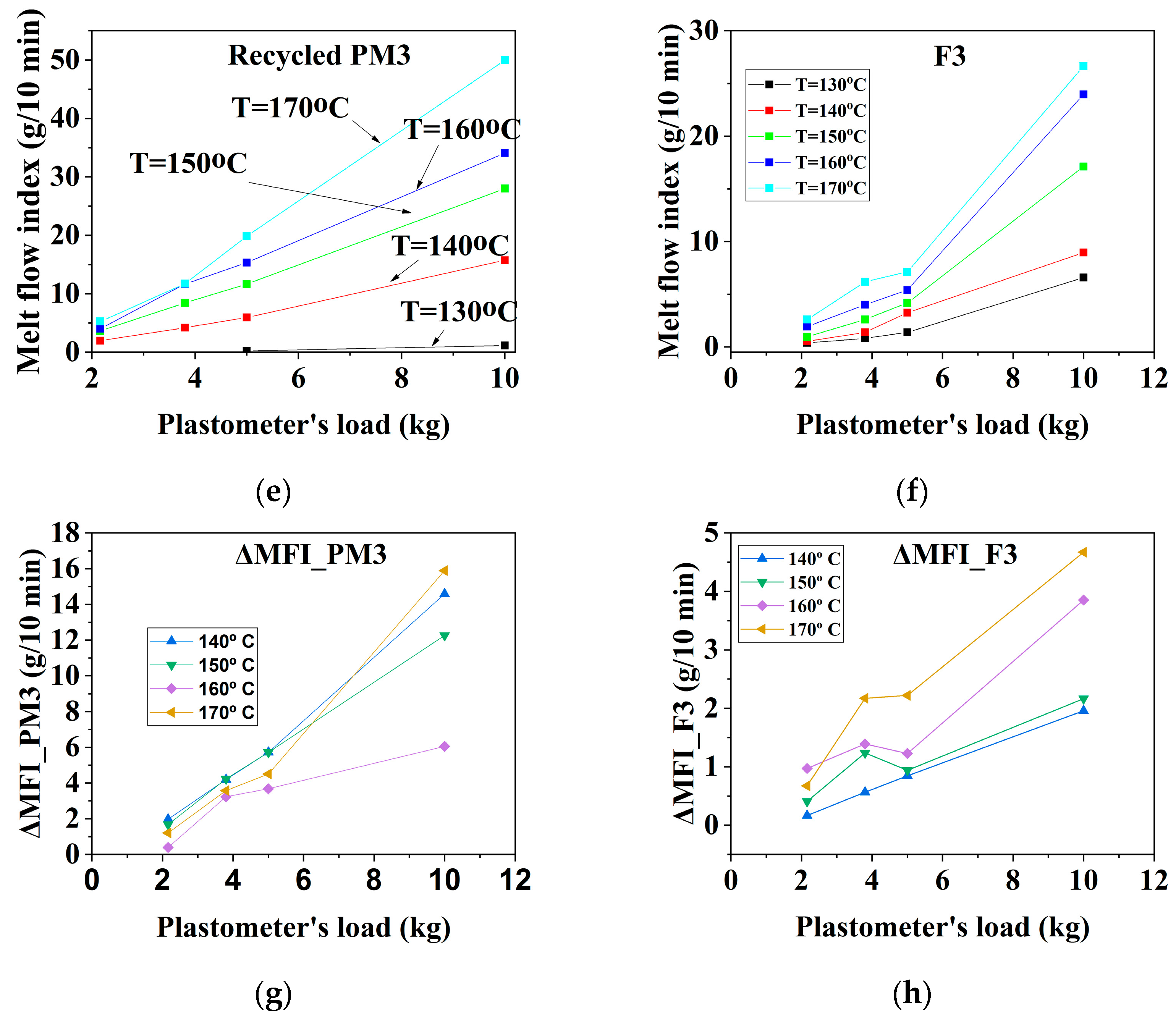
3.1.2. Mechanically Recycled Compound from PET-CW Formed during the Manufacture of Unprinted Films (PM3) and Primary Compound (F3) Used in this Production
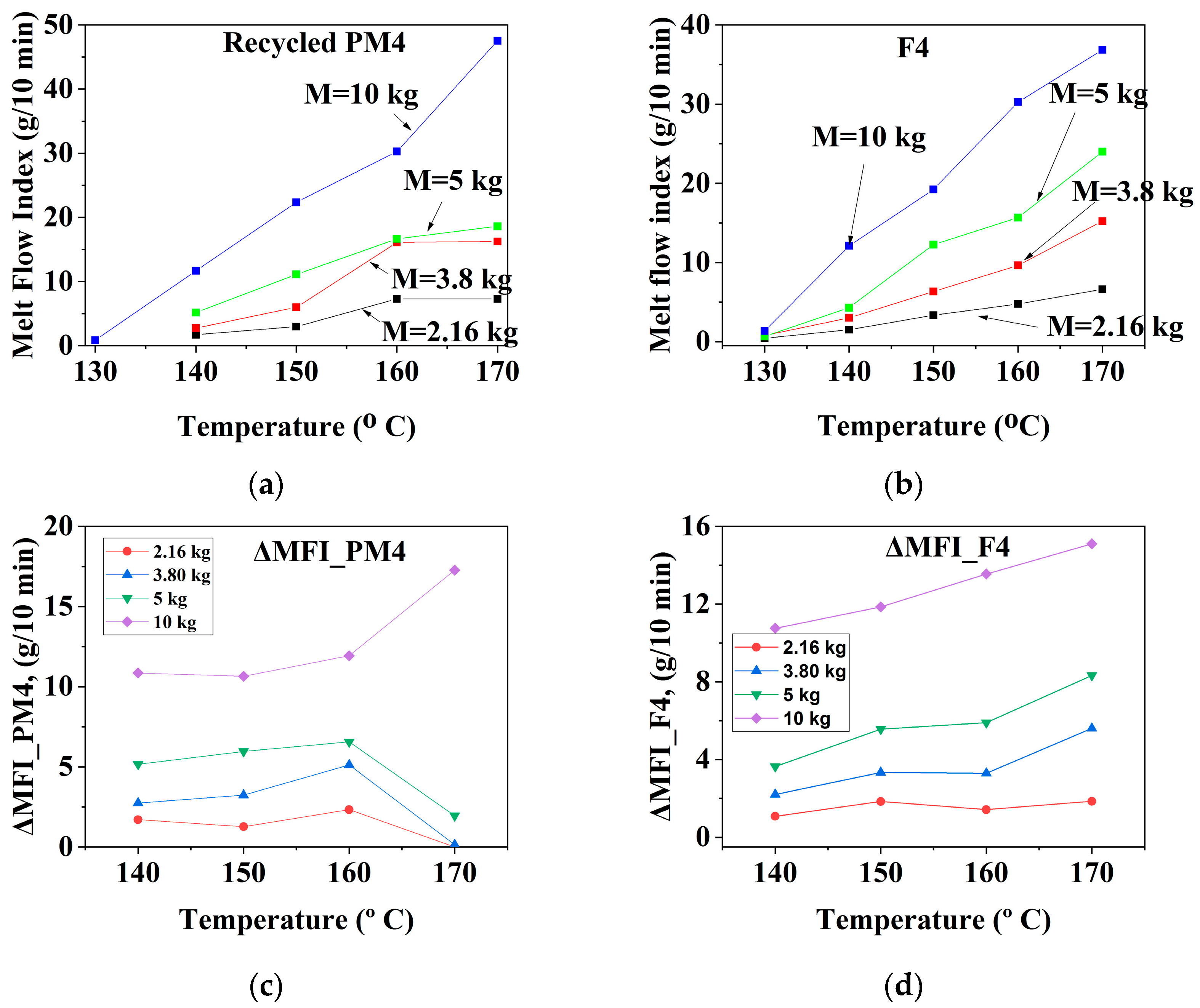
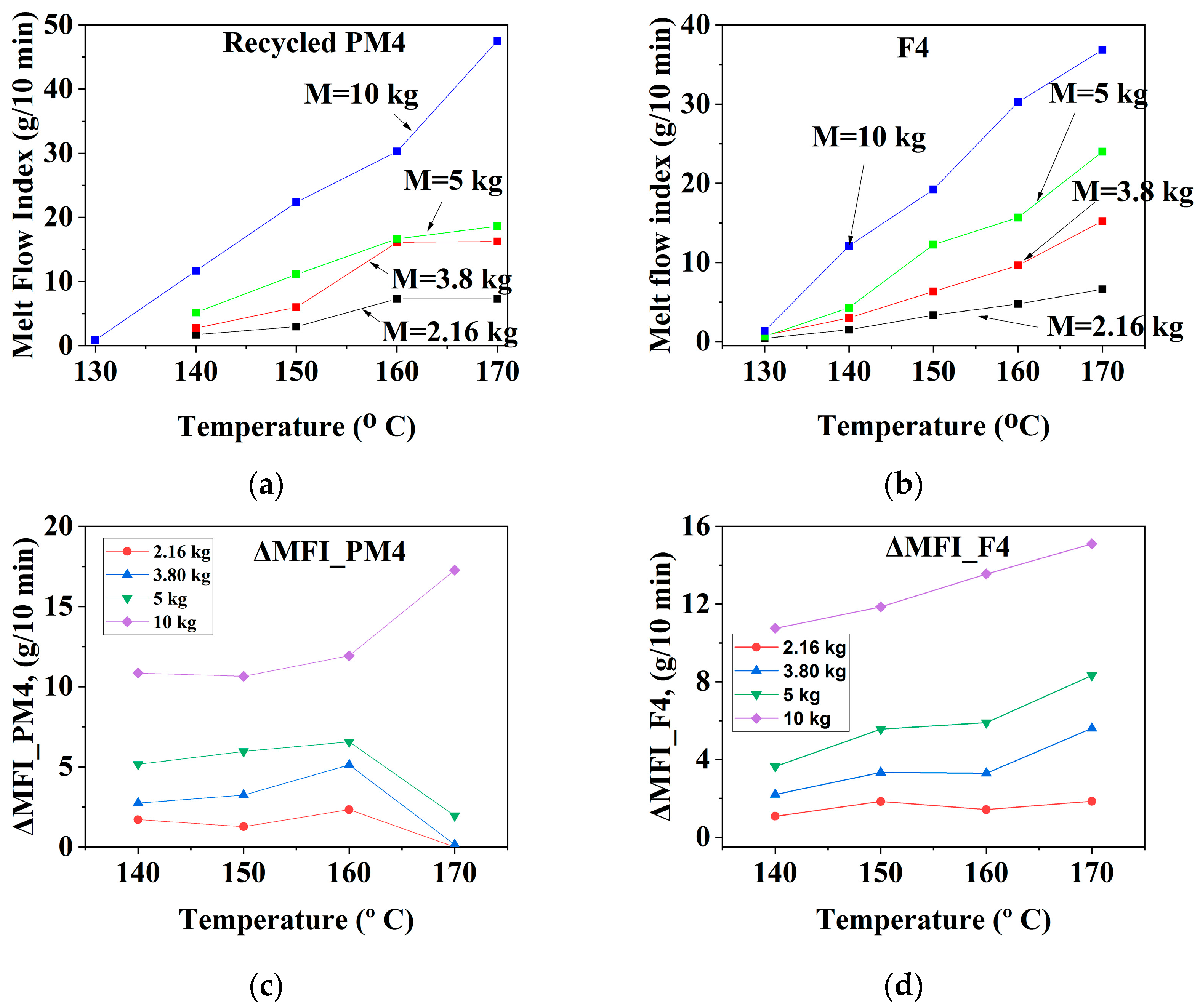
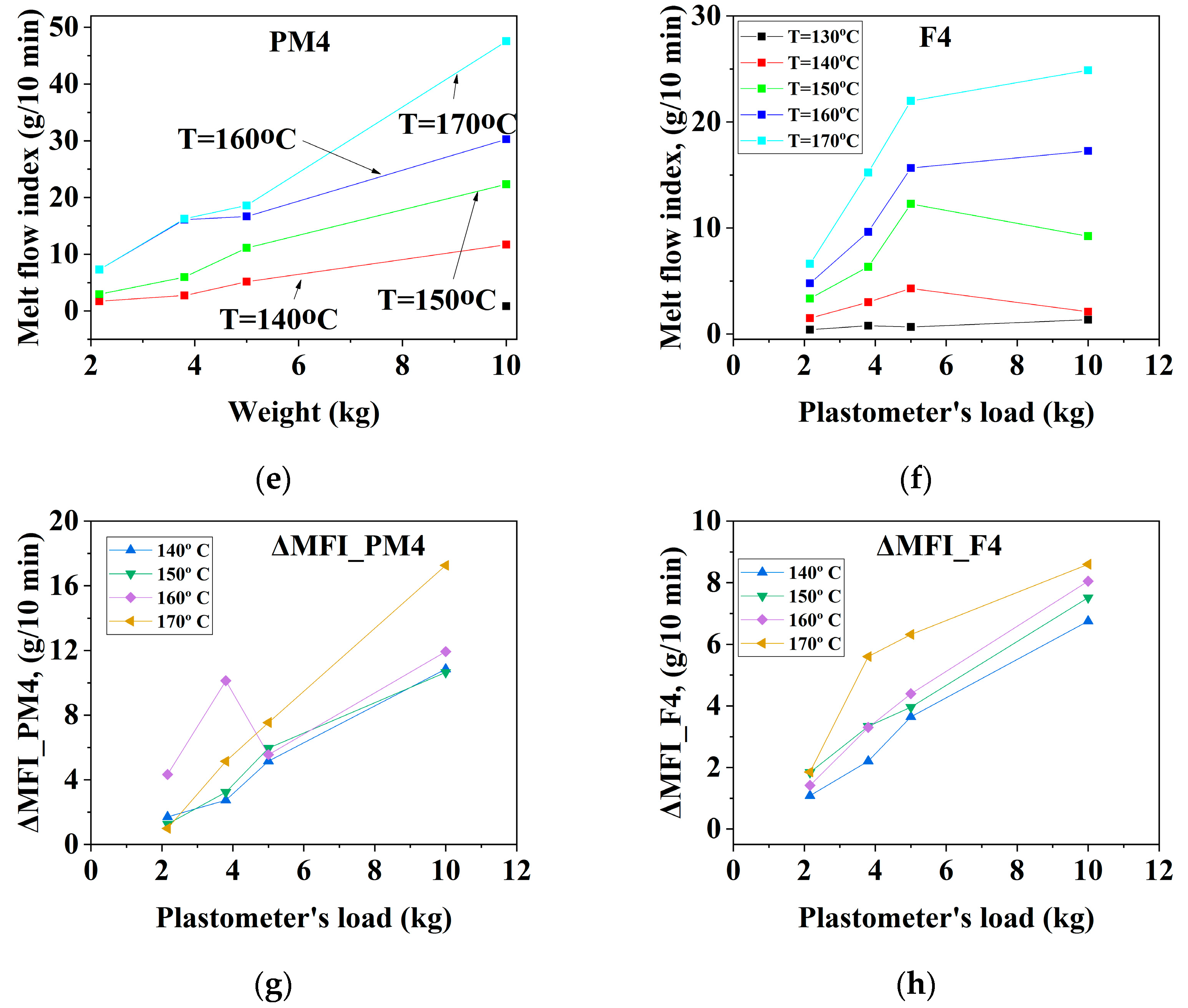
3.1.3. Mechanically Recycled Compounds from Pre-Consumer Waste from the Mass Manufacture of Green-Colored Film (PM4) and Primary Compound (F4) Used in this Production
3.2. Thermal Behavior
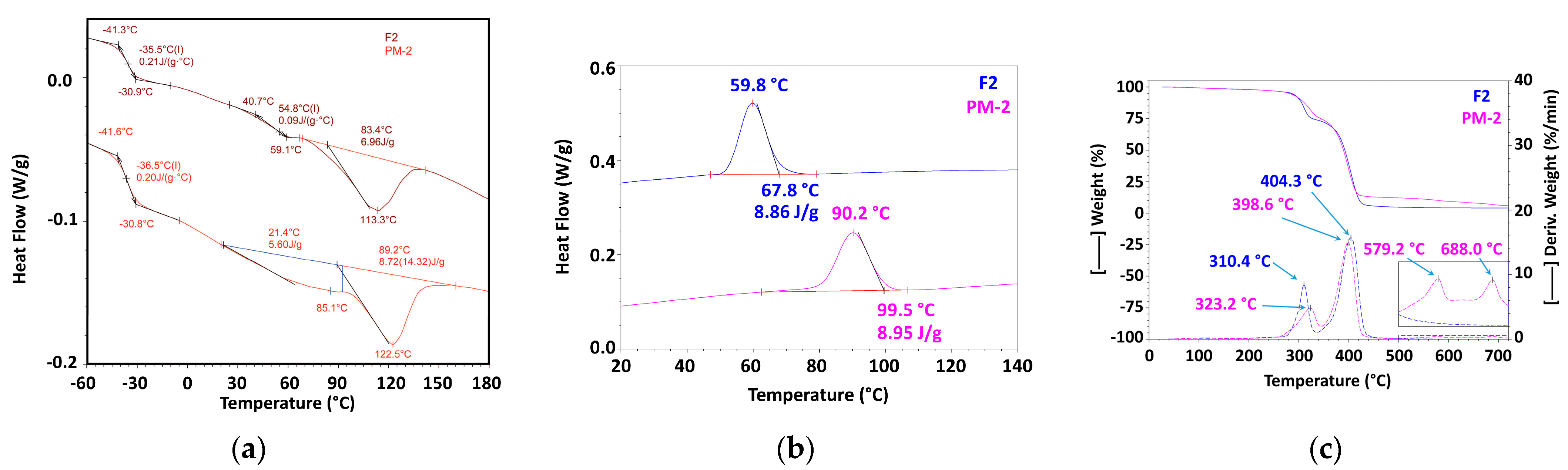
3.2.1. Mechanically Recycled Compounds from Pre-Consumer Waste Formed fromthe Manufacture of Films Flexographically Printed with Black (PM2) and Primary Pellets Used in this Production (F2) (Figure 5, Table 4, Table 5, Table 6, Table 7, Table 8, Table 9 and Table 10)

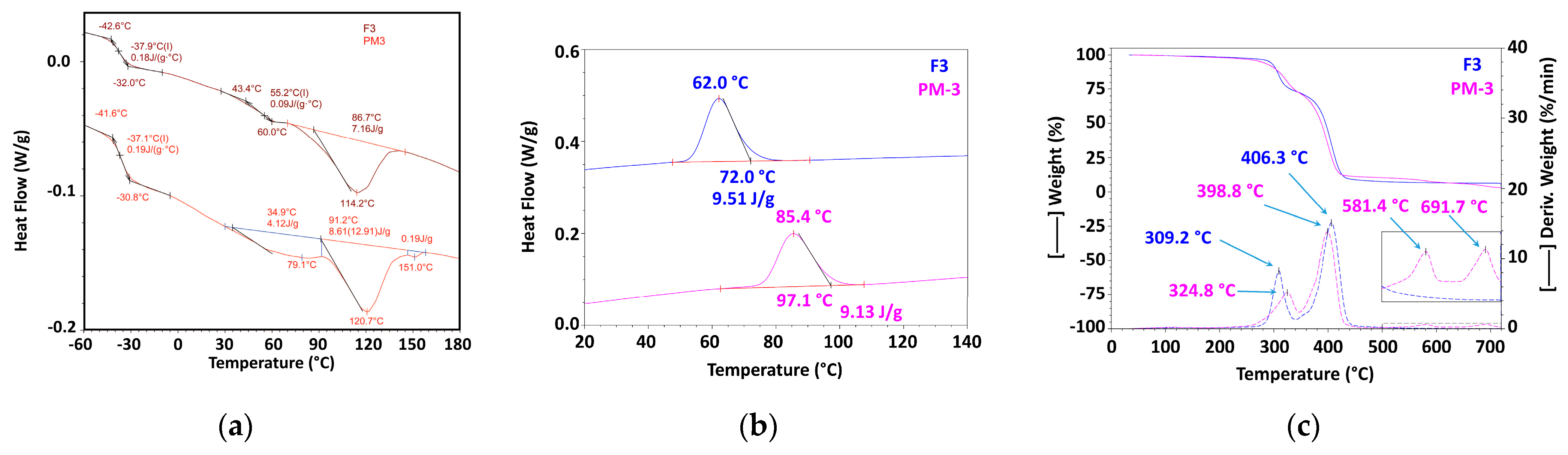
3.2.2. Mechanically Recycled Compounds from Pre-Consumer Waste Formed during the Manufacture of Non-Printed Films (PM3) and Primary Compound (F3) Used in this Production (Figure 6, Table 4, Table 5, Table 6, Table 7, Table 8, Table 9 and Table 10)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
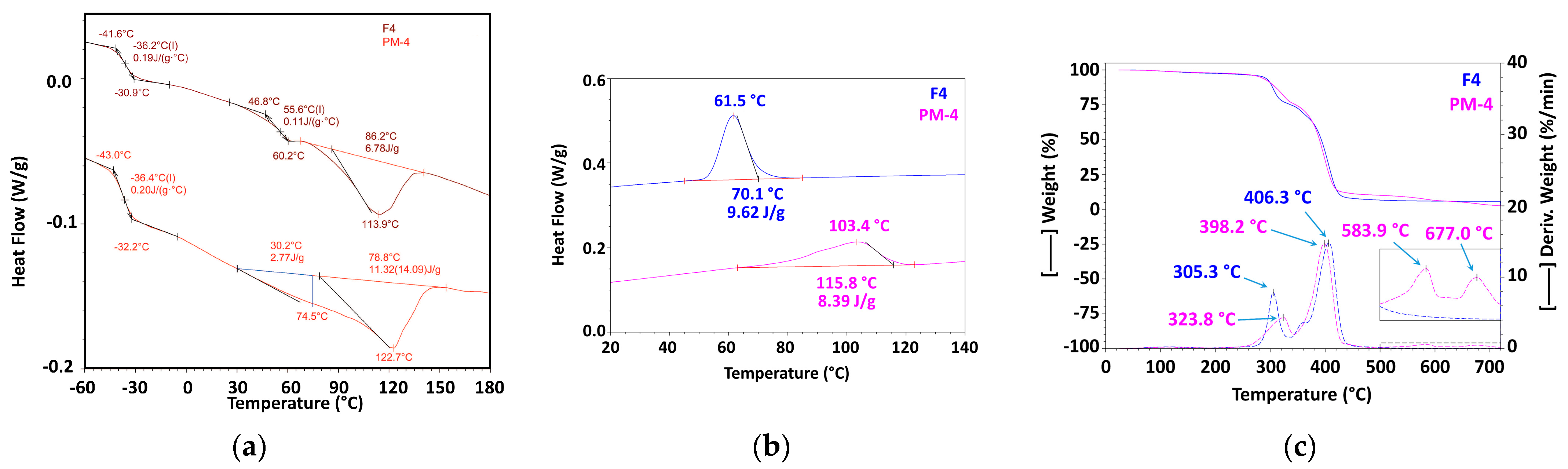
| 2nd Heating | Glass Transition 1 | Glass Transition2 | ||||||
|---|---|---|---|---|---|---|---|---|
| Onset | Tg | End | ΔCp | Onset | Tg | End | ΔCp | |
| °C | °C | °C | J/(g·°C) | °C | °C | °C | J/(g·°C) | |
| F2 | −41.3 | −35.5 | −30.9 | 0.21 | 40.7 | 54.8 | 59.1 | 0.09 |
| F3 | −42.6 | −37.9 | −32.0 | 0.18 | 43.4 | 55.2 | 60.0 | 0.09 |
| F4 | −41.6 | −36.2 | −30.9 | 0.19 | 46.8 | 55.6 | 60.2 | 0.11 |
| 2nd Heating | Enthalpy 2 | ||
|---|---|---|---|
| Onset2 | Tm2 | ΔH2 | |
| °C | °C | J/g | |
| F2 | 83.4 | 113.3 | 6.96 |
| F3 | 86.7 | 114.2 | 7.16 |
| F4 | 86.2 | 113.9 | 6.78 |
| 2nd Heating | Glass Transition | Enthalpy 1 | Enthalpy 2 | ΔH1 + ΔH2 | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Onset | Tg | End | ΔCp | Onset1 | Tm1 | ΔH1 | Onset2 | Tm2 | ΔH2 | ||
| °C | °C | °C | J/(g·°C) | °C | °C | J/g | °C | °C | J/g | J/g | |
| PM2 | −41.6 | −36.5 | −30.8 | 0.20 | 21.4 | 85.1 | 5.60 | 89.2 | 122.5 | 8.72 | 14.32 |
| PM3 | −41.6 | −37.1 | −30.8 | 0.19 | 34.9 | 79.1 | 4.12 | 91.2 | 120.7 | 8.61 | 12.72 |
| PM4 | −43.0 | −36.4 | −32.2 | 0.20 | 30.2 | 74.5 | 2.77 | 78.8 | 122.7 | 11.32 | 14.09 |
| Cooling | Crystallization | ||
|---|---|---|---|
| Onset | Tc | ΔHc | |
| °C | °C | J/g | |
| PM2 | 99.5 | 90.2 | 8.95 |
| PM3 | 97.1 | 85.4 | 9.13 |
| PM4 | 115.8 | 103.4 | 8.39 |
| Cooling | Crystallization | ||
|---|---|---|---|
| Onset | Tc | ΔHc | |
| °C | °C | J/g | |
| F2 | 67.8 | 59.8 | 8.86 |
| F3 | 72.0 | 62.0 | 9.51 |
| F4 | 70.1 | 61.5 | 9.62 |
| Code/Prop | RT-175 °C | 175–340 °C | 340–465 °C | 465–530 °C | 530–720 °C | Residue at 720 °C | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| Wt. Loss | Tmax | Wt. Loss | Tmax | Wt. Loss | Tmax | Wt. Loss | Wt. Loss | N2 | Air | |
| % | °C | % | °C | % | °C | % | % | % | % | |
| F2 | 1.50 | 108.9 | 24.55 | 310.4 | 68.64 | 404.3 | 0.81 | 0.44 | 4.05 | 0.24 |
| F3 | 1.35 | 128.1 | 25.37 | 309.2 | 65.02 | 406.3 | 1.26 | 0.77 | 6.23 | 0.41 |
| F4 | 2.31 | 118.4 | 22.90 | 305.3 | 67.43 | 406.3 | 1.11 | 0.67 | 5.58 | 0.37 |
| Code/Prop. | RT—175 °C | 175 °C–345 °C | 345 °C–485 °C | 485 °C–630 °C | 630 °C–750 °C | Residue at 750 °C | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Wt. Loss | Wt. Loss | Tmax | Wt. Loss | Tmax | Wt. Loss | Tmax | Wt. Loss | Tmax | N2 | Air | |
| % | % | °C | % | °C | % | °C | % | °C | % | % | |
| PM2 | 1.77 | 23.38 | 323.2 | 62.52 | 398.6 | 3.72 | 579.2 | 3.10 | 688.0 | 5.51 | 5.49 |
| PM3 | 1.80 | 25.54 | 324.8 | 62.14 | 398.8 | 4.21 | 581.4 | 3.96 | 691.7 | 2.35 | 2.19 |
| PM4 | 1.89 | 22.82 | 323.8 | 64.96 | 398.2 | 4.70 | 583.9 | 3.53 | 677 | 2.10 | 1.98 |
3.2.3. Mechanically Recycled Compound from Pre-Consumer Waste Generated fromthe Mass Manufacture of Green-Colored Films (PM4) and Primary Compound (F4) Used in this Production (Figure 7, Table 4, Table 5, Table 6, Table 7, Table 8, Table 9 and Table 10)
3.3. Chemical Structure
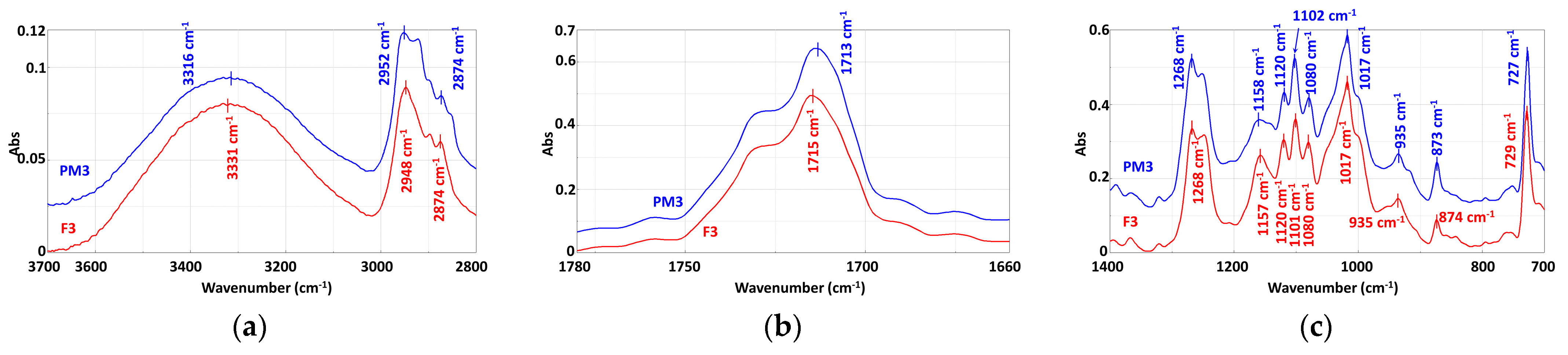
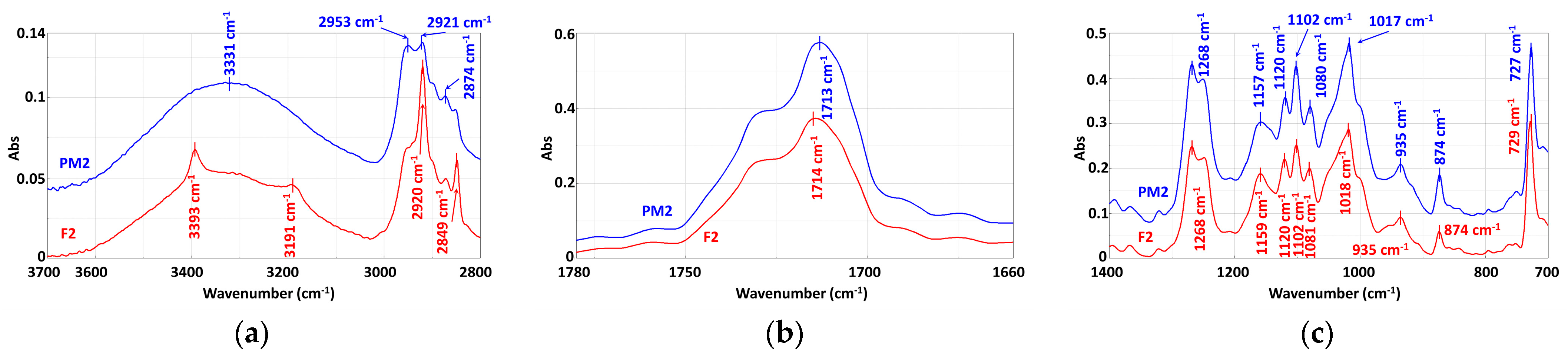
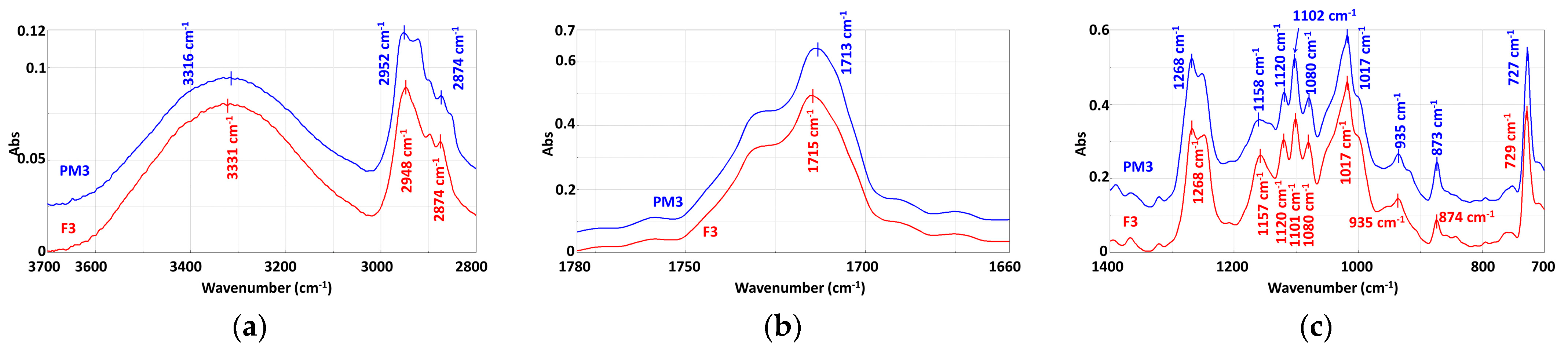
| Functional Group | Wavenumber, cm−1 |
|---|---|
| a. From compounds | |
| −OH –NH and/or –NH4+ | 3394 cm−1, 3335 cm−1, 3313 cm−1, 1631 cm−1, 652 cm−1, 647 cm−1, 615 cm−1, 611 cm−1 3200 cm−1 |
| –CH3 | 2953 cm−1, 2874 cm−1 |
| –CH din –CH2 | 2921 cm−1, 2850 cm−1 |
| C=O | 1754 cm−1, 1725 cm−1 shoulder, 1713 cm−1 |
| –C=C– unsaturated | 1646 cm−1, 1631 cm−1 |
| –C=C– unsaturated from the aromatic ring –Aromatic ring | 1578 cm−1505 cm−1 |
| –CH from –CH2 –CH2 and/or –O–CH | 1467 cm−1, 1454 cm−1, 1410 cm−1, 1392 cm−1, 1366 cm−1 1321 cm−1 |
| –C–C(O)–O | 1269 cm−1, 1252 cm−1 |
| –C–O–C and/or –(O)–OH –CH aromatic C–O | 1252 cm−1, 1160 cm−1, 1119 cm−1 1103 cm−1, 1080 cm−1, 868 cm−1 |
| –C–CH3 and/or C–OH –C–C –C–O –C–C– –CH2 of chain | 1080 cm−1, 1027 cm−1, 1018 cm−1, 935 cm−1, 759 cm−1 1018 cm−1 936 cm−1 868 cm−1 874 cm−1 729 cm−1 |
| b. From the compounds’ components | |
| Starch [23] | 3600–3300 cm−1 OH; 2931 cm−1 C–O C–O 1637–1458 cm−1 CH2 1385–1375 cm−1 C–H1149 cm−1 C–O–C 1200–800 cm−1 C–O 920 cm−1 856 cm−1 758 C cm−1–O–C ring vibration |
| PCL [24] | 3600 cm−1–3400 cm−1 -OH; 3000 cm−1–2800 cm−1–CH3, CH2, CH; C=O 1730 cm−1 |
| PBAT [24,25] | 3000 cm−1 alcohol OH; 1735 cm−1–1725 cm−1 C=O from ester; 1256 cm−1 C-O from ester; 901 cm−1–700 cm−1 benzene ring, 726 cm−1 4 adjacent –CH2– |
| Carbon black [26] | –COOH at 1720 cm−1, –C(O) –O–C(O), –(–C(=O)–O) 1735 cm−1, –C(O) –H 1700 cm−1 |
| Phthalocyanine [25] | NH AT 3289 cm−1 1006 cm−1, 3074 cm−1 3048 cm−1–CH asymmetric, 2923 cm−1 2854 cm−1–CH symmetric, 1609 cm−1 C–C in pyrrole, 1428 cm−1 1332 cm−1 C–C in isoindole 625 macro cycle ring 1517 cm−1 1490 cm−1 1478 cm−1 C–H in aryl 1184 cm−1 1164 cm−1 1070 cm−1 C–N in isoindole |
3.3.1. Mechanically Recycled Compounds from the Pre-Consumer Waste from the Manufacture of Films Flexographically Printed with Black (PM2) and from the Primary Pellets Used in this Production (F2) (Figure 8)
