Glycoprotein G-protein Coupled Receptors in Disease: Luteinizing Hormone Receptors and Follicle Stimulating Hormone Receptors


 and
and 
Abstract
1. Introduction
2. LHR and FSHR Structure and Function
2.1. Luteinizing Hormone Receptors (LHRs)
2.2. Follicle Stimulating Hormone Receptors (FSHRs)
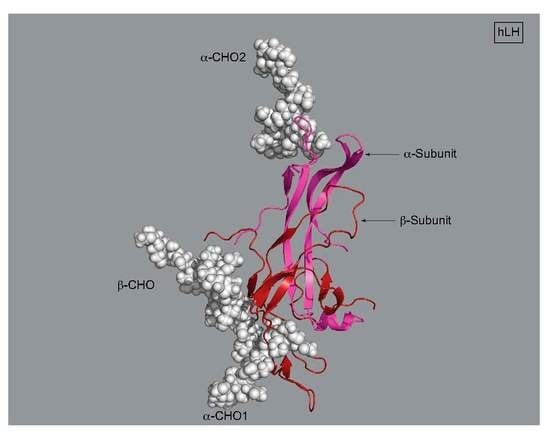
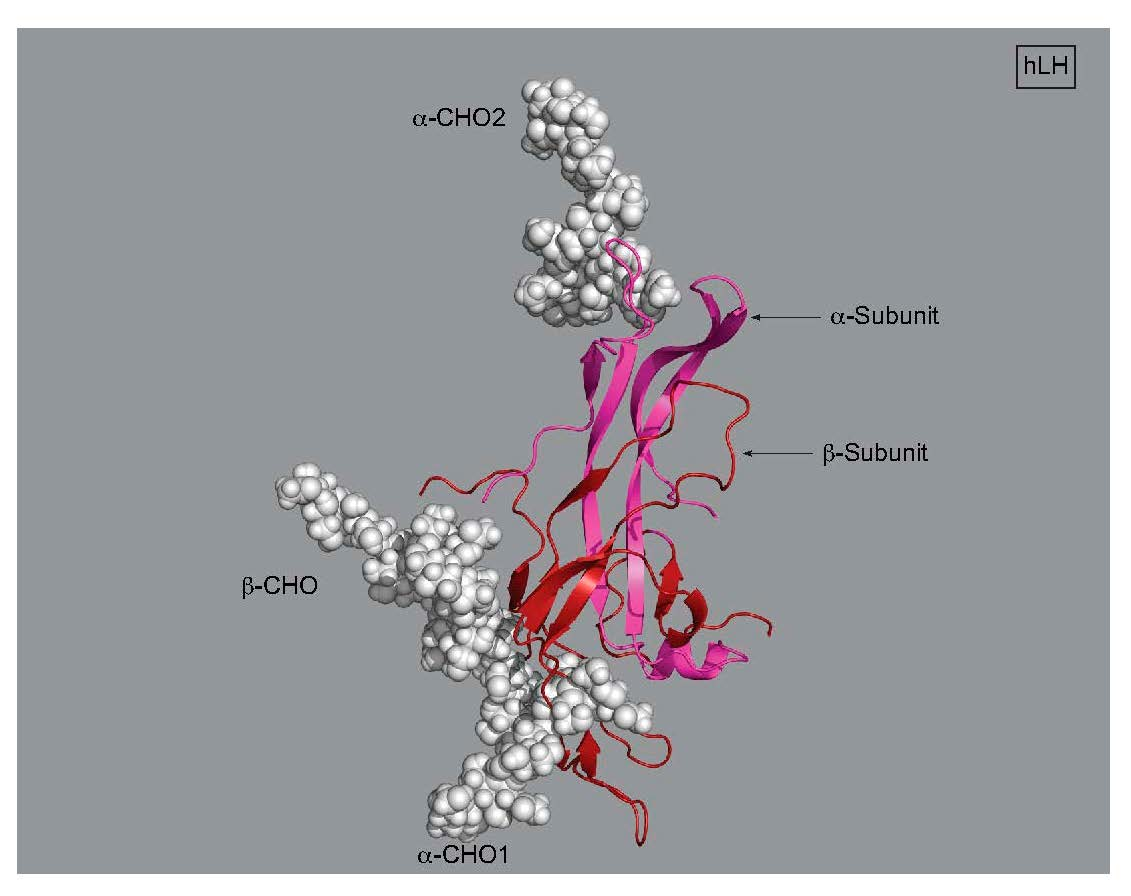
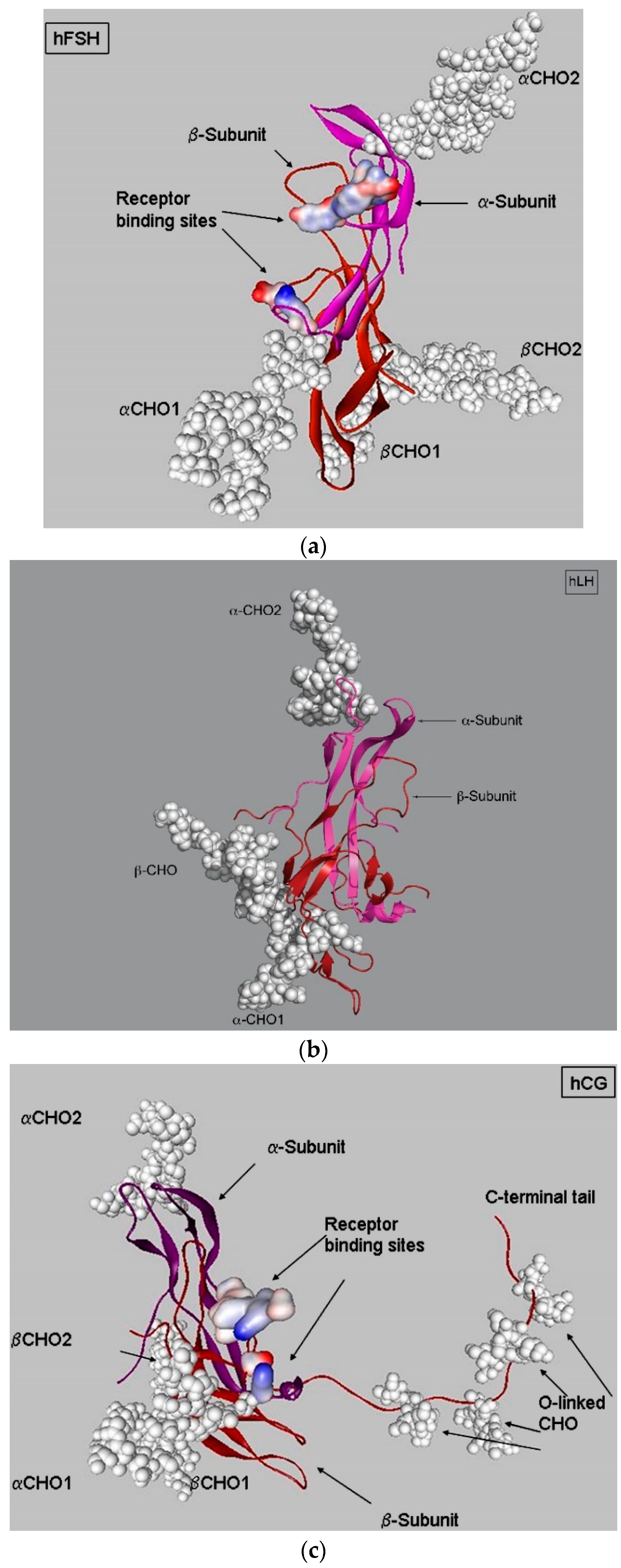
3. LH, hCG, and FSH Structures
4. Initiation of Signal Transduction: Effects of Receptor Aggregation on Receptor Function

{kind=link}
{kind=link}
{kind=link}
{kind=link}
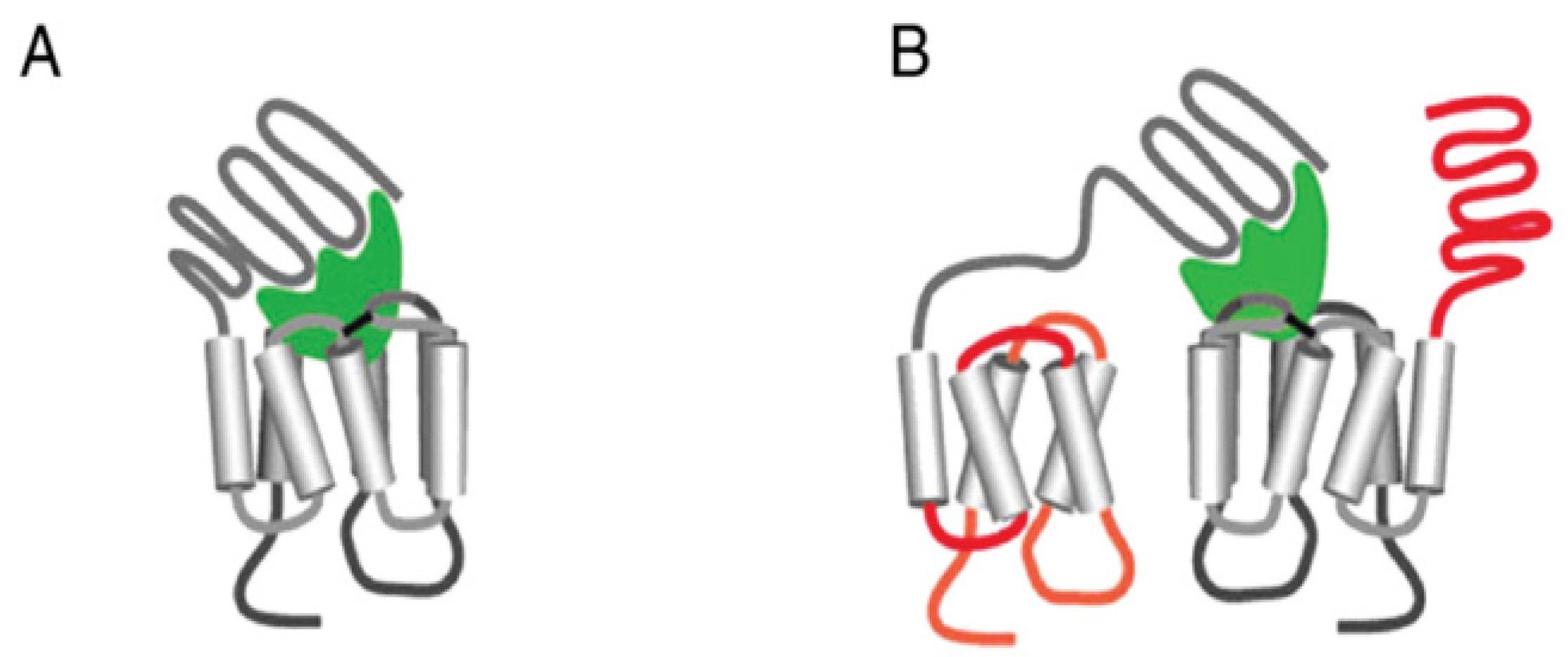{kind=link}
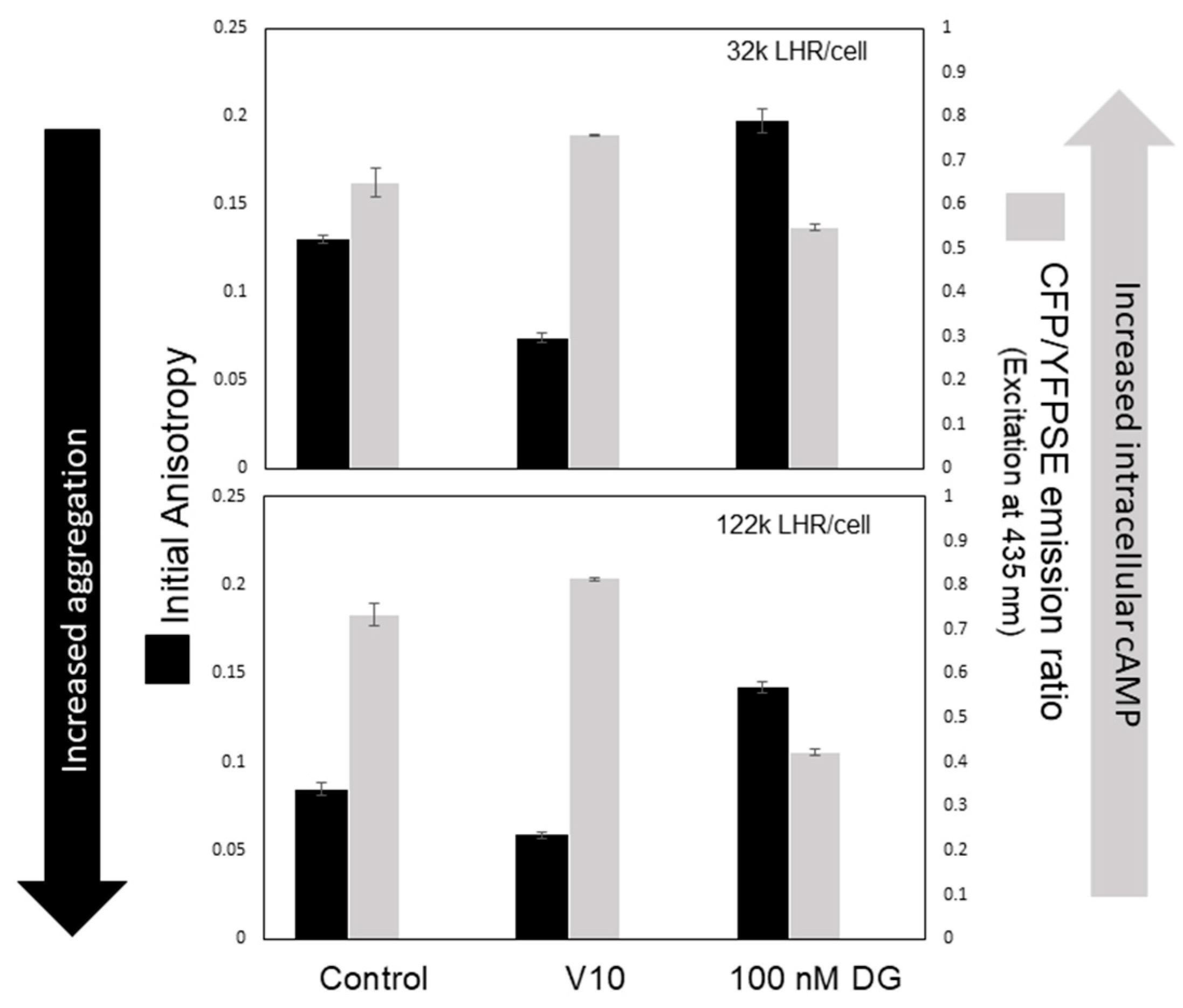{kind=link}
| Receptor | Cell Type/Tissue | Experimental Method (s) | Probe (s) | Result | Ref. |
|---|---|---|---|---|---|
| Rat LHRs | Granulosa cells | Formaldehyde fixation/light microscopy/autoradiography | Rabbit anti-hCG/FITC-goat anti rabbit IgG | hCG treatment produced small LHR clusters at 4 °C, larger clusters at 37 °C | [52] |
| Rat LHRs | Rat luteal cells | Electron microscopy | Ferritin-LH (FE-LH) | FE-LH treated LHR clusters at 37 °C | [53] |
| Porcine LHRs | Porcine granulosa cells | Fluorescence resonance energy transfer (FRET) measured using spectrofluorimetry | FITC-/TrITC-hCG or FITC-/TrITC-hCG | Positive FRET (4 °C) for LH and hCG probes. LH: Reduced FRET (37 °C), hCG: minimal FRET (37 °C) | [54] |
| Rat LHRs | CHO cells | Fluorescence recovery after photobleaching (FRAP) | LHR-GFP (C-terminus) | LH reduced the fraction of mobile LHRs at 37 °C. hCG produced visible, immobile LHR clusters | [55] |
| Rat LHRs | CHO cells | Fluorescence recovery after photobleaching (FRAP) | LHR-GFP (C-terminus) | hCG increases LHR clusters which must dissipate before receptors can signal | [56] |
| FRET | LHR-GFP/LHR-YFP | Immobile LHR clusters exhibit increased FRET | |||
| Porcine LHRs | Porcine follicle membranes | Confocal microscopy | TrITC-hCG | Active: LHRs in small clusters Desensitized: LHRs in large clusters | [46] |
| Time-resolved phosphorescence anisotropy | ErITC-hCG | Active: Small clusters, faster rotational correlation times Desensitized: larger clusters, slower rotational correlation times | |||
| FRET | FITC-hCG/TrITC-hCG | Active: Less FRET Desensitized: Increased FRET | |||
| Human LHRs | HEK 293 | Co-immunoprecipitation | c-myc-LHR (N-terminus)/ FLAG-LHR (N-terminus) | Coprecipitation of high molecular weight complexes from cells stably expressing LHRs. No detected change in complex molecular weight with hCG treatment. | [48] |
| Human LHRs | HEK 293 | Fluorescence cross-correlation spectroscopy (FCCS) | hLHRs-delExon10–GFP/hLHR-C131R–mCherry; hLHR-K605E–GFP/hLHR-C131R–mCherry | FCCS showed cross-correlation for each receptor combination. Trans-activation partially rescued hCG response (increased cAMP) but not LH response | [13] |
| Rat LHRs | HEK 293 | PALM super-resolution imaging | HA-WT-LHR, HA-LHRB-, FLAG-LHRS- (HA.11/FLAG Abs) | WT alone and LHRB- + LHRS- exhibited intermolecular interactions favoring the formation of LHR oligomers | [57] |
| Human FSHRs | HEK 293 | Imaging FRET | Anti-FSHR mAb-Alexa 588/Anti-FSHR An-Alexa 647 | Positive FRET for untreated/FSH-treated FSHRs | [18] |
| Co-immunoprecipitation | c-Myc-FSHR FLAG-FSHR | FSH oligomers form early in FSHR biosynthesis | |||
| Human FSHRs | X-ray Crystallography | Asna52-FSH or fully glycosylated FSH | FSHRs are a functional trimer when binding Asna52-FSH | [58] | |
| Human FSHRs | HEK293 | Fluorescence correlation spectroscopy/photon counting histogram analysis | Chimeric human-FSHR with rat LHR C terminus-EGFP | Human FSHR/LHR C-terminus chimeras are homodimers | [50] |
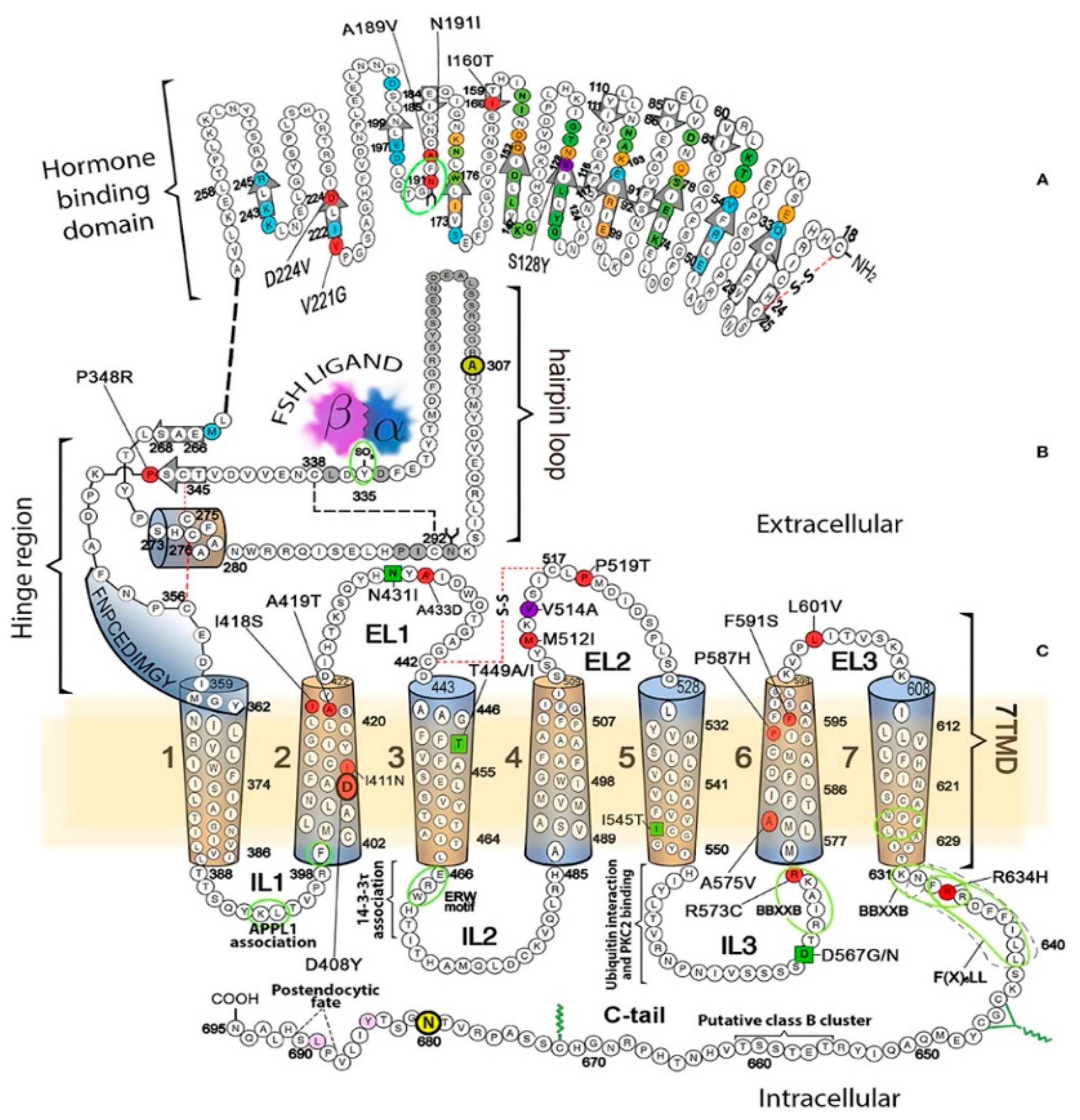
5. LHRs and FSHRs in Disease
| Receptor/Mutation | Homozygous/HeteRozygous | Phenotype | Reference |
|---|---|---|---|
| Follicle Stimulating Hormone Receptor | |||
| S128Y(T) | Spontaneous ovarian hyperstimulation syndrome during pregnancy, increased hCG, TSH response | [88] | |
| I61N | Heterozygous | Amenorrhea, infertility, early antral follicles, no cAMP | [89] |
| T449A | Spontaneous ovarian hyperstimulation syndrome during pregnancy, increased hCG, TSH response | [90] | |
| T449I | Spontaneous ovarian hyperstimulation syndrome during pregnancy, increased hCG, TSH response | [90] | |
| P519T | Failure of FSH to bind to FSHRs, hypergonadism | [91] | |
| D567N | Spontaneous ovarian hyperstimulation syndrome during pregnancy, increased hCG, TSH response, impaired FSHR desensitization, hypogonadotropic hypogonadism, precocious pseudopuberty | [90] | |
| N680S | Homozygous | PCOS, premature ovarian syndrome, high circulating FSH, decreased FSHR activity | [92] |
| P688T | Heterozygous | Amenorrhea, infertility, early antral follicles, decreased cAMP | [63] |
| Luteinizing Hormone Receptor | |||
| L10P | Signal peptide mutation causing micropenis, cryptorchidism | [93] | |
| Q18-L19ins9 | Signal peptide mutation causing severe Leydig cell hypoplasia | [94] | |
| I114F | Heterozygous | XY disorder of sexual development (XY DSD), Leydig cell hypoplasia, decreased LHRs, reduced signal transduction | [95] |
| C131R | Homozygous | Impaired cAMP response, micropenis, hypospadias, Hypoplastic phallus with hypospadias, XY DSD | [96] |
| V144F | homozygous | XY DSD | [97] |
| I152T | No Leydig cells, immature seminiferous tubules, impaired hormone binding, signal, genitalia with some virilization | [98] | |
| Q170Stop | Homozygous | Nonsense mutation causing primary amenorrhea | [99] |
| F194V | XY DSD, no cAMP signal | [100] | |
| N312S | Homozygous | Leydig cell hypoplasia in males and higher success rates for IVF pregnancy in females | [101] |
| Deletion between Y317 and S324 | Homozygous | Hypergonadism in males and Primary and secondary amenorrhea in females | [102] |
| Y317-S324 deletion | Homozygous | Males: Splice site mutation causing micropenis, delayed puberty, oligospermia Females: infertility with/without oligomenorrhea in females | [64] |
| C343S | Compound heterozygote | XY DSD | [103] |
| E354K | Homozygous | XY DSD, undescended testes in males and primary amenorrhea in females | [104] |
| L368P | Missense mutation causing precocious puberty, increased cAMP * | [105] | |
| I374T | Heterozygous | XY DSD, Leydig cell hypoplasia | [106] |
| T392I | Double homozygote | XY DSD, Leydig cell hypoplasia | [106] |
| M398T | Heterozygous | Familial male limited precocious puberty * | [107] |
| N400S | Homozygous | Infertility, empty follicle syndrome | [108] |
| I415T | Heterozygous | Leydig cell hypoplasia, micropenis, no cAMP production | [109] |
| L457R | Elevated cAMP, precocious puberty * | [12] | |
| T461I, exon 6A mutation | Compound heterozygote | XY DSD | [110] |
| L502P | XY DSD, Leydig cell hypoplasia | [111] | |
| Q525Stop | homozygous | Primary amenorrhea | [99] |
| I528Stop | Heterozygous | Leydig cell hypoplasia | [112] |
| I542L | Familial male limited precocious puberty * | [67] | |
| C543R | Compound heterozygote | XY DSD | [103] |
| C545Stop | Heterozygous | No cAMP, XY DSD | [113] |
| R554Stop | Homozygous | Males: XY DSD Females: small uterus, cystic ovary, primary and secondary amenorrhea | [114] |
| D564G | Heterozygous | Familial male limited precocious puberty * | [115] |
| A568V | Homozygous | Precocious puberty * | [105] |
| I575L | Heterozygous | Familial male limited precocious puberty * | [107] |
| D578G (H, E, Y) | Familial male precocious puberty, Leydig cell hyperplasia, precocious puberty * | [65,66,116,117,118,119] | |
| A593P | Homozygous | XY DSD, Leydig cell hypoplasia in males and primary amenorrhea, lack of breast development, infertility in females | [96] |
| I625K | Homozygous | Micropenis, no puberty, infertility | [96] |
| Exon 8 and S616Y deletion | Compound heterozygote | Leydig cell hypoplasia, micropenis, hypospadias | [120] |
| Exon 10 deletion | Homozygous | Hypogonadism, no puberty | [121] |
6. Turning the Signaling by Membrane-Expressed LHR Receptors on or off
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| LHR | Luteinizing hormone receptor |
| FSHR | follicle-stimulating hormone receptor |
| GPCR | G protein-coupled receptor |
| LH | luteinizing hormone |
| FSH | follicle-stimulating hormone |
| hCG | human chorionic gonadotropin |
| DG | deglycosylated human chorionic gonadotropin |
| COS | polycystic ovary disease |
References
- Narayan, P.; Ulloa-Aguirre, A.; Dias, J.A. Gonadotropin Hormones and Their Receptors. In Yen and Jaffe’s Reproductive Endocrinology, 8th ed.; Strauss, J.F., Barbieri, R.L., Eds.; Elsevier: Philadelphia, PA, USA, 2019; Chapter 2; pp. 25–57.e15. [Google Scholar]
- Casarini, L.; Huhtaniemi, I.; Simoni, M.; Rivero-Müller, A. Gonadotrophin Receptors. Endocrinol. Testis Male Reprod. 2017, 9, 123–168. [Google Scholar]
- Yung, Y.; Aviel-Ronen, S.; Maman, E.; Rubinstein, N.; Avivi, C.; Orvieto, R.; Hourvitz, A. Localization of luteinizing hormone receptor protein in the human ovary. Mol. Hum. Reprod. 2014, 20, 844–849. [Google Scholar] [CrossRef] [PubMed]
- Wahlström, T.; Huhtaniemi, I.; Hovatta, O.; Seppälä, M. Localization of luteinizing hormone, follicle-stimulating hormone, prolactin, and their receptors in human and rat testis using immunohistochemistry and radioreceptor assay. J. Clin. Endocrinol. Metab. 1983, 57, 825–830. [Google Scholar]
- Troppmann, B.; Kleinau, G.; Krause, G.; Gromoll, J. Structural and functional plasticity of the luteinizing hormone/choriogonadotrophin receptor. Hum. Reprod. Update 2013, 19, 583–602. [Google Scholar] [CrossRef]
- Narayan, P. Genetic models for the study of luteinizing hormone receptor function. Front. Endocrinol. 2015, 6, 152. [Google Scholar] [CrossRef]
- Maman, E.; Yung, Y.; Kedem, A.; Yerushalmi, G.M.; Konopnicki, S.; Cohen, B.; Dor, J.; Hourvitz, A. High expression of luteinizing hormone receptors messenger RNA by human cumulus granulosa cells is in correlation with decreased fertilization. Fertil. Steril. 2012, 97, 592–598. [Google Scholar] [CrossRef]
- Young, J.; McNeilly, A.S. Theca: The forgotten cell of the ovarian follicle. Reproduction 2010, 140, 489–504. [Google Scholar] [CrossRef]
- Zhu, X.; Gilbert, S.; Birnbaumer, M.; Birnbaumer, L. Dual signaling potential is common among Gs-coupled receptors and dependent on receptor density. Mol. Pharm. 1994, 46, 460–469. [Google Scholar]
- Borgbo, T.; Chrudimska, J.; Macek, M.; Jeppesen, J.; Bøtkjær, J.; Kristensen, S.; Macklon, K.; Ernst, E.; Hansen, L.; Andersen, C.Y. The polymorphic insertion of the luteinizing hormone receptor “insLQ” show a negative association to LHR gene expression and to the follicular fluid hormonal profile in human small antral follicles. Mol. Cell. Endocrinol. 2018, 460, 57–62. [Google Scholar] [CrossRef]
- Dufau, M.L. The luteinizing hormone receptor. Annu. Rev. Physiol. 1998, 60, 461–496. [Google Scholar] [CrossRef]
- Galet, C.; Ascoli, M. The differential binding affinities of the luteinizing hormone (LH)/choriogonadotropin receptor for LH and choriogonadotropin are dictated by different extracellular domain residues. Mol. Endocrinol. 2005, 19, 1263–1276. [Google Scholar] [CrossRef] [PubMed]
- Grzesik, P.; Kreuchwig, A.; Rutz, C.; Furkert, J.; Wiesner, B.; Schuelein, R.; Kleinau, G.; Gromoll, J.; Krause, G. Differences in signal activation by LH and hCG are mediated by the LH/CG receptor’s extracellular hinge region. Front. Endocrinol. 2015, 6, 140. [Google Scholar] [CrossRef] [PubMed]
- Grzesik, P.; Teichmann, A.; Furkert, J.; Rutz, C.; Wiesner, B.; Kleinau, G.; Schülein, R.; Gromoll, J.; Krause, G. Differences between lutropin-mediated and choriogonadotropin-mediated receptor activation. FEBS J. 2014, 281, 1479–1492. [Google Scholar] [CrossRef]
- Müller, T.; Gromoll, J.R.; Simoni, M. Absence of exon 10 of the human luteinizing hormone (LH) receptor impairs LH, but not human chorionic gonadotropin action. J. Clin. Endocrinol. Metab. 2003, 88, 2242–2249. [Google Scholar] [CrossRef] [PubMed]
- Bruysters, M.; Verhoef-Post, M.; Themmen, A.P. Asp330 and Tyr331 in the C-terminal cysteine-rich region of the luteinizing hormone receptor are key residues in hormone-induced receptor activation. J. Biol. Chem. 2008, 283, 25821–25828. [Google Scholar] [CrossRef]
- Mukherjee, S.; Gurevich, V.V.; Preninger, A.; Hamm, H.E.; Bader, M.-F.; Fazleabas, A.T.; Birnbaumer, L.; Hunzicker-Dunn, M. Aspartic acid 564 in the third cytoplasmic loop of the luteinizing hormone/choriogonadotropin receptor is crucial for phosphorylation-independent interaction with arrestin2. J. Biol. Chem. 2002, 277, 17916–17927. [Google Scholar] [CrossRef]
- Thomas, R.M.; Nechamen, C.A.; Mazurkiewicz, J.E.; Muda, M.; Palmer, S.; Dias, J.A. Follice-Stimulating Hormone Receptor Forms Oligomers and Shows Evidence of Carboxyl-Terminal Proteolytic Processing. Endocrinology 2007, 148, 1987–1995. [Google Scholar] [CrossRef][Green Version]
- Jiang, X.; Dias, J.A.; He, X. Structural biology of glycoprotein hormones and their receptors: Insights to signaling. Mol. Cell. Endocrinol. 2014, 382, 424–451. [Google Scholar] [CrossRef]
- Richards, J.S.; Pangas, S.A. The ovary: Basic biology and clinical implications. J. Clin. Investig. 2010, 120, 963–972. [Google Scholar] [CrossRef]
- Ulloa-Aguirre, A.; Zariñán, T.; Jardón-Valadez, E.; Gutiérrez-Sagal, R.; Dias, J.A. Structure-function relationships of the follicle-stimulating hormone receptor. Front. Endocrinol. 2018, 9, 707. [Google Scholar] [CrossRef]
- Riccetti, L.; De Pascali, F.; Gilioli, L.; Potì, F.; Giva, L.B.; Marino, M.; Tagliavini, S.; Trenti, T.; Fanelli, F.; Mezzullo, M. Human LH and hCG stimulate differently the early signalling pathways but result in equal testosterone synthesis in mouse Leydig cells in vitro. Reprod. Biol. Endocrinol. 2017, 15, 2. [Google Scholar] [CrossRef] [PubMed]
- Norman, A.W.; Litwack, G. Hormones; Academic Press: Cambridge, MA, USA, 1997. [Google Scholar]
- The Practice Committee of the American Society for Reproductive Medicine, Birmingham, Alabama. Gonadotropin preparations: Past, present, and future perspectives. Fertil. Steril. 2008, 90, S13–S20. [Google Scholar] [CrossRef] [PubMed]
- Lapthorn, A.; Harris, D.; Littlejohn, A.; Lustbader, J.; Canfield, R.; Machin, K.; Morgan, F.; Isaacs, N. Crystal structure of human chorionic gonadotropin. Nature 1994, 369, 455. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Lustbader, J.W.; Liu, Y.; Canfield, R.E.; Hendrickson, W.A. Structure of human chorionic gonadotropin at 2.6 Å resolution from MAD analysis of the selenomethionyl protein. Structure 1994, 2, 545–558. [Google Scholar] [CrossRef]
- Naylor, S.L.; Chin, W.W.; Goodman, H.M.; Lalley, P.A.; Grzeschik, K.-H.; Sakaguchi, A.Y. Chromosome assignment of genes encoding theα andΒ subunits of glycoprotein hormones in man and mouse. Somat. Cell Genet. 1983, 9, 757–770. [Google Scholar] [CrossRef]
- Ogiwara, K.; Fujimori, C.; Rajapakse, S.; Takahashi, T. Characterization of luteinizing hormone and luteinizing hormone receptor and their indispensable role in the ovulatory process of the medaka. PLoS ONE 2013, 8, e54482. [Google Scholar] [CrossRef]
- Potorac, I.; Rivero-Muller, A.; Trehan, A.; Kielbus, M.; Jozwiak, K.; Pralong, F.; Hafidi, A.; Thiry, A.; Ménagé, J.-J.; Huhtaniemi, I.T. A vital region for human glycoprotein hormone trafficking revealed by an LHB mutation. J. Endocrinol. 2016, 231, 197–207. [Google Scholar] [CrossRef]
- Pierce, J.G.; Parsons, T.F. Glycoprotein hormones: Structure and function. Annu. Rev. Biochem. 1981, 50, 465–495. [Google Scholar] [CrossRef]
- Ellinwood, W.; Nett, T.; Niswender, G. Maintenance of the corpus luteum of early pregnancy in the ewe. II. Prostaglandin secretion by the endometrium in vitro and in vivo. Biol. Reprod. 1979, 21, 845–856. [Google Scholar] [CrossRef]
- Boron, W.F.; Boulpaep, E.L. Medical Physiology E-Book; Elsevier Health Sciences: New York, NY, USA, 2016. [Google Scholar]
- Strauss, J.F.; Barbieri, R.L. Yen & Jaffe’s Reproductive Endocrinology E-Book: Physiology, Pathophysiology, and Clinical Management (Expert Consult-Online and Print); Elsevier Health Sciences: New York, NY, USA, 2013. [Google Scholar]
- Cole, L.A. Human Chorionic Gonadotropin (hCG); Elsevier: New York, NY, USA, 2014. [Google Scholar]
- Cole, L.A. Biological functions of hCG and hCG-related molecules. Reprod. Biol. Endocrinol. 2010, 8, 102. [Google Scholar] [CrossRef]
- Casarini, L.; Lispi, M.; Longobardi, S.; Milosa, F.; La Marca, A.; Tagliasacchi, D.; Pignatti, E.; Simoni, M. LH and hCG action on the same receptor results in quantitatively and qualitatively different intracellular signalling. PLoS ONE 2012, 7, e46682. [Google Scholar] [CrossRef] [PubMed]
- Minegishi, T.; Nakamura, K.; Ibuki, Y. Structure and Reguration of LH/CG Recepter. Endocr. J. 1993, 40, 275–287. [Google Scholar] [CrossRef][Green Version]
- Keutmann, H.T.; McIlroy, P.J.; Bergert, E.R.; Ryan, R.J. Chemically deglycosylated human chorionic gonadotropin subunits: Characterization and biological properties. Biochemistry 1983, 22, 3067–3072. [Google Scholar] [CrossRef] [PubMed]
- Dunkel, L.; Raivio, T.; Laine, J.; Holmberg, C. Circulating luteinizing hormone receptor inhibitor (s) in boys with chronic renal failure. Kidney Int. 1997, 51, 777–784. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Welt, C.; Schneyer, A. Differential regulation of inhibin B and inhibin a by follicle-stimulating hormone and local growth factors in human granulosa cells from small antral follicles. J. Clin. Endocrinol. Metab. 2001, 86, 330–336. [Google Scholar] [CrossRef] [PubMed]
- Papaleo, E.; Zaffagnini, S.; Munaretto, M.; Vanni, V.S.; Rebonato, G.; Grisendi, V.; Di Paola, R.; La Marca, A. Clinical application of a nomogram based on age, serum FSH and AMH to select the FSH starting dose in IVF/ICSI cycles: A retrospective two-centres study. Eur. J. Obstet. Gynecol. Reprod. Biol. 2016, 207, 94–99. [Google Scholar] [CrossRef]
- Behre, H.M. Clinical Use of FSH in Male Infertility. Front. Endocrinol. 2019, 10, 322. [Google Scholar] [CrossRef]
- Santi, D.; Crépieux, P.; Reiter, E.; Spaggiari, G.; Brigante, G.; Casarini, L.; Rochira, V.; Simoni, M. Follicle-stimulating Hormone (FSH) Action on Spermatogenesis: A Focus on Physiological and Therapeutic Roles. J. Clin. Med. 2020, 9, 1014. [Google Scholar] [CrossRef]
- Milligan, G. G protein-coupled receptor dimerisation: Molecular basis and relevance to function. Biochim. Biophys. Acta (BBA)-Biomembr. 2007, 1768, 825–835. [Google Scholar] [CrossRef]
- Ferré, S.; Casadó, V.; Devi, L.A.; Filizola, M.; Jockers, R.; Lohse, M.J.; Milligan, G.; Pin, J.-P.; Guitart, X. G protein–coupled receptor oligomerization revisited: Functional and pharmacological perspectives. Pharmacol. Rev. 2014, 66, 413–434. [Google Scholar] [CrossRef]
- Hunzicker-Dunn, M.; Barisas, B.G.; Song, J.; Roess, D.A. Membrane organization of luteinizing hormone receptors differs between actively signaling and desensitized receptors. J. Biol. Chem. 2003, 278, 42744–42749. [Google Scholar] [CrossRef] [PubMed]
- Ji, I.; Lee, C.; Jeoung, M.; Koo, Y.; Sievert, G.A.; Ji, T.H. Trans-activation of mutant follicle-stimulating hormone receptors selectively generates only one of two hormone signals. Mol. Endocrinol. 2004, 18, 968–978. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Tao, Y.-X.; Johnson, N.B.; Segaloff, D.L. Constitutive and agonist-dependent self-association of the cell surface human lutropin receptor. J. Biol. Chem. 2004, 279, 5904–5914. [Google Scholar] [CrossRef] [PubMed]
- Xing, Y.; Ross, S.; Berrnard, M.P.; Nederlof, M.; Scaduto, R.; Moyle, W. Lutropin receptors appear to bind human chorionic gonadotropin (hCG) as monomers. Endocrinology 2014, 383, 203–213. [Google Scholar]
- Mazurkiewicz, J.E.; Herrick-Davis, K.; Barroso, M.; Ulloa-Aguirre, A.; Lindau-Shepard, B.; Thomas, R.M.; Dias, J.A. Single-Molecule Analyses of Fully Functional Fluorescent Protein-Tagged Follitropin Receptor Reveal Homodimerization and Specific Heterodimerization with Lutropin Receptor. Biol. Reprod. 2015, 92, 100–101. [Google Scholar] [CrossRef]
- Fan, Q.; Hendrickson, W. Structure of human follicle-stimulating hormone in complex with its receptor. Nature 2005, 433, 269–277. [Google Scholar] [CrossRef]
- Amsterdam, A.; Berkowitz, A.; Nimrod, A.; Kohen, F. Aggregation of luteinizing hormone receptors in granulosa cells: A possible mechanism of desensitization to the hormone. Proc. Natl. Acad. Sci. USA 1980, 77, 3440–3444. [Google Scholar] [CrossRef]
- Luborsky, J.; Slater, W.; Behrman, H. Luteinizing Hormone (LH) Receptor Aggregation: Modification of Ferritin-LH Binding and Aggregation by Prostaglandin F2 α and Ferritin-LH. Endocrinology 1984, 115, 2217–2226. [Google Scholar] [CrossRef]
- Harmatz, D.; Ji, T.H.; Middaugh, C.R. Aggregation state of the gonadotropin receptor. Biochem. Biophys. Res. Commun. 1985, 127, 687–692. [Google Scholar] [CrossRef]
- Horvat, R.D.; Nelson, S.; Clay, C.M.; Barisas, B.G.; Roess, D.A. Intrinsically fluorescent luteinizing hormone receptor demonstrates hormone-driven aggregation. Biochem. Biophys. Res. Commun. 1999, 255, 382–385. [Google Scholar] [CrossRef]
- Horvat, R.D.; Barisas, B.G.; Roess, D.A. Luteinizing hormone receptors are self-associated in slowly diffusing complexes during receptor desensitization. Mol. Endocrinol. 2001, 15, 534–542. [Google Scholar] [CrossRef] [PubMed]
- Jonas, K.C.; Fanelli, F.; Huhtaniemi, I.T.; Hanyaloglu, A.C. Single molecule analysis of functionally asymmetric G protein-coupled receptor (GPCR) oligomers reveals diverse spatial and structural assemblies. J. Biol. Chem. 2015, 290, 3875–3892. [Google Scholar] [CrossRef]
- Jiang, X.; Fischer, D.; Chen, X.; McKenna, S.D.; Liu, H.; Sriraman, V.; Henry, N.Y.; Goutopoulos, A.; Arkinstall, S.; He, X. Evidence for follicle-stimulating hormone receptor as a functional trimer. J. Biol. Chem. 2014, 289, 14273–14282. [Google Scholar] [CrossRef]
- Ji, I.; Lee, C.; Song, Y.; Conn, P.M.; Ji, T.H. Cis-and trans-activation of hormone receptors: The LH receptor. Mol. Endocrinol. 2002, 16, 1299–1308. [Google Scholar] [CrossRef] [PubMed]
- Rivero-Müller, A.; Chou, Y.-Y.; Ji, I.; Lajic, S.; Hanyaloglu, A.C.; Jonas, K.; Rahman, N.; Ji, T.H.; Huhtaniemi, I. Rescue of defective G protein–coupled receptor function in vivo by intermolecular cooperation. Proc. Natl. Acad. Sci. USA 2010, 107, 2319–2324. [Google Scholar] [CrossRef] [PubMed]
- Szymańska, K.; Kałafut, J.; Przybyszewska, A.; Paziewska, B.; Adamczuk, G.; Kiełbus, M.; Rivero-Müller, A. FSHR trans-activation and oligomerization. Front. Endocrinol. 2018, 9, 760. [Google Scholar] [CrossRef] [PubMed]
- DiPilato, L.M.; Cheng, X.; Zhang, J. Fluorescent indicators of cAMP and Epac activation reveal differential dynamics of cAMP signaling within discrete subcellular compartments. Proc. Natl. Acad. Sci. USA 2004, 101, 16513–16518. [Google Scholar] [CrossRef]
- Achrekar, S.K.; Modi, D.N.; Meherji, P.K.; Patel, Z.M.; Mahale, S.D. Follicle stimulating hormone receptor gene variants in women with primary and secondary amenorrhea. J. Assist. Reprod. Genet. 2010, 27, 317–326. [Google Scholar] [CrossRef]
- Newton, C.L.; Anderson, R.C.; Katz, A.A.; Millar, R.P. Loss-of-function mutations in the human luteinizing hormone receptor predominantly cause intracellular retention. Endocrinology 2016, 157, 4364–4377. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Duranteau, L.; Carel, J.-C.; Monroe, J.; Doyle, D.A.; Shenker, A. Leydig-cell tumors caused by an activating mutation of the gene encoding the luteinizing hormone receptor. N. Engl. J. Med. 1999, 341, 1731–1736. [Google Scholar] [CrossRef] [PubMed]
- Shenker, A.; Laue, L.; Kosugi, S.; Merendino, J.J.; Minegishi, T.; Cutler, G.B. A constitutively activating mutation of the luteinizing hormone receptor in familial male precocious puberty. Nature 1993, 365, 652–654. [Google Scholar] [CrossRef]
- Laue, L.; Chan, W.-Y.; Hsueh, A.; Kudo, M.; Hsu, S.Y.; Wu, S.-M.; Blomberg, L.; Cutler, G. Genetic heterogeneity of constitutively activating mutations of the human luteinizing hormone receptor in familial male-limited precocious puberty. Proc. Natl. Acad. Sci. USA 1995, 92, 1906–1910. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.-M.; Leschek, E.W.; Rennert, O.M.; Chan, W.-Y. Luteinizing hormone receptor mutations in disorders of sexual development and cancer. Pediatric Pathol. Mol. Med. 2000, 19, 21–40. [Google Scholar] [CrossRef]
- Qiao, J.; Han, B. Diseases caused by mutations in luteinizing hormone/chorionic gonadotropin receptor. Prog. Mol. Biol. Transl. Sci. 2019, 161, 69–89. [Google Scholar] [PubMed]
- Puttabyatappa, M.; Padmanabhan, V. Ovarian and extra-ovarian mediators in the development of polycystic ovary syndrome. J. Mol. Endocrinol. 2018, 61, R161–R184. [Google Scholar] [CrossRef]
- Zou, J.; Wu, D.; Liu, Y.; Tan, S. Association of luteinizing hormone/choriogonadotropin receptor gene polymorphisms with polycystic ovary syndrome risk: A meta-analysis. Gynecol. Endocrinol. 2019, 35, 81–85. [Google Scholar] [CrossRef]
- Huang, W.; Cao, Y.; Shi, L. Effects of FSHR polymorphisms on premature ovarian insufficiency in human beings: A meta-analysis. Reprod. Biol. Endocrinol. 2019, 17, 80. [Google Scholar] [CrossRef]
- Gheorghiu, M. Actualities in mutations of luteinizing hormone (LH) and follicle-stimulating hormone (FSH) receptors. Acta Endocrinol. 2019, 5, 139–142. [Google Scholar]
- Zhang, Q.; Madden, N.E.; Wong, A.S.T.; Chow, B.K.C.; Lee, L.T.O. The role of endocrine G protein-coupled receptors in ovarian cancer progression. Front. Endocrinol 2017, 8, 66. [Google Scholar] [CrossRef]
- Perales-Puchalt, A.; Svoronos, N.; Rutkowski, M.R.; Allegrezza, M.J.; Tesone, A.J.; Payne, K.K.; Wickramasinghe, J.; Nguyen, J.M.; O’Brien, S.W.; Gumireddy, K. Follicle-stimulating hormone receptor is expressed by most ovarian cancer subtypes and is a safe and effective immunotherapeutic target. Clin. Cancer Res. 2017, 23, 441–453. [Google Scholar] [CrossRef]
- Lenhard, M.; Lennerová, T.; Ditsch, N.; Kahlert, S.; Friese, K.; Mayr, D.; Jeschke, U. Opposed roles of follicle-stimulating hormone and luteinizing hormone receptors in ovarian cancer survival. Histopathology 2011, 58, 990–994. [Google Scholar] [CrossRef] [PubMed]
- Zheng, W.; Lu, J.J.; Luo, F.; Zheng, Y.; Feng, Y.-J.; Felix, J.C.; Lauchlan, S.C.; Pike, M.C. Ovarian epithelial tumor growth promotion by follicle-stimulating hormone and inhibition of the effect by luteinizing hormone. Gynecol. Oncol. 2000, 76, 80–88. [Google Scholar] [CrossRef] [PubMed]
- Mariani, S.; Salvatori, L.; Basciani, S.; Arizzi, M.; Franco, G.; Petrangeli, E.; Spera, G.; Gnessi, L. Expression and Cellular Localization of Follicle-Stimulating Hormone Receptor in Normal Human Prostate, Benign Prostatic Hyperplasia and Prostate Cancer. J. Urol. 2006, 175, 2072–2077. [Google Scholar] [CrossRef]
- Papadimitriou, K.; Kountourakis, P.; Kottorou, A.E.; Antonacopoulou, A.G.; Rolfo, C.; Peeters, M.; Kalofonos, H.P. Follicle-Stimulating Hormone Receptor (FSHR): A Promising Tool in Oncology? Mol. Diagn. Ther. 2016, 20, 523–530. [Google Scholar] [CrossRef]
- Ghinea, N. Vascular Endothelial FSH Receptor, a Target of Interest for Cancer Therapy. Endocrinology 2018, 159, 3268–3274. [Google Scholar] [CrossRef]
- Bi, W.; Shao, S.; Li, Z.; Ruan, Y.; Luan, S.; Dong, Z.; Wang, J.; Wu, S.; Guo, T.; Ma, S.; et al. FSHR ablation induces depression-like behaviors. Acta Pharmacol. Sin. 2020, 43, 1033–1040. [Google Scholar] [CrossRef] [PubMed]
- Movsas, T.Z.; Sigler, R.; Muthusamy, A. Elimination of signaling by the luteinizing hormone receptor reduces ocular VEGF and retinal vascularization during mouse eye development. Curr. Eye Res. 2018, 43, 1286–1289. [Google Scholar] [CrossRef]
- Lin, J.; Li, X.; Yuan, F.; Lin, L.; Cook, C.L.; Rao, C.V.; Lei, Z. Genetic ablation of luteinizing hormone receptor improves the amyloid pathology in a mouse model of Alzheimer disease. J. Neuropathol. Exp. Neurol. 2010, 69, 253–261. [Google Scholar] [CrossRef]
- Bird, J.; Li, X.; Lei, Z.; Sanfilippo, J.; Yussman, M.; Rao, C.V. Luteinizing hormone and human chorionic gonadotropin decrease type 2 5α-reductase and androgen receptor protein levels in women’s skin. J. Clin. Endocrinol. Metab. 1998, 83, 1776–1782. [Google Scholar] [CrossRef]
- Pabon, J.; Bird, J.; Li, X.; Huang, Z.; Lei, Z.; Sanfilippo, J.; Yussman, M.; Rao, C.V. Human skin contains luteinizing hormone/chorionic gonadotropin receptors. J. Clin. Endocrinol. Metab. 1996, 81, 2738–2741. [Google Scholar]
- Rao, C.V. An overview of the past, present, and future of nongonadal LH/hCG actions in reproductive biology and medicine. Semin. Reprod. Med. 2001, 19, 7–18. [Google Scholar] [CrossRef] [PubMed]
- Bertone-Johnson, E.R.; Virtanen, J.K.; Niskanen, L.; Nurmi, T.; Ronkainen, K.; Voutilainen, S.; Mursu, J.; Kauhanen, J.i.; Tuomainen, T.-P. Association of follicle-stimulating hormone levels and risk of type 2 diabetes in older postmenopausal women. Menopause 2017, 24, 796–802. [Google Scholar] [CrossRef] [PubMed]
- Topdagi Yilmaz, E.P.; Yapca, O.E.; Topdagi, Y.E.; Kaya Topdagi, S.; Kumtepe, Y. Spontaneous ovarian Hyperstimulation syndrome with FSH receptor gene mutation: Two rare case reports. Case Rep. Obstet. Gynecol. 2018, 2018, 9294650. [Google Scholar] [CrossRef]
- Khor, S.; Lyu, Q.; Kuang, Y.; Lu, X. Novel FSHR variants causing female resistant ovary syndrome. Mol. Genet. Genom. Med. 2020, 8, e1082. [Google Scholar] [CrossRef]
- Montanelli, L.; Delbaere, A.; Di Carlo, C.; Nappi, C.; Smits, G.; Vassart, G.; Costagliola, S. A mutation in the follicle-stimulating hormone receptor as a cause of familial spontaneous ovarian hyperstimulation syndrome. J. Clin. Endocrinol. Metab. 2004, 89, 1255–1258. [Google Scholar] [CrossRef]
- Meduri, G.; Touraine, P.; Beau, I.; Lahuna, O.; Desroches, A.; Vacher-Lavenu, M.; