Precision Medicine in Rare Diseases

 ,
, 
Abstract
1. Precision Medicine in Rare Diseases
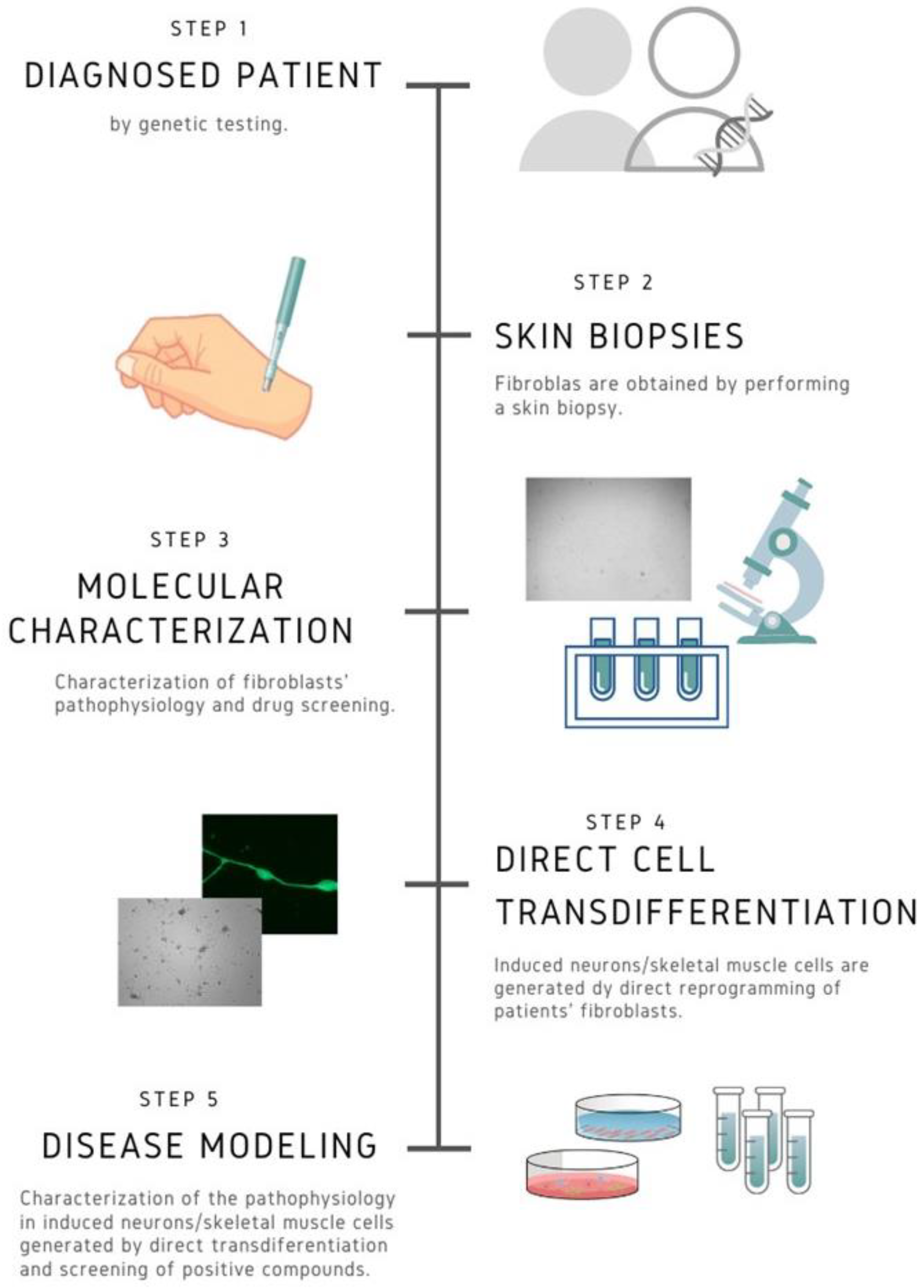
2. Fibroblast Cultures and Transdifferentiation
2.1. Two Transdifferentiation Approaches (Direct vs. Indirect): Advantages and Drawbacks
2.1.1. Human Induced Pluripotent Stem Cells (iPSCs) Model
2.1.2. Direct Reprogramming
2.2. Advantages and Disadvantages of Direct Reprogramming
2.3. Applications of Direct Reprogramming
3. Braincure/Mitocure/Myocure Platforms
3.1. Braincure Platform
3.1.1. Strategy for the Identification of Effective Treatments for Neurodegeneration Associated with Pantothenate Kinase (PKAN)
3.1.2. Precision Medicine in PKAN
3.2. Mitocure Platform
Mitocure-KAT6A Platform
3.3. Myocure Platform
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ashley, E.A. Towards precision medicine. Nat. Rev. Genet. 2016, 17, 507–522. [Google Scholar] [CrossRef]
- Schee Genannt, H.S.; Mahlmann, L.; Leyens, L.; Reumann, M.; Brand, A. Personalized Medicine: What’s in it for Rare Diseases? Adv. Exp. Med. Biol. 2017, 1031, 387–404. [Google Scholar] [CrossRef]
- Anderson, R.H.; Francis, K.R. Modeling rare diseases with induced pluripotent stem cell technology. Mol. Cell. Probes 2018, 40, 52–59. [Google Scholar] [CrossRef] [PubMed]
- Kelaini, S.; Cochrane, A.; Margariti, A. Direct reprogramming of adult cells: Avoiding the pluripotent state. Stem Cells Cloning Adv. Appl. 2014, 7, 19–29. [Google Scholar] [CrossRef]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef] [PubMed]
- Freel, B.A.; Sheets, J.N.; Francis, K.R. iPSC modeling of rare pediatric disorders. J. Neurosci. Methods 2020, 332, 108533. [Google Scholar] [CrossRef] [PubMed]
- Borger, D.K.; McMahon, B.; Roshan Lal, T.; Serra-Vinardell, J.; Aflaki, E.; Sidransky, E. Induced pluripotent stem cell models of lysosomal storage disorders. Dis. Models Amp. Mech. 2017, 10, 691–704. [Google Scholar] [CrossRef] [PubMed]
- Doss, M.X.; Sachinidis, A. Current Challenges of iPSC-Based Disease Modeling and Therapeutic Implications. Cells 2019, 8, 403. [Google Scholar] [CrossRef] [PubMed]
- Suhr, S.T.; Chang, E.A.; Tjong, J.; Alcasid, N.; Perkins, G.A.; Goissis, M.D.; Ellisman, M.H.; Perez, G.I.; Cibelli, J.B. Mitochondrial rejuvenation after induced pluripotency. PLoS ONE 2010, 5, e14095. [Google Scholar] [CrossRef]
- Van Haute, L.; Spits, C.; Geens, M.; Seneca, S.; Sermon, K. Human embryonic stem cells commonly display large mitochondrial DNA deletions. Nat. Biotechnol. 2013, 31, 20–23. [Google Scholar] [CrossRef]
- Drouin-Ouellet, J.; Pircs, K.; Barker, R.A.; Jakobsson, J.; Parmar, M. Direct Neuronal Reprogramming for Disease Modeling Studies Using Patient-Derived Neurons: What Have We Learned? Front. Neurosci. 2017, 11, 530. [Google Scholar] [CrossRef] [PubMed]
- Vierbuchen, T.; Ostermeier, A.; Pang, Z.P.; Kokubu, Y.; Sudhof, T.C.; Wernig, M. Direct conversion of fibroblasts to functional neurons by defined factors. Nature 2010, 463, 1035–1041. [Google Scholar] [CrossRef] [PubMed]
- Pang, Z.P.; Yang, N.; Vierbuchen, T.; Ostermeier, A.; Fuentes, D.R.; Yang, T.Q.; Citri, A.; Sebastiano, V.; Marro, S.; Sudhof, T.C.; et al. Induction of human neuronal cells by defined transcription factors. Nature 2011, 476, 220–223. [Google Scholar] [CrossRef] [PubMed]
- Passeri, E.; Wilson, A.M.; Primerano, A.; Kondo, M.A.; Sengupta, S.; Srivastava, R.; Koga, M.; Obie, C.; Zandi, P.P.; Goes, F.S.; et al. Enhanced conversion of induced neuronal cells (iN cells) from human fibroblasts: Utility in uncovering cellular deficits in mental illness-associated chromosomal abnormalities. Neurosci. Res. 2015, 101, 57–61. [Google Scholar] [CrossRef] [PubMed]
- Pfisterer, U.; Wood, J.; Nihlberg, K.; Hallgren, O.; Bjermer, L.; Westergren-Thorsson, G.; Lindvall, O.; Parmar, M. Efficient induction of functional neurons from adult human fibroblasts. Cell Cycle 2011, 10, 3311–3316. [Google Scholar] [CrossRef]
- Caiazzo, M.; Dell’Anno, M.T.; Dvoretskova, E.; Lazarevic, D.; Taverna, S.; Leo, D.; Sotnikova, T.D.; Menegon, A.; Roncaglia, P.; Colciago, G.; et al. Direct generation of functional dopaminergic neurons from mouse and human fibroblasts. Nature 2011, 476, 224–227. [Google Scholar] [CrossRef]
- Ladewig, J.; Koch, P.; Brustle, O. Leveling Waddington: The emergence of direct programming and the loss of cell fate hierarchies. Nat. Rev. Mol. Cell Biol. 2013, 14, 225–236. [Google Scholar] [CrossRef]
- Yoo, A.S.; Sun, A.X.; Li, L.; Shcheglovitov, A.; Portmann, T.; Li, Y.; Lee-Messer, C.; Dolmetsch, R.E.; Tsien, R.W.; Crabtree, G.R. MicroRNA-mediated conversion of human fibroblasts to neurons. Nature 2011, 476, 228–231. [Google Scholar] [CrossRef]
- Mollinari, C.; Zhao, J.; Lupacchini, L.; Garaci, E.; Merlo, D.; Pei, G. Transdifferentiation: A new promise for neurodegenerative diseases. Cell Death Dis. 2018, 9, 830. [Google Scholar] [CrossRef]
- Richner, M.; Victor, M.B.; Liu, Y.; Abernathy, D.; Yoo, A.S. MicroRNA-based conversion of human fibroblasts into striatal medium spiny neurons. Nat. Protoc. 2015, 10, 1543–1555. [Google Scholar] [CrossRef]
- Victor, M.B.; Richner, M.; Hermanstyne, T.O.; Ransdell, J.L.; Sobieski, C.; Deng, P.Y.; Klyachko, V.A.; Nerbonne, J.M.; Yoo, A.S. Generation of human striatal neurons by microRNA-dependent direct conversion of fibroblasts. Neuron 2014, 84, 311–323. [Google Scholar] [CrossRef] [PubMed]
- Xue, Y.; Ouyang, K.; Huang, J.; Zhou, Y.; Ouyang, H.; Li, H.; Wang, G.; Wu, Q.; Wei, C.; Bi, Y.; et al. Direct conversion of fibroblasts to neurons by reprogramming PTB-regulated microRNA circuits. Cell 2013, 152, 82–96. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, H.F.; Terry, A.; Beretta, C.; Pereira, C.F.; Leleu, M.; Chen, Z.F.; Kelly, C.; Merkenschlager, M.; Fisher, A.G. REST selectively represses a subset of RE1-containing neuronal genes in mouse embryonic stem cells. Development 2009, 136, 715–721. [Google Scholar] [CrossRef] [PubMed]
- Ladewig, J.; Mertens, J.; Kesavan, J.; Doerr, J.; Poppe, D.; Glaue, F.; Herms, S.; Wernet, P.; Kogler, G.; Muller, F.J.; et al. Small molecules enable highly efficient neuronal conversion of human fibroblasts. Nat. Methods 2012, 9, 575–578. [Google Scholar] [CrossRef]
- Hu, W.; Qiu, B.; Guan, W.; Wang, Q.; Wang, M.; Li, W.; Gao, L.; Shen, L.; Huang, Y.; Xie, G.; et al. Direct Conversion of Normal and Alzheimer’s Disease Human Fibroblasts into Neuronal Cells by Small Molecules. Cell Stem Cell 2015, 17, 204–212. [Google Scholar] [CrossRef]
- Liu, M.L.; Zang, T.; Zou, Y.; Chang, J.C.; Gibson, J.R.; Huber, K.M.; Zhang, C.L. Small molecules enable neurogenin 2 to efficiently convert human fibroblasts into cholinergic neurons. Nat. Commun. 2013, 4, 2183. [Google Scholar] [CrossRef]
- Wang, P.; Zhang, H.L.; Li, W.; Sha, H.; Xu, C.; Yao, L.; Tang, Q.; Tang, H.; Chen, L.; Zhu, J. Generation of patient-specific induced neuronal cells using a direct reprogramming strategy. Stem Cells Dev. 2014, 23, 16–23. [Google Scholar] [CrossRef]
- Hsu, Y.C.; Chen, S.L.; Wang, Y.J.; Chen, Y.H.; Wang, D.Y.; Chen, L.; Chen, C.H.; Chen, H.H.; Chiu, I.M. Signaling adaptor protein SH2B1 enhances neurite outgrowth and accelerates the maturation of human induced neurons. Stem Cells Transl. Med. 2014, 3, 713–722. [Google Scholar] [CrossRef]
- Xu, Z.; Jiang, H.; Zhong, P.; Yan, Z.; Chen, S.; Feng, J. Direct conversion of human fibroblasts to induced serotonergic neurons. Mol. Psychiatry 2016, 21, 62–70. [Google Scholar] [CrossRef]
- Mertens, J.; Paquola, A.C.M.; Ku, M.; Hatch, E.; Bohnke, L.; Ladjevardi, S.; McGrath, S.; Campbell, B.; Lee, H.; Herdy, J.R.; et al. Directly Reprogrammed Human Neurons Retain Aging-Associated Transcriptomic Signatures and Reveal Age-Related Nucleocytoplasmic Defects. Cell Stem Cell 2015, 17, 705–718. [Google Scholar] [CrossRef]
- Huh, C.J.; Zhang, B.; Victor, M.B.; Dahiya, S.; Batista, L.F.; Horvath, S.; Yoo, A.S. Maintenance of age in human neurons generated by microRNA-based neuronal conversion of fibroblasts. eLife 2016, 5. [Google Scholar] [CrossRef] [PubMed]
- Horvath, S. DNA methylation age of human tissues and cell types. Genome Biol. 2013, 14, R115. [Google Scholar] [CrossRef] [PubMed]
- Miura, K.; Okada, Y.; Aoi, T.; Okada, A.; Takahashi, K.; Okita, K.; Nakagawa, M.; Koyanagi, M.; Tanabe, K.; Ohnuki, M.; et al. Variation in the safety of induced pluripotent stem cell lines. Nat. Biotechnol. 2009, 27, 743–745. [Google Scholar] [CrossRef] [PubMed]
- Torper, O.; Pfisterer, U.; Wolf, D.A.; Pereira, M.; Lau, S.; Jakobsson, J.; Bjorklund, A.; Grealish, S.; Parmar, M. Generation of induced neurons via direct conversion in vivo. Proc. Natl. Acad. Sci. USA 2013, 110, 7038–7043. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.; Zi, X.; Schulz, V.P.; Cheng, J.; Zhong, M.; Koochaki, S.H.; Megyola, C.M.; Pan, X.; Heydari, K.; Weissman, S.M.; et al. Nonstochastic reprogramming from a privileged somatic cell state. Cell 2014, 156, 649–662. [Google Scholar] [CrossRef]
- Li, Z.; Rana, T.M. A kinase inhibitor screen identifies small-molecule enhancers of reprogramming and iPS cell generation. Nat. Commun. 2012, 3, 1085. [Google Scholar] [CrossRef]
- Fishman, V.S.; Shnayder, T.A.; Orishchenko, K.E.; Bader, M.; Alenina, N.; Serov, O.L. Cell divisions are not essential for the direct conversion of fibroblasts into neuronal cells. Cell Cycle 2015, 14, 1188–1196. [Google Scholar] [CrossRef]
- Masserdotti, G.; Gillotin, S.; Sutor, B.; Drechsel, D.; Irmler, M.; Jorgensen, H.F.; Sass, S.; Theis, F.J.; Beckers, J.; Berninger, B.; et al. Transcriptional Mechanisms of Proneural Factors and REST in Regulating Neuronal Reprogramming of Astrocytes. Cell Stem Cell 2015, 17, 74–88. [Google Scholar] [CrossRef]
- Price, J.D.; Park, K.Y.; Chen, J.; Salinas, R.D.; Cho, M.J.; Kriegstein, A.R.; Lim, D.A. The Ink4a/Arf locus is a barrier to direct neuronal transdifferentiation. J. Neurosci. 2014, 34, 12560–12567. [Google Scholar] [CrossRef]
- Drouin-Ouellet, J.; Lau, S.; Brattas, P.L.; Rylander Ottosson, D.; Pircs, K.; Grassi, D.A.; Collins, L.M.; Vuono, R.; Andersson Sjoland, A.; Westergren-Thorsson, G.; et al. REST suppression mediates neural conversion of adult human fibroblasts via microRNA-dependent and -independent pathways. EMBO Mol. Med. 2017, 9, 1117–1131. [Google Scholar] [CrossRef]
- Zhang, Q.J.; Li, J.J.; Lin, X.; Lu, Y.Q.; Guo, X.X.; Dong, E.L.; Zhao, M.; He, J.; Wang, N.; Chen, W.J. Modeling the phenotype of spinal muscular atrophy by the direct conversion of human fibroblasts to motor neurons. Oncotarget 2017, 8, 10945–10953. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.M.; Choi, W.J.; Oh, K.W.; Xue, Y.; Choi, J.Y.; Kim, S.H.; Nahm, M.; Kim, Y.E.; Lee, J.; Noh, M.Y.; et al. Directly converted patient-specific induced neurons mirror the neuropathology of FUS with disrupted nuclear localization in amyotrophic lateral sclerosis. Mol. Neurodegener 2016, 11, 8. [Google Scholar] [CrossRef] [PubMed]
- Bavamian, S.; Mellios, N.; Lalonde, J.; Fass, D.M.; Wang, J.; Sheridan, S.D.; Madison, J.M.; Zhou, F.; Rueckert, E.H.; Barker, D.; et al. Dysregulation of miR-34a links neuronal development to genetic risk factors for bipolar disorder. Mol. Psychiatry 2015, 20, 573–584. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Xue, Y.; Ridley, S.; Zhang, D.; Rezvani, K.; Fu, X.D.; Wang, H. Direct reprogramming of Huntington’s disease patient fibroblasts into neuron-like cells leads to abnormal neurite outgrowth, increased cell death, and aggregate formation. PLoS ONE 2014, 9, e109621. [Google Scholar] [CrossRef] [PubMed]
- Puschmann, A.; Fiesel, F.C.; Caulfield, T.R.; Hudec, R.; Ando, M.; Truban, D.; Hou, X.; Ogaki, K.; Heckman, M.G.; James, E.D.; et al. Heterozygous PINK1 p.G411S increases risk of Parkinson’s disease via a dominant-negative mechanism. Brain 2017, 140, 98–117. [Google Scholar] [CrossRef] [PubMed]
- Santambrogio, P.; Dusi, S.; Guaraldo, M.; Rotundo, L.I.; Broccoli, V.; Garavaglia, B.; Tiranti, V.; Levi, S. Mitochondrial iron and energetic dysfunction distinguish fibroblasts and induced neurons from pantothenate kinase-associated neurodegeneration patients. Neurobiol. Dis. 2015, 81, 144–153. [Google Scholar] [CrossRef]
- Xiao, D.; Liu, X.; Zhang, M.; Zou, M.; Deng, Q.; Sun, D.; Bian, X.; Cai, Y.; Guo, Y.; Liu, S.; et al. Direct reprogramming of fibroblasts into neural stem cells by single non-neural progenitor transcription factor Ptf1a. Nat. Commun. 2018, 9, 2865. [Google Scholar] [CrossRef]
- Barker, R.A.; Barrett, J.; Mason, S.L.; Bjorklund, A. Fetal dopaminergic transplantation trials and the future of neural grafting in Parkinson’s disease. Lancet Neurol. 2013, 12, 84–91. [Google Scholar] [CrossRef]
- Kefalopoulou, Z.; Politis, M.; Piccini, P.; Mencacci, N.; Bhatia, K.; Jahanshahi, M.; Widner, H.; Rehncrona, S.; Brundin, P.; Bjorklund, A.; et al. Long-term clinical outcome of fetal cell transplantation for Parkinson disease: Two case reports. JAMA Neurol. 2014, 71, 83–87. [Google Scholar] [CrossRef]
- Torper, O.; Gotz, M. Brain repair from intrinsic cell sources: Turning reactive glia into neurons. Prog. Brain Res. 2017, 230, 69–97. [Google Scholar] [CrossRef]
- Buffo, A.; Vosko, M.R.; Erturk, D.; Hamann, G.F.; Jucker, M.; Rowitch, D.; Gotz, M. Expression pattern of the transcription factor Olig2 in response to brain injuries: Implications for neuronal repair. Proc. Natl. Acad. Sci. USA 2005, 102, 18183–18188. [Google Scholar] [CrossRef] [PubMed]
- Gascon, S.; Masserdotti, G.; Russo, G.L.; Gotz, M. Direct Neuronal Reprogramming: Achievements, Hurdles, and New Roads to Success. Cell Stem Cell 2017, 21, 18–34. [Google Scholar] [CrossRef] [PubMed]
- Niu, W.; Zang, T.; Zou, Y.; Fang, S.; Smith, D.K.; Bachoo, R.; Zhang, C.L. In vivo reprogramming of astrocytes to neuroblasts in the adult brain. Nat. Cell Biol. 2013, 15, 1164–1175. [Google Scholar] [CrossRef] [PubMed]
- Niu, W.; Zang, T.; Smith, D.K.; Vue, T.Y.; Zou, Y.; Bachoo, R.; Johnson, J.E.; Zhang, C.L. SOX2 reprograms resident astrocytes into neural progenitors in the adult brain. Stem Cell Rep. 2015, 4, 780–794. [Google Scholar] [CrossRef]
- Brulet, R.; Matsuda, T.; Zhang, L.; Miranda, C.; Giacca, M.; Kaspar, B.K.; Nakashima, K.; Hsieh, J. NEUROD1 Instructs Neuronal Conversion in Non-Reactive Astrocytes. Stem Cell Rep. 2017, 8, 1506–1515. [Google Scholar] [CrossRef]
- Guo, Z.; Zhang, L.; Wu, Z.; Chen, Y.; Wang, F.; Chen, G. In vivo direct reprogramming of reactive glial cells into functional neurons after brain injury and in an Alzheimer’s disease model. Cell Stem Cell 2014, 14, 188–202. [Google Scholar] [CrossRef]
- Su, Z.; Niu, W.; Liu, M.L.; Zou, Y.; Zhang, C.L. In vivo conversion of astrocytes to neurons in the injured adult spinal cord. Nat. Commun. 2014, 5, 3338. [Google Scholar] [CrossRef]
- Wang, L.L.; Su, Z.; Tai, W.; Zou, Y.; Xu, X.M.; Zhang, C.L. The p53 Pathway Controls SOX2-Mediated Reprogramming in the Adult Mouse Spinal Cord. Cell Rep. 2016, 17, 891–903. [Google Scholar] [CrossRef]
- Heinrich, C.; Bergami, M.; Gascon, S.; Lepier, A.; Vigano, F.; Dimou, L.; Sutor, B.; Berninger, B.; Gotz, M. Sox2-mediated conversion of NG2 glia into induced neurons in the injured adult cerebral cortex. Stem Cell Rep. 2014, 3, 1000–1014. [Google Scholar] [CrossRef]
- Grande, A.; Sumiyoshi, K.; Lopez-Juarez, A.; Howard, J.; Sakthivel, B.; Aronow, B.; Campbell, K.; Nakafuku, M. Environmental impact on direct neuronal reprogramming in vivo in the adult brain. Nat. Commun. 2013, 4, 2373. [Google Scholar] [CrossRef]
- Gascon, S.; Murenu, E.; Masserdotti, G.; Ortega, F.; Russo, G.L.; Petrik, D.; Deshpande, A.; Heinrich, C.; Karow, M.; Robertson, S.P.; et al. Identification and Successful Negotiation of a Metabolic Checkpoint in Direct Neuronal Reprogramming. Cell Stem Cell 2016, 18, 396–409. [Google Scholar] [CrossRef]
- Rivetti di Val Cervo, P.; Romanov, R.A.; Spigolon, G.; Masini, D.; Martin-Montanez, E.; Toledo, E.M.; La Manno, G.; Feyder, M.; Pifl, C.; Ng, Y.H.; et al. Induction of functional dopamine neurons from human astrocytes in vitro and mouse astrocytes in a Parkinson’s disease model. Nat. Biotechnol. 2017, 35, 444–452. [Google Scholar] [CrossRef]
- Di Meo, I.; Tiranti, V. Classification and molecular pathogenesis of NBIA syndromes. Eur. J. Paediatr. Neurol. 2018, 22, 272–284. [Google Scholar] [CrossRef]
- Dang, T.N.; Bishop, G.M.; Dringen, R.; Robinson, S.R. The putative heme transporter HCP1 is expressed in cultured astrocytes and contributes to the uptake of hemin. Glia 2010, 58, 55–65. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Cordoba, M.; Fernandez Khoury, A.; Villanueva-Paz, M.; Gomez-Navarro, C.; Villalon-Garcia, I.; Suarez-Rivero, J.M.; Povea-Cabello, S.; de la Mata, M.; Cotan, D.; Talaveron-Rey, M.; et al. Pantothenate Rescues Iron Accumulation in Pantothenate Kinase-Associated Neurodegeneration Depending on the Type of Mutation. Mol. Neurobiol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Andreotti, G.; Cabeza de Vaca, I.; Poziello, A.; Monti, M.C.; Guallar, V.; Cubellis, M.V. Conformational response to ligand binding in phosphomannomutase2: Insights into inborn glycosylation disorder. J. Biol. Chem. 2014, 289, 34900–34910. [Google Scholar] [CrossRef]
- Goldin, E.; Zheng, W.; Motabar, O.; Southall, N.; Choi, J.H.; Marugan, J.; Austin, C.P.; Sidransky, E. High throughput screening for small molecule therapy for Gaucher disease using patient tissue as the source of mutant glucocerebrosidase. PLoS ONE 2012, 7, e29861. [Google Scholar] [CrossRef] [PubMed]
- Newton, C.L.; Whay, A.M.; McArdle, C.A.; Zhang, M.; van Koppen, C.J.; van de Lagemaat, R.; Segaloff, D.L.; Millar, R.P. Rescue of expression and signaling of human luteinizing hormone G protein-coupled receptor mutants with an allosterically binding small-molecule agonist. Proc. Natl. Acad. Sci. USA 2011, 108, 7172–7176. [Google Scholar] [CrossRef] [PubMed]
- Andreotti, G.; Guarracino, M.R.; Cammisa, M.; Correra, A.; Cubellis, M.V. Prediction of the responsiveness to pharmacological chaperones: Lysosomal human alpha-galactosidase, a case of study. Orphanet. J. Rare Dis. 2010, 5, 36. [Google Scholar] [CrossRef]
- Alvarez-Cordoba, M.; Villanueva-Paz, M.; Villalon-Garcia, I.; Povea-Cabello, S.; Suarez-Rivero, J.M.; Talaveron-Rey, M.; Abril-Jaramillo, J.; Vintimilla-Tosi, A.B.; Sanchez-Alcazar, J.A. Precision medicine in pantothenate kinase-associated neurodegeneration. Neural Regen. Res. 2019, 14, 1177–1185. [Google Scholar] [CrossRef]
- Hay Mele, B.; Citro, V.; Andreotti, G.; Cubellis, M.V. Drug repositioning can accelerate discovery of pharmacological chaperones. Orphanet J. Rare Dis. 2015, 10, 55. [Google Scholar] [CrossRef]
- Maitra, R.; Hamilton, J.W. Altered biogenesis of deltaF508-CFTR following treatment with doxorubicin. Cell. Phys. Biochem. 2007, 20, 465–472. [Google Scholar] [CrossRef] [PubMed]
- Rigat, B.; Mahuran, D. Diltiazem, a L-type Ca(2+) channel blocker, also acts as a pharmacological chaperone in Gaucher patient cells. Mol. Genet. Metab. 2009, 96, 225–232. [Google Scholar] [CrossRef] [PubMed]
- Bendikov-Bar, I.; Maor, G.; Filocamo, M.; Horowitz, M. Ambroxol as a pharmacological chaperone for mutant glucocerebrosidase. Blood Cells Mol. Dis. 2013, 50, 141–145. [Google Scholar] [CrossRef] [PubMed]
- Porto, C.; Ferrara, M.C.; Meli, M.; Acampora, E.; Avolio, V.; Rosa, M.; Cobucci-Ponzano, B.; Colombo, G.; Moracci, M.; Andria, G.; et al. Pharmacological enhancement of alpha-glucosidase by the allosteric chaperone N-acetylcysteine. Mol. Ther. 2012, 20, 2201–2211. [Google Scholar] [CrossRef] [PubMed]
- Maegawa, G.H.; Tropak, M.; Buttner, J.; Stockley, T.; Kok, F.; Clarke, J.T.; Mahuran, D.J. Pyrimethamine as a potential pharmacological chaperone for late-onset forms of GM2 gangliosidosis. J. Biol. Chem. 2007, 282, 9150–9161. [Google Scholar] [CrossRef] [PubMed]
- Martin, G.M.; Chen, P.C.; Devaraneni, P.; Shyng, S.L. Pharmacological rescue of trafficking-impaired ATP-sensitive potassium channels. Front. Physiol. 2013, 4, 386. [Google Scholar] [CrossRef]
- Ishihara, K.; Okuyama, S.; Kumano, S.; Iida, K.; Hamana, H.; Murakoshi, M.; Kobayashi, T.; Usami, S.; Ikeda, K.; Haga, Y.; et al. Salicylate restores transport function and anion exchanger activity of missense pendrin mutations. Hear. Res. 2010, 270, 110–118. [Google Scholar] [CrossRef]
- Sharma, L.K.; Subramanian, C.; Yun, M.K.; Frank, M.W.; White, S.W.; Rock, C.O.; Lee, R.E.; Jackowski, S. A therapeutic approach to pantothenate kinase associated neurodegeneration. Nat. Commun. 2018, 9, 4399. [Google Scholar] [CrossRef]
- Strafella, C.; Caputo, V.; Galota, M.R.; Zampatti, S.; Marella, G.; Mauriello, S.; Cascella, R.; Giardina, E. Application of Precision Medicine in Neurodegenerative Diseases. Front. Neurol. 2018, 9, 701. [Google Scholar] [CrossRef]
- Gorman, G.S.; Chinnery, P.F.; DiMauro, S.; Hirano, M.; Koga, Y.; McFarland, R.; Suomalainen, A.; Thorburn, D.R.; Zeviani, M.; Turnbull, D.M. Mitochondrial diseases. Nat. Rev. Dis. Primers 2016, 2, 16080. [Google Scholar] [CrossRef] [PubMed]
- Chinnery, P.; Majamaa, K.; Turnbull, D.; Thorburn, D. Treatment for mitochondrial disorders. Cochrane Database Syst. Rev. 2006. [Google Scholar] [CrossRef]
- Marroquin, L.D.; Hynes, J.; Dykens, J.A.; Jamieson, J.D.; Will, Y. Circumventing the Crabtree effect: Replacing media glucose with galactose increases susceptibility of HepG2 cells to mitochondrial toxicants. Toxicol. Sci. Off. J. Soc. Toxicol. 2007, 97, 539–547. [Google Scholar] [CrossRef]
- Robinson, B.H.; Petrova-Benedict, R.; Buncic, J.R.; Wallace, D.C. Nonviability of cells with oxidative defects in galactose medium: A screening test for affected patient fibroblasts. Biochem. Med. Metab. Biol. 1992, 48, 122–126. [Google Scholar] [CrossRef]
- Saada, A. The use of individual patient’s fibroblasts in the search for personalized treatment of nuclear encoded OXPHOS diseases. Mol. Genet. Metab. 2011, 104, 39–47. [Google Scholar] [CrossRef] [PubMed]
- Saada, A. Mitochondria: Mitochondrial OXPHOS dysfunction ex vivo—The use of primary fibroblasts. Int. J. Biochem. Cell Biol. 2014, 104, 39–47. [Google Scholar] [CrossRef]
- Gilissen, C.; Hehir-Kwa, J.Y.; Thung, D.T.; van de Vorst, M.; van Bon, B.W.; Willemsen, M.H.; Kwint, M.; Janssen, I.M.; Hoischen, A.; Schenck, A.; et al. Genome sequencing identifies major causes of severe intellectual disability. Nature 2014, 511, 344–347. [Google Scholar] [CrossRef]
- Moeschler, J.B.; Shevell, M.; Committee on Genetics. Comprehensive evaluation of the child with intellectual disability or global developmental delays. Pediatrics 2014, 134, e903–e918. [Google Scholar] [CrossRef]
- Fahrner, J.A.; Bjornsson, H.T. Mendelian disorders of the epigenetic machinery: Tipping the balance of chromatin states. Annu. Rev. Genom. Hum. Genet. 2014, 15, 269–293. [Google Scholar] [CrossRef]
- Avvakumov, N.; Cote, J. The MYST family of histone acetyltransferases and their intimate links to cancer. Oncogene 2007, 26, 5395–5407. [Google Scholar] [CrossRef]
- Voss, A.K.; Collin, C.; Dixon, M.P.; Thomas, T. Moz and retinoic acid coordinately regulate H3K9 acetylation, Hox gene expression, and segment identity. Dev. Cell 2009, 17, 674–686. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, J.; Goudie, D.; Blair, E.; Chandler, K.; Joss, S.; McKay, V.; Green, A.; Armstrong, R.; Lees, M.; Kamien, B.; et al. KAT6A Syndrome: Genotype-phenotype correlation in 76 patients with pathogenic KAT6A variants. Genet. Med. Off. J. Am. Coll. Med. Genet. 2018. [Google Scholar] [CrossRef] [PubMed]
- Boycott, K.M.; Innes, A.M. 39th Annual David W. Smith Workshop on Malformations and Morphogenesis: Abstracts of the 2018 Annual Meeting. Am. J. Med. Genet. Part A 2019, 6, 674–746. [Google Scholar] [CrossRef] [PubMed]
- North, K.N.; Wang, C.H.; Clarke, N.; Jungbluth, H.; Vainzof, M.; Dowling, J.J.; Amburgey, K.; Quijano-Roy, S.; Beggs, A.H.; Sewry, C.; et al. Approach to the diagnosis of congenital myopathies. Neuromuscul. Disord. 2014, 24, 97–116. [Google Scholar] [CrossRef] [PubMed]
- Claeys, K.G. Congenital myopathies: An update. Dev. Med. Child Neurol. 2020, 62, 297–302. [Google Scholar] [CrossRef]
- Shy, G.M.; Engel, W.K.; Somers, J.E.; Wanko, T. Nemaline Myopathy. A New Congenital Myopathy. Brain 1963, 86, 793–810. [Google Scholar] [CrossRef]
- Sanoudou, D.; Beggs, A.H. Clinical and genetic heterogeneity in nemaline myopathy--a disease of skeletal muscle thin filaments. Trends Mol. Med. 2001, 7, 362–368. [Google Scholar] [CrossRef]
- Yamamoto, D.L.; Vitiello, C.; Zhang, J.; Gokhin, D.S.; Castaldi, A.; Coulis, G.; Piaser, F.; Filomena, M.C.; Eggenhuizen, P.J.; Kunderfranco, P.; et al. The nebulin SH3 domain is dispensable for normal skeletal muscle structure but is required for effective active load bearing in mouse. J. Cell Sci. 2013, 126, 5477–5489. [Google Scholar] [CrossRef]
- Gonorazky, H.D.; Bonnemann, C.G.; Dowling, J.J. The genetics of congenital myopathies. Handb. Clin. Neurol. 2018, 148, 549–564. [Google Scholar] [CrossRef]
- Bar-Nur, O.; Gerli, M.F.M.; Di Stefano, B.; Almada, A.E.; Galvin, A.; Coffey, A.; Huebner, A.J.; Feige, P.; Verheul, C.; Cheung, P.; et al. Direct Reprogramming of Mouse Fibroblasts into Functional Skeletal Muscle Progenitors. Stem Cell Rep. 2018, 10, 1505–1521. [Google Scholar] [CrossRef]
- Bello, L.; Pegoraro, E. Genetic diagnosis as a tool for personalized treatment of Duchenne muscular dystrophy. Acta Myol. 2016, 35, 122–127. [Google Scholar] [PubMed]


{kind=link}
| NBIA Subtype | Gene | Chromosome Location | Biological Process | Associated Diseases |
|---|---|---|---|---|
| Pantothenate kinase-associated neurodegeneration (PKAN) | PANK2 | 20p13 | Regulation of CoA biosynthesis | Neurodegeneration with brain iron accumulation 1 Classic PKAN Atypical PKAN HARP syndrome |
| Phospholipase A2 group VI-associated neurodegeneration (PLAN) | PLA2G6 | 22q13.1 | Membrane remodeling | Neurodegeneration with brain iron accumulation 2 Infantile Neuroaxonal Dystrophy Atypical Neuroaxonal Dystrophy Parkinson disease 14 |
| Mitochondrial-membrane proteins-associated neurodegeneration (MPAN) | C19ORF12 | 19q12 | Apoptotic process Autophagy Mitochondrial calcium ion homeostasis Response to oxidative stress | Neurodegeneration with brain iron accumulation 4 Spastic paraplegia 43 |
| Beta-propeller protein-associated neurodegeneration (BPAN) | WDR45 | Xp11.23 | Autophagy | Neurodegeneration with brain iron accumulation 5 Static encephalopathy of childhood with neurodegeneration in adulthood (SENDA) |
| Fatty acid hydrolase-associated neurodegeneration (FHAN) | FAG2H | 16q23.1 | Lipid biosynthesis | Spastic paraplegia 35 Leukodystrophy |
| Gene | Genome | Biological Process | Associated Diseases |
|---|---|---|---|
| MT-ND1 | mtDNA | Electron transport | Leber hereditary optic neuropathy (LHON) Mitochondrial complex I deficiency, mitochondrial type 3 Leigh syndrome, MELAS syndrome Diabetes mellitus, non-insulin-dependent (NIDDM) |
| MT-ND3 | mtDNA | Electron transport | Leigh syndrome Mitochondrial complex I deficiency, mitochondrial type 1 Parkinson disease |
| NDUFS1 | nDNA | Electron transport | Mitochondrial complex I deficiency, nuclear type 5 |
| COX15 | nDNA | Electron transport Proton transmembrane transport Heme biosynthesis process | Mitochondrial complex IV deficiency, nuclear type 6 |
| GFM1 | nDNA | Protein biosynthesis | Combined oxidative phosphorylation deficiency 1 |
| OPA1 | nDNA | Apoptosis Sensory transduction Vision | Behr syndrome Optic atrophy 1 Optic atrophy plus syndrome Mitochondrial DNA depletion syndrome 14 (encephalocardiomyopathic type) |
| LIPT01 | nDNA | Nitrogen compound metabolic process Protein modification process Lipid metabolic process | Lipoyltransferase 1 deficiency |
| COQ7 | nDNA | Ubiquinone biosynthesis | Coenzyme Q10 deficiency, primary, 8 |
| NDUFV1 | nDNA | Electron transport | Mitochondrial complex I deficiency, nuclear type 4 |
| NDUFAF6 | nDNA | Mitochondrial respiratory chain complex I assembly | Mitochondrial complex I deficiency, nuclear type 17 Fanconi renotubular syndrome 5 |
| NDUFS4 | nDNA | Electron transport | Mitochondrial complex I deficiency, nuclear type 1 |
| Gene | Cytogenetic Location | Biological Process | Associated Diseases |
|---|---|---|---|
| NEB | 2q23.3 | Actin binding | Nemaline myopathy 2 |
| ACTA1 | 1q42.13 | Actin filament polymerization and assembly | Nemaline myopathy 3 Myopathy, congenital, with fiber-type disproportion 1 Myopathy, actin, congenital, with excess of thin myofilaments Myopathy, actin, congenital, with cores Myopathy, scapulohumeroperoneal |
| TPM2 | 9p13.3 | Actin filament organization | Nemaline myopathy 4 Cap myopathy 2 Arthrogryposis, distal, type 2B4 Arthrogryposis, distal, type 1A |
| TPM3 | 1q21.3 | Actin filament organization | Nemaline myopathy 1 Cap myopathy 1 Myopathy, congenital, with fiber-type disproportion |
| TNNT1 | 19q13.42 | Muscle filament signaling | Nemaline myopathy 5 |
| CFL2 | 14q13.1 | Actin filament depolymerization | Nemaline myopathy 7 |
| LMOD3 | 3p14.1 | Actin filament organization | Nemaline myopathy 10 |
| KBTBD13 | 15q22.31 | Post-translational process modifications Ubiquitin-proteasome pathway | Nemaline myopathy 6 |
| KLHL40 | 3p22.1 | Ubiquitin conjugation pathway | Nemaline myopathy 8 |
| KLHL41 | 2q31.1 | Myofibril assembly Post-translational protein modification Ubiquitin-proteasome pathway | Nemaline myopathy 9 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Villalón-García, I.; Álvarez-Córdoba, M.; Suárez-Rivero, J.M.; Povea-Cabello, S.; Talaverón-Rey, M.; Suárez-Carrillo, A.; Munuera-Cabeza, M.; Sánchez-Alcázar, J.A. Precision Medicine in Rare Diseases. Diseases 2020, 8, 42. https://doi.org/10.3390/diseases8040042
Villalón-García I, Álvarez-Córdoba M, Suárez-Rivero JM, Povea-Cabello S, Talaverón-Rey M, Suárez-Carrillo A, Munuera-Cabeza M, Sánchez-Alcázar JA. Precision Medicine in Rare Diseases. Diseases. 2020; 8(4):42. https://doi.org/10.3390/diseases8040042
Chicago/Turabian StyleVillalón-García, Irene, Mónica Álvarez-Córdoba, Juan Miguel Suárez-Rivero, Suleva Povea-Cabello, Marta Talaverón-Rey, Alejandra Suárez-Carrillo, Manuel Munuera-Cabeza, and José Antonio Sánchez-Alcázar. 2020. "Precision Medicine in Rare Diseases" Diseases 8, no. 4: 42. https://doi.org/10.3390/diseases8040042
APA StyleVillalón-García, I., Álvarez-Córdoba, M., Suárez-Rivero, J. M., Povea-Cabello, S., Talaverón-Rey, M., Suárez-Carrillo, A., Munuera-Cabeza, M., & Sánchez-Alcázar, J. A. (2020). Precision Medicine in Rare Diseases. Diseases, 8(4), 42. https://doi.org/10.3390/diseases8040042