3.3.2. Mechanically Recycled Compounds from Pre-Consumer Waste Formed from Manufacture of Films (PM3) and Primary Pellets (F3) Used in this Production (Figure 9)
3.3.3. Mechanically Recycled Compound from Pre-Consumer Waste Formed from Mass Manufacture of Colored Film (PM4) and Primary Compound (F4) Used in this Production (Figure 10)

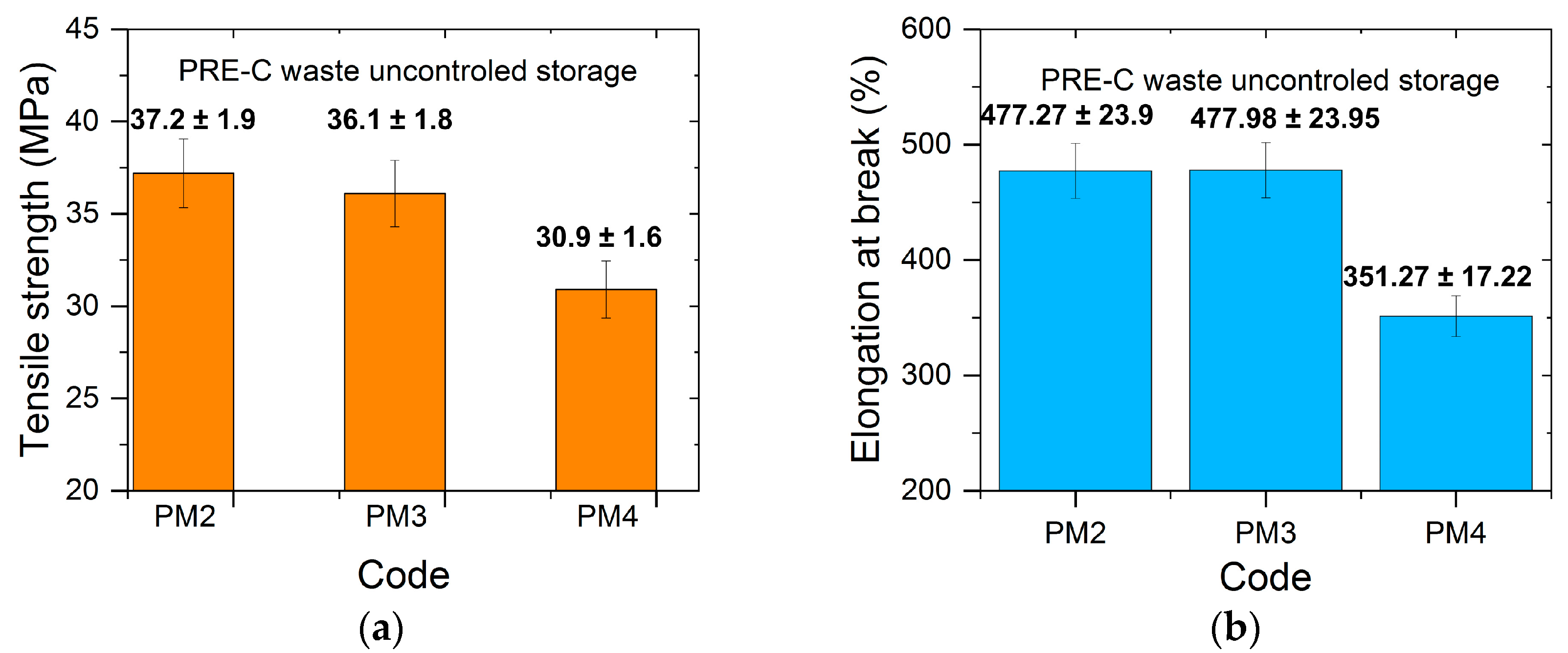
3.4. Mechanical Properties
4. Discussion
- the FTIR changes, which may have been a consequence of degradation via the breaking of macromolecular chains and of intermolecular bonds with more or less involvement of functional groups corresponding to each polymer from the studied compounds;
- the 2–5-times greater melt fluidity of all the mechanically recycled compounds, most likely being due to the PRE-CW degradation through chain breaking during storage, with the molecular weight decreasing;
- the melting of mechanically recycled polymers in successive processes, over the entire range of positive temperatures up to 120 °C, compared to a single melt, which had a relatively narrow end with a maximum at almost 120 °C. This behavior could be a result of the compositional de-mixing following the chain breaking and the alteration of the intermolecular interactions because of the storage degradation of the PET-CW;
- the crystallization of mechanically recycled compounds at 20 °C–35 °C higher temperatures than the primary compounds, possibly because of a large number of short chains appearing via chain breaking, which had a greater mobility and crystallized much more easily.
- the thermal degradation of the recycled compounds with the formation of slightly more stable compounds with the movement of the TGA degradation steps towards higher values and with a greater amount of residue at the end of the process, at 750 °C (stable compounds in air).
5. Conclusions
- 1.
- It was demonstrated that the pre-consumer waste (PRE-CW) from the manufacture of multilayer packaging films starting from starch-, PCL-, and PBAT-based compounds cannot be stored before mechanical recycling, especially in uncontrolled conditions.In spaces without ventilation, during the hot, rainy summers with alternating scorching heat and rain in these unventilated spaces, conditions similar to composting are created. Temperatures around 50 °C–60 °C enable enzymatic hydrolysis on the PET-CW surface, probably at the glycosidic linkages from starch and the esters from PCL (a polymer with a melting temperature of 60 °C) and PBAT. Once the sitesof destruction are created, then it propagates into the mass. Storage time accelerates the destruction that is also possibly due to the physical aging, which in turn can be enhanced by chemical degradation.
- 2.
- The degradation was highlighted by:
- the FTIR changes, which may be the consequence of some destruction processes, with the breaking of the macromolecular chains’, destruction of intermolecular bonds and/or more or less involvement of functional groups corresponding to each polymer from the studied compounds;
- the 2–5-times greater melt fluidity of all the mechanically recycled compounds, which was most likely due to the PET-CW degradation during storage with the molecular weight decreasing through chain breaking;
- the melting of mechanically recycled compounds in successive processes over the entire range of positive temperatures up to 120 °C, compared to melting with a single, relatively narrow endotherm with a maximum at almost 120 °C. This behavior can be a result of the partial compositional de-mixing following the alteration of the intermolecular interactions and/or the chain breaking, and of the storage degradation of PET-CW;
- the crystallization of mechanically recycled compounds at 20 °C–35 °C higher temperatures than the primary compounds, which was possible because of the large number of short chains appearing via chain breaking, which had a greater mobility and crystallized much more easily;
- the thermal degradation of recycled compounds with the formation of slightly more stable compounds with the movement of the TGA temperature steps towards higher values and with greater amounts of residues at the end of the process, at 750 °C (stable compounds in air).
- 3.
- The coloring did not introduce spectacular differences between the studied properties of the mechanically recycled pellets compared to the primary ones, possibly because of the very small amounts of dyes used. The flexographic printing with carbon black did not induce variations other than those recorded in the case for the uncolored recycled compound.
- 4.
- In order to make the mechanical recycling of starch- and biodegradable-polyester-based compounds for packaging films more efficient, the PET-CW must be immediately mechanically recycled without storage. This method avoids both chemical degradation and the physical aging, which can reciprocally potentiate each other much more the longer the storage time.
- 5.
- Mechanical recycling can also be made more efficient by drying the primary granules prior to mechanical recycling, and/or through the optimal selection of working conditions to ensure protection (low and medium temperatures, medium shear speeds) and the immediate recycling of waste.
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Horrocks, M. Post-Industrial Plastic Recycling Succes Story. Available online: https://blog.erema.com/post-industrial-plastic (accessed on 24 February 2020).
- European Bioplastics. PLA in the Waste Stream, 18 December 2017. Available online: https://www.european-bioplastics.org/pla-in-the-waste-stream/ (accessed on 6 January 2023).
- Wojnowska-Baryła, I.; Kulikowska, D.; Bernat, K. Effect of bio-based products on waste management. Sustainability 2020, 12, 2088. [Google Scholar] [CrossRef]
- Sesotec GmbH. Recycling Rate, Recyclate Content and the Impact on the Plastics Industry. Available online: https://www.sesotec.com/emea/en/resources/blog/recycling-rate-recyclate-content-and-the-impact-on-the-plastics-industry (accessed on 6 January 2023).
- Elsayed, H.; Farag, M.; Megahed, H.; Mehanny, S. Influence of flax fibers on properties of starch-based composites. In Proceedings of the ASME 2012 International Mechanical Engineering Congress and Exposition, IMECE 2012, Houston, TX, USA, 9–15 November 2012; American Society of Mechanical Engineers: New York, NY, USA, 2012; pp. 1397–1408. [Google Scholar]
- Mehanny, S.; Abu-El Magd, E.E.; Sorbara, S.; Navarro, J.; Gil-San-millan, R. Spanish poplar biomass as a precursor for nanocellulose extraction. Appl. Sci. 2021, 11, 6863. [Google Scholar] [CrossRef]
- Mohamed, H.E.-M.; Justin, K.; Sea-Hyun, L.; Mahmoud, F.; Sherif, M. Potential of lignin valorization with emphasis on bioepoxy production. In Lignin-Chemistry, Structure, and Application; Arpit, S., Jaya, T., Eds.; IntechOpen: Rijeka, Croatia, 2022; p. 4. [Google Scholar]
- Rusu, M.; Goldenberg-Şerban, N.; Ivan, G. Valorificarea Resurselor Polimerice Secundare; Editura Tehnică: Louisville, KY, USA, 1989. [Google Scholar]
- Dimonie, D. Materiale Polimerice Multifazice Micro-si Nano-Structurate; Politehnica Press: Bucuresti, Romania, 2019; ISBN 978-606-515-893-1. [Google Scholar]
- Grima, S.; Bellon-Maurel, V.; Feuilloley, P.; Silvestre, F. Aerobic biodegradation of polymers in solid-state conditions: A review of environmental and physicochemical parameter settings in laboratory simulations. J. Polym. Environ. 2000, 8, 183–195. [Google Scholar] [CrossRef]
- Cangialosi, D.; Boucher, V.M.; Alegría, A.; Colmenero, J. Physical aging in polymers and polymer nanocomposites: Recent results and open questions. Soft Matter 2013, 9, 8619–8630. [Google Scholar] [CrossRef]
- Chen, C.; Du, Y.; Zuo, G.; Chen, F.; Liu, K.; Zhang, L. Effect of storage condition on the physico-chemical properties of corn–wheat starch/zein edible bilayer films. R. Soc. Open Sci. 2020, 7, 191777. [Google Scholar] [CrossRef]
- Cowie, J.M.G.; Arrighi, V. Physical aging of polymer blends. In Polymer Blends Handbook; Utracki, L.A., Wilkie, C.A., Eds.; Springer: Dordrecht, The Netherlands, 2014; pp. 1357–1394. [Google Scholar]
- Frieberg, B.R.; Glynos, E.; Sakellariou, G.; Tyagi, M.; Green, P.F. Effect of molecular stiffness on the physical aging of polymers. Macromolecules 2020, 53, 7684–7690. [Google Scholar] [CrossRef]
- Minguez, R.; Barrenetxea, L.; Solaberrieta, E.; Lizundia, E. A simple approach to understand the physical aging in polymers. Eur. J. Phys. 2019, 40, 015502. [Google Scholar] [CrossRef]
- Verdu, J. Effect of aging on the mechanical properties of polymeric materials. J. Macromol. Sci. Part A 1994, 31, 1383–1398. [Google Scholar] [CrossRef]
- Doina, D.; Mircea, F.; Mihai, D.; Alina, M.; Nicoleta, D. Micro and nano structuring as method to enhance the functional properties of starch-based polymeric materials. In Starch; Martins Ochubiojo, E., Ed.; IntechOpen: Rijeka, Croatia, 2021; p. 10. [Google Scholar]
- Dimonie, D.; Petrache, M.; Damian, C.; Anton, L.; Musat, M.; Dima, Ş.-O.; Jinescu, C.; Maria, R. New evidences on the process sensitivity of some renewable blends based on starch considering their melt rheological properties. Int. J. Polym. Sci. 2016, 2016, 7873120. [Google Scholar] [CrossRef]
- Dimonie, D.; Damian, C.; Trusca, R.; Rapa, M. Some aspects conditioning the achieving of filaments for 3D printing from physical modified corn starch. Mater. Plast. 2019, 56, 351–359. [Google Scholar] [CrossRef]
- Dimonie, D.; Musat, M.; Doncea, S.M.; Damian, C.M.; Anton, L.; Vasile, E.; Trusca, R.; Râpă, M. Controlling the melt resistance to flow as a possibility of improving the miscibility and the time behavior of some blends based on starch. Int. J. Polym. Sci. 2015, 2015, 582901. [Google Scholar] [CrossRef]
- Song, J.H.; Murphy, R.J.; Narayan, R.; Davies, G.B.H. Biodegradable and compostable alternatives to conventional plastics. Philos. Trans. R. Soc. B Biol. Sci. 2009, 364, 2127–2139. [Google Scholar] [CrossRef] [PubMed]
- Mehanny, S.; Ibrahim, H.; Darwish, L.; Farag, M.; El-Habbak, A.-H.M.; El-Kashif, E. Effect of environmental conditions on date palm fiber composites. In Date Palm Fiber Composites: Processing, Properties and Applications; Midani, M., Saba, N., Alothman, O.Y., Eds.; Springer: Singapore, 2020; pp. 287–320. [Google Scholar]
- Schyns, Z.O.G.; Shaver, M.P. Mechanical recycling of packaging plastics: A review. Macromol. Rapid Commun. 2021, 42, 2000415. [Google Scholar] [CrossRef] [PubMed]
- Cross, M.M. Viscosity, molecular weight and chain entanglement. Polymer 1970, 11, 238–244. [Google Scholar] [CrossRef]
- Mezger, T.G. The Rheology Handbook; Vincentz Network: Hanover, Germany, 2021. [Google Scholar]
- Morris, B.A. 5-rheology of polymer melts. In The Science and Technology of Flexible Packaging; Morris, B.A., Ed.; William Andrew Publishing: Oxford, UK, 2017; pp. 121–147. [Google Scholar]
- Jalali, A.; Huneault, M.A.; Elkoun, S. Effect of thermal history on nucleation and crystallization of poly(lactic acid). J. Mater. Sci. 2016, 51, 7768–7779. [Google Scholar] [CrossRef]
- Deng, Y.; Yu, C.; Wongwiwattana, P.; Thomas, N.L. Optimising ductility of poly(lactic acid)/poly(butylene adipate-co-terephthalate) blends through co-continuous phase morphology. J. Polym. Environ. 2018, 26, 3802–3816. [Google Scholar] [CrossRef]
- Tiptipakorn, S.; Keungputpong, N.; Phothiphiphit, S.; Rimdusit, S. Effects of polycaprolactone molecular weights on thermal and mechanical properties of polybenzoxazine. J. Appl. Polym. Sci. 2015, 132. [Google Scholar] [CrossRef]
- Dimonie, D.; Radu, S.; Doncea, S.; Pop, F.S.; Petre, C.; Dumitriu, I.; Fierascu, R. The miscibility estimation of some nanocomposites based on starch. e-Polymers 2011, 11. [Google Scholar] [CrossRef]
- Dimonie, D.; Dimonie, M.; Vasilievici, G. Polyolefines mechanical recycling considering the carbonyl variation during the first life (i). Mater. Plast. 2007, 44, 361. [Google Scholar]
- Dimonie, D.; Dimonie, M.; Vasilievici, G. Polyolefines mechanical recycling considering the carbonyl variation during the first life (ii). Mater. Plast. 2008, 45, 91. [Google Scholar]
- Liu, X.; Wang, Y.; Yu, L.; Tong, Z.; Chen, L.; Liu, H.; Li, X. Thermal degradation and stability of starch under different processing conditions. Starch-Stärke 2013, 65, 48–60. [Google Scholar] [CrossRef]
- Merino, D.; A Alvarez, V. In-soil biodegradation behavior of chitosan-coated phosphorylated starch films. Adv. Mater. Lett. 2019, 10, 907–912. [Google Scholar] [CrossRef]
- Ahimbisibwe, M.; Banadda, N.; Seay, J.; Nabuuma, B.; Atwijukire, E.; Wembabazi, E.; Nuwamanya, E. Influence of weather and purity of plasticizer on degradation of cassava starch bioplastics in natural environmental conditions. J. Agric. Chem. Environ. 2019, 8, 237–250. [Google Scholar] [CrossRef]
- Rocha, T.d.S.; Carneiro, A.P.d.A.; Franco, C.M.L. Effect of enzymatic hydrolysis on some physicochemical properties of root and tuber granular starches. Food Sci. Technol. 2010, 30, 544–551. [Google Scholar] [CrossRef]
- Liu, W.-C.; Halley, P.J.; Gilbert, R.G. Mechanism of degradation of starch, a highly branched polymer, during extrusion. Macromolecules 2010, 43, 2855–2864. [Google Scholar] [CrossRef]
- Liu, X.; Xiao, X.; Liu, P.; Yu, L.; Li, M.; Zhou, S.; Xie, F. Shear degradation of corn starches with different amylose contents. Food Hydrocoll. 2017, 66, 199–205. [Google Scholar] [CrossRef]
- Greenwood, C.T. The thermal degradation of starch. In Advances in Carbohydrate Chemistry; Wolfrom, M.L., Tipson, R.S., Eds.; Academic Press: Cambridge, MA, USA, 1967; Volume 22, pp. 483–515. [Google Scholar]
- Cesur, S. The effects of additives on the biodegradation of polycaprolactone composites. J. Polym. Environ. 2018, 26, 1425–1444. [Google Scholar] [CrossRef]
- Heimowska, A.; Morawska, M.; Bocho-Janiszewska, A. Biodegradation of poly(ε-caprolactone) in natural water environments. Pol. J. Chem. Technol. 2017, 19, 120–126. [Google Scholar] [CrossRef]
- Mudhoo, A.; Mohee, R.; Unmar, G.D.; Sharma, S.K. Degradation of biodegradable and green polymers in the composting environment. In A Handbook of Applied Biopolymer Technology: Synthesis, Degradation and Applications; Royal Society of Chemistry: London, UK, 2011; pp. 332–364. [Google Scholar]
- Rutkowska, M.; Krasowska, K.; Heimowska, A.; Steinka, I.; Janik, H.; Haponiuk, J.; Karlsson, S. Biodegradation of modified poly(ε-caprolactone) in different environments. Pol. J. Environ. Stud. 2002, 11, 413–420. [Google Scholar]
- Bartnikowski, M.; Dargaville, T.R.; Ivanovski, S.; Hutmacher, D.W. Degradation mechanisms of polycaprolactone in the context of chemistry, geometry and environment. Prog. Polym. Sci. 2019, 96, 1–20. [Google Scholar] [CrossRef]
- Ferreira, F.V.; Cividanes, L.S.; Gouveia, R.F.; Lona, L.M.F. An overview on properties and applications of poly(butylene adipate-co-terephthalate)–pbat based composites. Polym. Eng. Sci. 2019, 59, E7–E15. [Google Scholar] [CrossRef]
- Chaves, R.P.; Fechine, G.J.M. Thermo stabilisation of poly (butylene adipate-co-terephthalate). Polimeros 2016, 26, 102–105. [Google Scholar] [CrossRef]
- Encalada, K.; Aldás, M.B.; Proaño, E.; Valle, V. An overview of starch-based biopolymers and their biodegradability. Cienc. Ing. 2018, 39, 245–258. [Google Scholar]
- Iwamoto, A.; Tokiwa, Y. Effect of the phase structure on biodegradability of polypropylene/poly(ε-caprolactone) blends. J. Appl. Polym. Sci. 1994, 52, 1357–1360. [Google Scholar] [CrossRef]
- Jayasekara, R.; Harding, I.; Bowater, I.; Lonergan, G. Biodegradability of a selected range of polymers and polymer blends and standard methods for assessment of biodegradation. J. Polym. Environ. 2005, 13, 231–251. [Google Scholar] [CrossRef]
- Sugih, A.K. Synthesis and Properties of Starch Based Biomaterials; University Library Groningen [Host]: Groningen, The Netherlands, 2008. [Google Scholar]
- Vikman, M.; Hulleman, S.H.D.; Van Der Zee, M.; Myllärinen, P.; Feil, H. Morphology and enzymatic degradation of thermoplastic starch–polycaprolactone blends. J. Appl. Polym. Sci. 1999, 74, 2594–2604. [Google Scholar] [CrossRef]
- Kliem, S.; Kreutzbruck, M.; Bonten, C. Review on the biological degradation of polymers in various environments. Materials 2020, 13, 4586. [Google Scholar] [CrossRef]
- La Fuente, C.I.A.; Maniglia, B.C.; Tadini, C.C. Biodegradable polymers: A review about biodegradation and its implications and applications. Packag. Technol. Sci. 2023, 36, 81–95. [Google Scholar] [CrossRef]
- Lucas, N.; Bienaime, C.; Belloy, C.; Queneudec, M.; Silvestre, F.; Nava-Saucedo, J.-E. Polymer biodegradation: Mechanisms and estimation techniques–A review. Chemosphere 2008, 73, 429–442. [Google Scholar] [CrossRef] [PubMed]
- Samir, A.; Ashour, F.H.; Hakim, A.A.A.; Bassyouni, M. Recent advances in biodegradable polymers for sustainable applications. NPJ Mater. Degrad. 2022, 6, 68. [Google Scholar] [CrossRef]
- Si, S.; Tang, Q.; Li, X. The accelerated thermo-oxidative aging characteristics of wood fiber/polycaprolactone composite: Effect of temperature, humidity and time. J. Renew. Mater. 2021, 9, 2209. [Google Scholar] [CrossRef]
- Bossard, F.; Pillin, I.; Aubry, T.; Grohens, Y. Rheological characterization of starch derivatives/polycaprolactone blends processed by reactive extrusion. Polym. Eng. Sci. 2008, 48, 1862–1870. [Google Scholar] [CrossRef]
- White, J.R. Polymer ageing: Physics, chemistry orengineering? Time toreflect. Comptes Rendus Chim. 2006, 9, 1396–1408. [Google Scholar] [CrossRef]
- Caddeo, C.; Ackermann, J.; Mattoni, A. A theoretical perspective on the thermodynamic stability of polymer blends for solar cells: From experiments to predictive modeling. Solar RRL 2022, 6, 2200172. [Google Scholar] [CrossRef]
- Wolf, B.A. Polymer incompatibility caused by different molecular architectures: Modeling via chain connectivity and conformational relaxation. Macromol. Theory Simul. 2010, 19, 36–43. [Google Scholar] [CrossRef]

| Code | Fraction of PRE-CW after Color Separation | Coloring | MFI x, g/10 min |
|---|---|---|---|
| PM2 | Flexographic printing with black | Carbon black | 10.1 |
| PM3 | Unprinted, uncolored | - | 14.2 |
| PM4 | Bulk coloring | Phthalocyanine blue | 14.8 |
| Code\Properties | Properties of Primary Compounds 2x | ||||
|---|---|---|---|---|---|
| Density (ρ) g/cm3 | Fluidity (MFI) 3x, g/10 min | Melting Temp., °C | Break Tensile Strength, (σbr), MPa | Elongation at Break, (ε), % | |
| F2 | 1.29 | 3 | 118 | 30 | 450 |
| F3 | 1.27 | 4.5 | 117 | 35 | 485 |
| F4 | 1.28 | 3 | 115 | 30 | 390 |
| CODE | The Variation of the Fluidity Increment in the Ranges of 130 °C–170 °C and 2.16–10 kg Load for Mechanically Recycled Pre-Consumer Wastes and Primary Compounds | |||
|---|---|---|---|---|
| g/10 min | g/10 min | |||
| 2.16 kg–5 kg | 10 kg | 2.16 kg–5 kg | 10 kg | |
| I. Films with surfaceflexographically printed with black pigments | ||||
| PM2 | 1–8 | 13–13.5 | 140–150 °C: 2–5 160–170 °C: 2–6 | 140–150 °C: 9–10 160–170 °C: 2–14 |
| F2 | 1–4 | 140–150 °C: 12 160–170 °C: 1–4 | 140–150 °C: 1.5–4.5 160–170 °C: 1–2 | 140–150 °C: 1–2 160–170 °C: 10–11 |
| II. Colorless film | ||||
| PM3 | 1–6 | 13.5–16 | 0.5–6 | 4–16 |
| F3 | 0.5–2 | 7.5–9.5 | 0.2–2 | 1.5–4.5 |
| III. Mass colored films (green pigments) | ||||
| PM4 | 1–6 | 11.5–18 | 1–6.5 | 5.5–18 |
| F4 | 0.5–5.5 | 11–15 | 0.9–6 | 5–8.5 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dimonie, D.; Dragne, M.; Trica, B.; Nicolae, C.-A.; Raduly, M.; Doncea, S.; Ladaniuc, M.; Mustatea, A.; Miu, F.; Soare, L.; et al. New Biodegradable Materials for Re-Thought Packaging from Pre-Consumer Wastes by Controlling the Storage Time as Method to Increase the Mechanical Recycling Efficiency. Materials 2023, 16, 1503. https://doi.org/10.3390/ma16041503
Dimonie D, Dragne M, Trica B, Nicolae C-A, Raduly M, Doncea S, Ladaniuc M, Mustatea A, Miu F, Soare L, et al. New Biodegradable Materials for Re-Thought Packaging from Pre-Consumer Wastes by Controlling the Storage Time as Method to Increase the Mechanical Recycling Efficiency. Materials. 2023; 16(4):1503. https://doi.org/10.3390/ma16041503
Chicago/Turabian StyleDimonie, Doina, Mihail Dragne, Bogdan Trica, Cristian-Andi Nicolae, Monica Raduly, Sanda Doncea, Magda Ladaniuc, Alina Mustatea, Florentina Miu, Laurentiu Soare, and et al. 2023. "New Biodegradable Materials for Re-Thought Packaging from Pre-Consumer Wastes by Controlling the Storage Time as Method to Increase the Mechanical Recycling Efficiency" Materials 16, no. 4: 1503. https://doi.org/10.3390/ma16041503
APA StyleDimonie, D., Dragne, M., Trica, B., Nicolae, C.-A., Raduly, M., Doncea, S., Ladaniuc, M., Mustatea, A., Miu, F., Soare, L., & Georgescu, T. (2023). New Biodegradable Materials for Re-Thought Packaging from Pre-Consumer Wastes by Controlling the Storage Time as Method to Increase the Mechanical Recycling Efficiency. Materials, 16(4), 1503. https://doi.org/10.3390/ma16041503