
A Nitrate-Transforming Bacterial Community Dominates in the Miscanthus Rhizosphere on Nitrogen-Deficient Volcanic Deposits of Miyake-jima

 , ,
, ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Site Description and Sample Collection
2.2. Soil Chemical Properties Analysis
2.3. Sample Pretreatment of Miscanthus
2.4. Genome Sequencing, Assembly, and Annotation of Metagenomic Contigs
2.5. Isolation of the Miscanthus Root-Associated Bacteria
2.6. Terminal-Restriction Fragment Length Polymorphism (T-RFLP) Profiling
2.7. Amplified Ribosomal DNA Restriction Analysis (ARDRA)
2.8. Identification of Representative Strains of Each OTU Based on the 16S rRNA Gene
2.9. Measurement of the Potential Activity of Denitrification
2.10. Phylogenetic Analysis
2.11. Accession Number
3. Results
3.1. Chemical Properties of Substrates in Site IG7
3.2. Metagenome Assembly and Nitrogen Cycle-Related Gene Abundance
3.3. Diversity and Taxonomy of Cultured Bacteria with Potential Nitrogen Transformation in the Miscanthus Rhizosphere
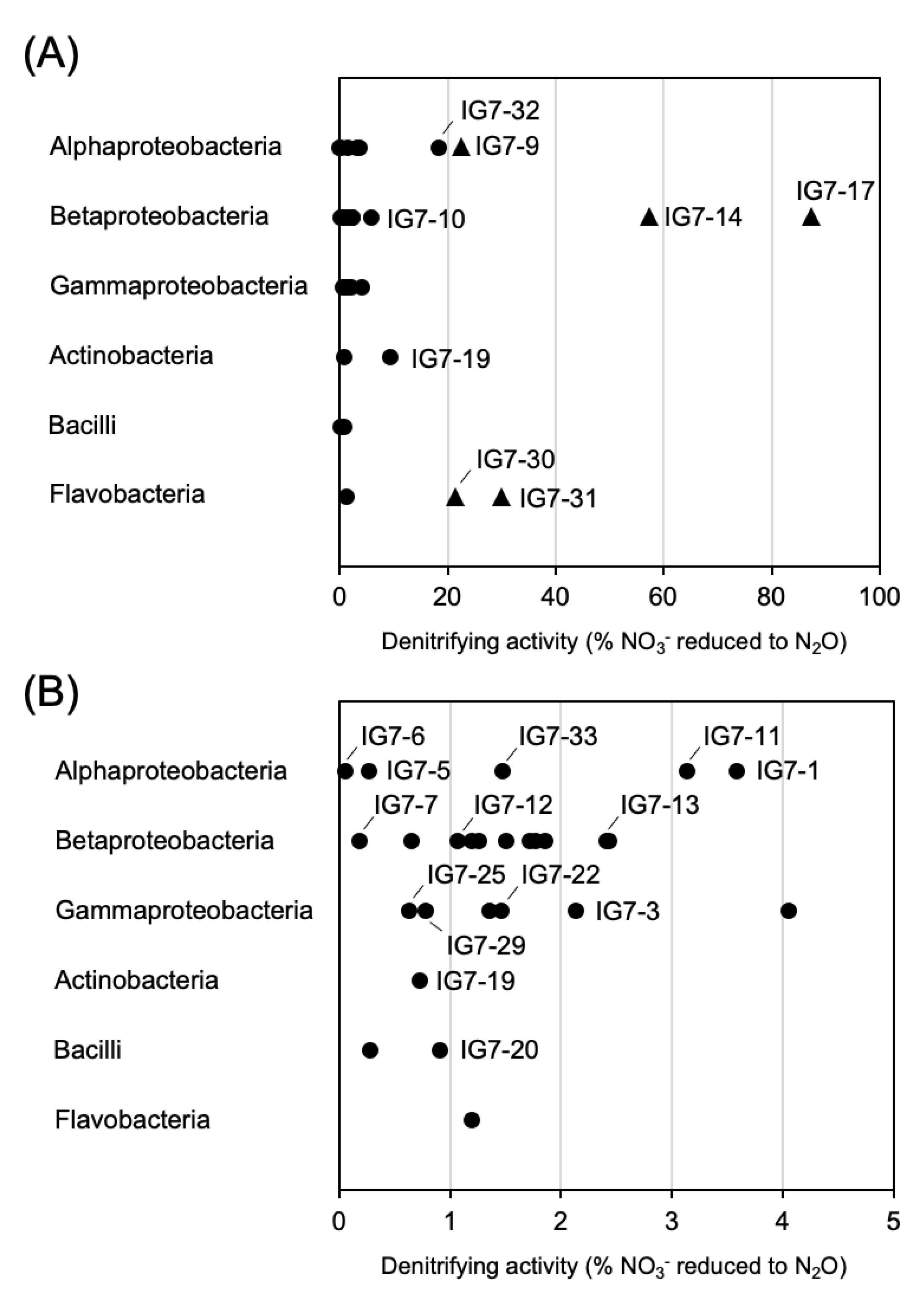
3.4. Denitrifying Activity
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kandeler, E.; Deiglmayr, K.; Tscherko, D.; Bru, D.; Pilippot, L. Abundance of narG, nirS, nirK, and nosZ Genes of Denitrifying Bacteria during Primary Sucessions of a Glacier Foreland. Appl. Environ. Microbiol. 2006, 72, 5957–5962. [Google Scholar] [CrossRef] [PubMed]
- Fujimura, R.; Kim, S.W.; Sato, Y.; Oshima, K.; Hattori, M.; Kamijo, T.; Ohta, H. Unique Pioneer Microbial Communities Exposed to Volcanic Sulfur Dioxide. Sci. Rep. 2016, 6, 19687. [Google Scholar] [CrossRef] [PubMed]
- Kato, T.; Kamijo, T.; Hatta, T.; Tamura, K.; Higashi, T. Initial Soil Formation Processes of Volcanogenous Regosols (Scoriaciuos) from Miyake-jima Island Japan. Soil Sci. Plant Nutr. 2005, 51, 291–301. [Google Scholar] [CrossRef]
- Sato, Y.; Hosokawa, K.; Fujimura, R.; Nishizawa, T.; Kamijo, T.; Ohta, H. Nitrogenase Activity (Acetylene Reduction) of an Iron-oxidizing Leptospirillum Strain Cultured as a Pioneer Microbe from a Recent Volcanic Deposit on Miyake-jima, Japan. Microbes Environ. 2009, 24, 291–296. [Google Scholar] [CrossRef]
- Fujimura, R.; Sato, Y.; Nishizawa, T.; Nanba, K.; Oshima, K.; Hattori, M.; Kamijo, T.; Ohta, H. Analysis of Early Bacterial Communities on Volcanic Deposits on the Island of Miyake (Miyake-jima) Japan: A 6-year Study at a Fixed Site. Microbes Environ. 2012, 27, 19–29. [Google Scholar] [CrossRef]
- Guo, Y.; Fujimura, R.; Sato, Y.; Suda, W.; Kim, S.W.; Oshima, K.; Hattori, M.; Kamijo, T.; Narisawa, K.; Ohta, H. Characterization of Early Microbial Communities on Volcanic Deposits along a Vegetation Gradient on the Island of Miyake, Japan. Microbes Environ. 2014, 29, 38–49. [Google Scholar] [CrossRef]
- Lathifah, A.N.; Guo, Y.; Sakagami, N.; Suda, W.; Higuchi, M.; Nishizawa, T.; Prijambada, I.; Ohta, H. Comparative Characterization of Bacterial Communities in Moss-covered and Unvegetated Volcanic Deposits of Mount Merapi, Indonesia. Microbes Environ. 2019, 34, 268–277. [Google Scholar] [CrossRef]
- Guo, Y.; Nishizawa, T.; Sakagami, N.; Fujimura, R.; Kamijo, T.; Ohta, H. Root Bacteriome of a Pioneer Grass Miscanthus condensatus along Restored Vegetation on Recent Miyake-jima Volcanic Deposits. Rhizosphere 2021, 19, 100422. [Google Scholar] [CrossRef]
- Stewart, J.R.; Toma, Y.; Fernández, F.G.; Nishiwaki, A.; Yamada, T.; Bollero, G. The Ecology and Agronomy of Miscanthus sinensis, A Specie Important to Bioenergy Crop Development, in Its Native Range in Japan: A Review. GCB-Bioenergy 2009, 1, 126–153. [Google Scholar] [CrossRef]
- An, G.H.; Miyakawa, S.; Kawahara, A.; Osaki, M.; Ezawa, T. Community Structure of Arbuscular Mycorrhizal Fungi Associated with Pioneer Grass Species Miscanthus sinensis in Acid Sulfate Soils: Habitat Segregation along pH Gradients. Soil Sci. Plant Nutr. 2008, 54, 517–528. [Google Scholar] [CrossRef]
- Kawahara, A.; An, G.H.; Miyakawa, S.; Sonoda, J.; Ezawa, T. Nestedness in Arbuscular Mycorrhizal Fungal Communities along Soil pH Gradient in Early Primary Succession: Acid-Tolerant Fungi Are pH Generalists. PLoS ONE 2016, 11, e0165035. [Google Scholar] [CrossRef] [PubMed]
- Pandey, C.B.; Kumar, U.; Kaviraj, M.; Minick, K.J.; Mishra, A.K.; Singh, J.S. DNRA: A Short-Circuit in Biological N-cycling to Conserve Nitrogen in Terrestrial Ecosystems. Sci. Total Environ. 2020, 738, 139710. [Google Scholar] [CrossRef] [PubMed]
- Tiedje, J.M. Ecology of Denitrification and Dissimilatory Nitrate Reduction to Ammonium. In Biology of Anaerobic Microorganisms; Zehnder, A.J.B., Ed.; John Wiley and Sons: New York, NY, USA, 1988; pp. 179–244. [Google Scholar]
- Zumft, W.G. Cell Biology and Molecular Basis of Denitrification. Microbiol. Mol. Biol. Rev. 1997, 61, 533–616. [Google Scholar] [PubMed]
- Rich, J.J.; Heichen, R.S.; Bottomley, P.J.; Cromack Jr., K.; Myrold, D.D. Community Composition and Functioning of Denitrifying Bacteria from Adjacent Meadow and Forest Soils. Appl. Environ. Microbiol. 2003, 69, 5974–5982. [Google Scholar] [CrossRef] [PubMed]
- Silver, W.L.; Herman, D.J.; Firestone, M.K. Dissimilatory Nitrate Reduction to Ammonium in Upland Tropical Forest Soils. Ecology 2001, 82, 2410–2416. [Google Scholar] [CrossRef]
- Isobe, K.; Ohte, N. Ecological Perspectives on Microbes Involved in N-Cycling. Microbes Environ. 2014, 29, 4–16. [Google Scholar] [CrossRef]
- Kato, T.; Higashi, T.; Kamijo, T.; Tamura, K. Several Chemical and Mineralogical Properties of Volcanic Ash Samples Erupted in 2000 from Miyake Island. Pedrogist 2000, 46, 14–21. (In Japanese) [Google Scholar]
- Kamijo, T.; Hashiba, K. Island Ecosystem and Vegetation Dynamics before and after the 2000-year Eruption on Miyake-jima Ssland, Japan, with Implications for Conservation of the Island’s Ecosystem. Glob. Environ. Res. 2003, 7, 69–78. [Google Scholar]
- Yoshinaga, S.; Abe, K.; Okamoto, T. Effects of Soil Chemical Properties on Forest Decline in Miyake Island, Japan. Phyton 2005, 45, 429–435. [Google Scholar]
- Kamijo, T.; Kawagoe, M.; Kato, T.; Kiyohara, Y.; Matsuda, M.; Hashiba, K.; Shimada, K. Destruction and Recovery of Vegetation Caused by the 2000-year Eruption on Miyake-jima Island Japan. J. Disaster Res. 2008, 3, 226–236. [Google Scholar] [CrossRef]
- Uritskiy, G.V.; DiRuggiero, J.; Taylor, J. MetaWRAP—A Flexible Pipeline for Genome-resolved Metagenomic Data Analysis. Microbiome 2018, 6, 158. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Liu, C.M.; Luo, R.; Sadakane, K.; Lam, T.W. MEGAHIT: An Ultra-fast Single-node Solution for Large and Complex Metagenomics Assembly via Succinct de Bruijn Graph. Bioinformatics 2015, 31, 1674–1676. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Lomsadze, A.; Borodovsky, M. Ab initio Gene Identification in Metagenomic Sequences. Nucleic Acids Res. 2010, 38, e132. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Sato, Y.; Morishima, K. BlastKOALA and GhostKOALA: KEGG Tools for Functional Characterization of Genome and Metagenome Sequences. J. Mol. Biol. 2016, 428, 726–731. [Google Scholar] [CrossRef]
- Kwak, M.J.; Kong, H.G.; Choi, K.; Kwon, S.K.; Song, J.Y.; Lee, J.; Lee, P.A.; Choi, S.Y.; Seo, M.; Lee, H.J.; et al. Rhizosphere Microbiome Structure Alters to Enable Wilt Resistance in Tomato. Nat. Biotechnol. 2018, 36, 1100–1109. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Aligment/Map Format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Kang, D.D.; Li, F.; Kirton, E.; Thomas, A.; Egan, R.; An, H.; Wang, Z. MetaBAT 2: An Adaptive Binning Algorithm for Robust and Efficient Genome Reconstruction from Metagenome Assemblies. PeerJ 2019, 7, e7359. [Google Scholar] [CrossRef]
- Tago, K.; Ishii, S.; Nishizawa, T.; Otsuka, S.; Senoo, K. Phylogenetic and Functional Diversity of Denitrifying Bacteria Isolated from Various Rice Paddy and Rice-soybean Rotation Fields. Microbes Environ. 2011, 26, 30–35. [Google Scholar] [CrossRef]
- Nishizawa, T.; Uei, Y.; Tago, K.; Isobe, K.; Otsuka, S.; Senoo, K. Taxonomic Composition of Denitrifying Bacterial Isolates is Different among Three Rice Paddy Field Soils in Japan. Soil Sci. Plant Nutr. 2013, 59, 305–310. [Google Scholar] [CrossRef]
- Ashida, N.; Ishii, S.; Hayano, S.; Tago, K.; Tsuji, T.; Yoshimura, Y.; Otsuka, S.; Senoo, K. Isolation of Functional Single Cells from Environments Using a Micromanipulator: Application to Study Denitrifying Bacteria. Appl. Microbiol. Biotechnol. 2009, 85, 1211–1217. [Google Scholar] [CrossRef]
- Puri, R.R.; Dangi, S.R.; Dhungana, S.A.; Itoh, K. Diversity and Plant Growth Promoting Ability of Culturable Endophytic Bacteria in Nepalese Sweet Potato. Adv. Microbiol. 2018, 8, 734–761. [Google Scholar] [CrossRef]
- Nishizawa, T.; Komatsuzaki, M.; Kaneko, N.; Ohta, H. Archaeal Diversity of Upland Rice Field Soils Assessed by the Terminal Restriction Fragment Length Polymorphism Method Combined with Real Time Quantitative-PCR and a Clone Library Analysis. Microbes Environ. 2008, 23, 237–243. [Google Scholar] [CrossRef] [PubMed]
- Culman, S.W.; Bukowski, R.; Gauch, G.H.; Cadillo-Quiroz, H.; Buckley, D.H. T-REX: Software for the Processing and Analysis of T-RFLP Data. BMC Bioinform. 2009, 10, 171. [Google Scholar] [CrossRef] [PubMed]
- R Development Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2011; ISBN 3-900051-07-0. Available online: http://www.R-project.org/ (accessed on 24 November 2022).
- Weidner, S.; Arnold, W.; Pühler, A. Diversity of Uncultured Microorganisms Associated with the Seagrass Halophila stipulacea Estimated by Restriction Fragment Length Polymorphism Analysis of PCR-Amplified 16S rRNA Genes. Appl. Environ. Microbiol. 1996, 62, 766–771. [Google Scholar] [CrossRef] [PubMed]
- Tiedje, J.M. Methods of Soil Analysis, Part 2: Microbiological and Biochemical properties. In Denitrifiers; Weaver, R.W., Angle, J., Bottomley, P.J., Eds.; Soil Science Society of America: Madison, WI, USA, 1994; pp. 245–267. [Google Scholar]
- Cole, J.R.; Wang, Q.; Fish, J.A.; Chai, B.; McGarrell, D.M.; Sun, Y.; Brown, C.T.; Porras-Alfaro, A.; Kuske, C.R.; Tiedje, J.M. Ribosomal Database Project: Data and Tools for High Throughput rRNA Analysis. Nucleic Acids Res. 2014, 42, D633–D642. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, T.; Kawahara, M.; Minamisawa, K. Novel Endophytic Nitrogen-fixing Clostridia from the Grass Miscanthus sinensis as Revealed by Terminal Restriction Fragment Length Polymorphism Analysis. Appl. Environ. Microbiol. 2004, 70, 6580–6586. [Google Scholar] [CrossRef]
- Tsuyuzaki, S. Miscanthus sinensis Grassland is an Indicator Plant Community to Predict Forest Regeneration and Development on Ski Slopes in Japan. Ecol. Indic. 2004, 5, 109–115. [Google Scholar] [CrossRef]
- Huang, C.L.; Sarkar, R.; Hsu, T.W.; Yang, C.F.; Chien, C.H.; Chang, W.C.; Chiang, T.Y. Endophytic Microbiome of Biofuel Plant Miscanthus sinensis (Poaceae) Interacts with Environmental Gradients. Microb. Ecol. 2020, 80, 133–144. [Google Scholar] [CrossRef]
- Anderson, C.R.; Peterson, M.E.; Frampton, R.A.; Bulman, S.R.; Keenan, S.; Curtin, D. Rapid Increases in Soil pH Solubilise Organic Matter, Dramatically Increase Denitrification Potential and Strongly Stimulate Microorganisms from the Fimicutes Phylum. PeerJ. 2018, 6, e6090. [Google Scholar] [CrossRef]
- Daly, E.J.; Hernandez-Ramirez, G. Sources and Priming of Soil N2O and CO2 Production: Nitrogen and Stimulated Exudate Additions. Soil Biol. Biochem. 2020, 149, 107942. [Google Scholar] [CrossRef]
- Dixon, R.; Kahn, D. Genetic Regulation of Biological Nitrogen Fixation. Nat. Rev. Microbiol. 2004, 2, 621–631. [Google Scholar] [CrossRef]
- Tsang, J.; Hoover, T.R. Themes and Variations: Regulation of RpoN-Dependent Flagellar Genes across Diverse Bacterial Species. Scientifica 2014, 2014, 681754. [Google Scholar] [CrossRef] [PubMed]
- Berendsen, R.L.; Pieterse, C.M.J.; Bakker, P.A.H.M. The Rhizosphere Microbiome and Plant Health. Trends Plant Sci. 2012, 17, 478–486. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; AlMomin, S.; Al-Aqeel, H.; Al-Salameen, F.; Nair, S.; Shajan, A. Metagenomic Analysis of Rhizosphere Microflora of Oil-contaminated Soil Planted with Barley and Alfalfa. PLoS ONE 2018, 13, e0202127. [Google Scholar] [CrossRef]
- Marasco, R.; Rolli, E.; Ettoumi, B.; Vigani, G.; Mapelli, F.; Borin, S.; Abou-Hadid, A.F.; El-Behairy, U.A.; Sorlini, C.; Cherif, A.; et al. A Drought Resistance-promoting Microbiome is Selected by Root System under Desert Farming. PLoS ONE 2012, 7, e48479. [Google Scholar] [CrossRef] [PubMed]
- Morley, N.; Baggs, E.M. Carbon and Oxygen Control of N2O and N2 Production during Nitrate Reduction. Soil Biol. Biochem. 2010, 42, 1864–1871. [Google Scholar] [CrossRef]
- Van den Berg, E.M.; Boleij, M.; Kuenen, J.G.; Kleerebezem, R.; van Loosdrecht, M.C.M. DNRA and Denitrification Coexist over a Broad Range of Acetate/N-NO3− Ratios, in a Chemostat Enrichment Culture. Front. Microbiol. 2016, 7, 1842. [Google Scholar] [CrossRef]
- Van den Berg, E.M.; Elisário, M.P.; Kuenen, J.G.; Kleerebezem, R.; van Loosdrecht, M.C.M. Fermentative Bacteria Influence the Competition between Denitrifiers and DNRA Bacteria. Front. Microbiol. 2017, 8, 1684. [Google Scholar] [CrossRef]
- Rütting, T.; Boeckx, P.; Müller, C.; Klemedtsson, L. Assessment of the Importance of Dissimilatory Nitrate Reduction to Ammonium for the Terrestrial Nitrogen Cycle. Biogeosciences 2011, 8, 1779–1791. [Google Scholar] [CrossRef]
- Wang, J.; Wang, L.; Feng, X.; Hu, H.; Cai, Z.; Müller, C.; Zhang, J. Soil N Transformations and Its Controlling Factors in Temperate Grasslands in China: A Study from 15N Tracing Experiment to Literature Synthesis. J. Geophys. Res. Biogeosci. 2016, 121, 2949–2959. [Google Scholar] [CrossRef]
- Yang, W.H.; Ryals, R.A.; Cusack, D.F.; Silver, W.L. Cross-biome Assessment of Gross Soil Nitrogen Cycling in California Ecosystems. Soil Biol. Biochem. 2017, 107, 144–155. [Google Scholar] [CrossRef]
- Tiedje, J.M.; Sexstone, A.J.; Myrold, D.D.; Robinson, J.A. Denitrification: Ecological Niches, Competition and Survival. Antonie Leeuwenhoek 1983, 48, 569–583. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Han, J.G.; Ma, Z.W.; Wang, Q.; Li, L.H. Effect of Carbon Source on Dissimilatory Nitrate Reduction to Ammonium in Coastal Wetland Sediments. J. Soil Sci. Plant Nutr. 2016, 16, 337–349. [Google Scholar]
- Nishizawa, T.; Tago, K.; Oshima, K.; Hattori, M.; Ishii, S.; Otsuka, S.; Senoo, K. Complete Genome Sequence of the Denitrifying and N2O-Reducing Bacterium Azoarcus sp. Strain KH32C. J. Bacteriol. 2011, 194, 1255. [Google Scholar] [CrossRef]
- Poehlein, A.; Kusian, B.; Friedrich, B.; Daniel, R.; Bowien, B. Complete Genome Sequence of the Type Strain Cupriavidus necator N-1. J. Bacteriol. 2011, 193, 5017. [Google Scholar] [CrossRef]
- Cole, J.A.; Brown, C.M. Nitrate Reduction to Ammonia by Fermentative Bacteria: A Short Circuit in the Biological Nitrogen Cycle. FEMS Microbiol. Lett. 1980, 7, 65–72. [Google Scholar] [CrossRef]
- De Graaf, M.C.C.; Bobbink, R.; Roelofs, J.G.M.; Verbeek, P.J.M. Differential Effects of Ammonium and Nitrate on Three Heathland Species. Plant Ecol. 1998, 135, 185–196. [Google Scholar] [CrossRef]
- Yan, L.; Xu, X.; Xia, J. Higher Response of Terrestrial Plant Growth to Ammonium than Nitrate Addition. Biogeosci. Discuss. 2018, preprint. [Google Scholar] [CrossRef]
- Lang, E.; Jagnow, G. Fungi of a Forest Soil Nitrifying at Low pH Values. FEMS Microbiol. Ecol. 1986, 2, 257–265. [Google Scholar] [CrossRef]
- Martikainen, P.J. Heterotrophic Nitrification—An Eternal Mystery in the Nitrogen Cycle. Soil Biol. Biochem. 2022, 168, 108611. [Google Scholar] [CrossRef]
- Gubry-Rangin, C.; Nicol, G.W.; Prosser, J.I. Archaea rather than Bacteria Control Nitrification in Two Agricultural Acidic Soils. FEMS Microbiol. Ecol. 2010, 74, 566–574. [Google Scholar] [CrossRef] [PubMed]
- Humbert, S.; Tarnawski, S.; Fromin, N.; Mallet, M.P.; Aragno, M.; Zopfi, J. Molecular Detection of Annamox Bacteria in Terrestrial Ecosystems: Distribution and Diversity. ISME J. 2010, 4, 450–454. [Google Scholar] [CrossRef] [PubMed]
- Sato, Y.; Ohta, H.; Yamagishi, T.; Guo, Y.; Nishizawa, T.; Rahman, M.H.; Kuroda, H.; Kato, T.; Saito, M.; Yoshinaga, I.; et al. Detection of Anammox Activity and 16S rRNA Genes in Ravine Paddy Field Soil. Microbes Environ. 2012, 27, 316–319. [Google Scholar] [CrossRef]
- Xi, D.; Bai, R.; Zhang, L.; Fang, Y. Contribution of Anammox to Nitrogen Removal in Two Temperate Forest Soils. Appl. Environ. Microbiol. 2016, 82, 4602–4612. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sampling Date/Layer | Soil Chemical Properties | ||||||
|---|---|---|---|---|---|---|---|
| TC g/kg Dry Soil | TN g/kg Dry Soil | C/N Ratio | NO3− g/kg Dry Soil | NH4+ g/kg Dry Soil | pH (H2O) | Water Content (%) | |
| Mar. 2016 | |||||||
| A layer | n.a. | n.a. | n.a. | n.a. | n.a. | n.a. | n.a. |
| C layer | 0.4 | 0.1 | 4.0 | 0.02 | <0.01 | 5.1 | 25.1 |
| Sep. 2017 | |||||||
| A layer | 8.1 | 0.2 | 40.5 | n.d. | n.d. | 5.0 | 19.5 |
| C layer | 0.2 | N.D. | n.a. | n.d. | n.d. | 4.9 | 10.9 |
| Sep. 2018 | |||||||
| A layer | 27.7 | 1.4 | 19.8 | 0.01 | 0.06 | 5.0 | 31.2 |
| C1 layer | 1.2 | 0.2 | 6.0 | <0.01 | 0.02 | 4.9 | 11.8 |
| C2 layer | 0.6 | 0.1 | 6.0 | <0.01 | 0.01 | 4.7 | 9.7 |
| Selected Key Function * and Protein | Gene Name | Gene Count | Gene Copies per 1M Reads |
|---|---|---|---|
| DNA replication and recombination | |||
| DNA gyrase | gyrA† | 738 | 122.6 |
| gyrB† | 721 | 148.1 | |
| DNA polymerase III subunit | dnaN† | 370 | 68.1 |
| Recombinase | recA† | 372 | 66.2 |
| Transcription | |||
| DNA-directed RNA polymerase, alpha subunit | rpoA† | 306 | 104.4 |
| DNA-directed RNA polymerase, beta subunit | rpoB† | 1000 | 218.7 |
| DNA-directed RNA polymerase, beta subunit | rpoC† | 1036 | 277.0 |
| DNA-directed RNA polymerase, omega subunit | rpoZ | 120 | 36.2 |
| RNA polymerase primary sigma factor | rpoD† | 793 | 131.8 |
| RNA polymerase sigma-54 factor | rpoN (ntrA) | 380 | 74.4 |
| Extracytoplasmic function (ECF) sigma factors | rpoE | 1256 | 160.4 |
| Cell division | |||
| Transpeptidase | ftsI† | 538 | 120.6 |
| GTPase | ftsZ† | 368 | 68.1 |
| Two-component regulatory system | |||
| Chemotaxis protein, methyltransferase | cheR | 298 | 54.5 |
| Purine-binding chemotaxis protein | cheW | 242 | 41.3 |
| Chemotaxis protein | cheX | 36 | 4.1 |
| Response regulator | cheY | 281 | 42.1 |
| Secretion system | |||
| Type III | yscV (escV) | 49 | 8.0 |
| Type IV | virB4 | 155 | 21.8 |
| virD4 | 215 | 22.7 | |
| Type VI | vgrG | 905 | 183.9 |
| hcp | 385 | 97.7 | |
| icmF (vasK) | 784 | 124.5 | |
| Stringent response | |||
| ppGpp synthase/hydrolase | spoT (relA) | 545 | 104.5 |
| Transporter | |||
| Ammonium transporter superfamily | amt | 640 | 92.8 |
| Culture Conditions | Samples | Diversity Index * | ||
|---|---|---|---|---|
| T-RF Types | E | H’ | ||
| Aerobic | Lateral roots | 19.5 ± 5.1 | 0.70 ± 0.09 | 2.06 ± 0.43 |
| Main roots | 21.5 ± 3.1 | 0.82 ± 0.04 | 2.50 ± 0.23 | |
| Anaerobic | Lateral roots | 14.0 ± 3.4 | 0.79 ± 0.05 | 2.08 ± 0.30 |
| Main roots | 14.5 ± 2.4 | 0.76 ± 0.05 | 2.01 ± 0.16 | |
| Anaerobic with antibiotic | Lateral roots | 13.3 ± 2.6 | 0.76 ± 0.06 | 1.95 ± 0.28 |
| Main roots | 13.8 ± 2.9 | 0.76 ± 0.06 | 1.98 ± 0.31 | |
| OTUs | Strain Name | Phylogenetic Relationship | ||
|---|---|---|---|---|
| Closest Organism | Accession no. | S_ab Score * | ||
| 01 | IG7-1 | Agrobacterium radiobacter | AB247582 | 0.995 |
| 02 | IG7-2 | Achromobacter sp. | HQ256543 | 0.555 |
| 03 | IG7-3 | Rahnella aquatilis | KJ781940 | 0.993 |
| 04 | IG7-4 | Flavobacterium johnsoniae | AB681010 | 0.984 |
| 05 | IG7-5 | Starkeya sp. | KC904964 | 0.958 |
| 06 | IG7-6 | Rhizobium sp. | KF465952 | 0.955 |
| 07 | IG7-7 | Alcaligenes faecalis | AJ509012 | 0.990 |
| 08 | IG7-8 | Bacillus cereus | CM000739 | 0.994 |
| 09 | IG7-9 | Agrobacterium radiobacter | AB247582 | 0.968 |
| 10 | IG7-10 | Paraburkholderia unamae | KM974658 | 0.967 |
| 11 | IG7-11 | Agrobacterium radiobacter | AB247582 | 0.993 |
| 12 | IG7-12 | Mitsuaria chitosanitabida | AB006851 | 0.990 |
| 13 | IG7-13 | Duganella sp. | HQ829837 | 0.917 |
| 14 | IG7-14 | Cupriavidus sp. | JN128831 | 0.972 |
| 15 | IG7-15 | Enterobacteriaceae bacterium | CP003938 | 0.511 |
| 16 | IG7-16 | Oxalobacteraceae bacterium | DQ337591 | 0.984 |
| 17 | IG7-17 | Cupriavidus sp. | JN226398 | 0.990 |
| 18 | IG7-18 | Cupriavidus sp. | HE662654 | 0.969 |
| 19 | IG7-19 | Leifsonia sp. | KT462730 | 0.794 |
| 20 | IG7-20 | Bacillus cereus | JN579711 | 0.920 |
| 21 | IG7-21 | Mycobacterium sp. | AY188086 | 1.000 |
| 22 | IG7-22 | Stenotrophomonas maltophilia | KR007965 | 0.840 |
| 23 | IG7-23 | Alcaligenes faecalis | AJ509012 | 0.985 |
| 24 | IG7-24 | Achromobacter denitrificans | FJ810080 | 0.990 |
| 25 | IG7-25 | Klebsiella sp. | HQ204298 | 0.973 |
| 26 | IG7-26 | Beta-proteobacterium | AB607287 | 0.713 |
| 27 | IG7-27 | Achromobacter xylosoxidans | JN381516 | 0.507 |
| 28 | IG7-28 | Pseudomonas mendocina | KP324955 | 0.365 |
| 29 | IG7-29 | Stenotrophomonas maltophilia | GQ360071 | 0.993 |
| 30 | IG7-30 | Flavobacterium johnsoniae | AB681010 | 0.974 |
| 31 | IG7-31 | Flavobacterium johnsoniae | AB078043 | 0.848 |
| 32 | IG7-32 | Bosea sp. | AB542375 | 0.988 |
| 33 | IG7-33 | Agrobacterium sp. | KF465838 | 0.985 |
| 34 | IG7-34 | Achromobacter sp. | KF059272 | 0.979 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arsyadi, A.; Guo, Y.; Ebihara, A.; Sakagami, N.; Sakoda, M.; Tago, K.; Kamijo, T.; Ohta, H.; Nishizawa, T. A Nitrate-Transforming Bacterial Community Dominates in the Miscanthus Rhizosphere on Nitrogen-Deficient Volcanic Deposits of Miyake-jima. Microorganisms 2023, 11, 260. https://doi.org/10.3390/microorganisms11020260
Arsyadi A, Guo Y, Ebihara A, Sakagami N, Sakoda M, Tago K, Kamijo T, Ohta H, Nishizawa T. A Nitrate-Transforming Bacterial Community Dominates in the Miscanthus Rhizosphere on Nitrogen-Deficient Volcanic Deposits of Miyake-jima. Microorganisms. 2023; 11(2):260. https://doi.org/10.3390/microorganisms11020260
Chicago/Turabian StyleArsyadi, Ahmad, Yong Guo, Akiko Ebihara, Nobuo Sakagami, Midori Sakoda, Kanako Tago, Takashi Kamijo, Hiroyuki Ohta, and Tomoyasu Nishizawa. 2023. "A Nitrate-Transforming Bacterial Community Dominates in the Miscanthus Rhizosphere on Nitrogen-Deficient Volcanic Deposits of Miyake-jima" Microorganisms 11, no. 2: 260. https://doi.org/10.3390/microorganisms11020260
APA StyleArsyadi, A., Guo, Y., Ebihara, A., Sakagami, N., Sakoda, M., Tago, K., Kamijo, T., Ohta, H., & Nishizawa, T. (2023). A Nitrate-Transforming Bacterial Community Dominates in the Miscanthus Rhizosphere on Nitrogen-Deficient Volcanic Deposits of Miyake-jima. Microorganisms, 11(2), 260. https://doi.org/10.3390/microorganisms11020260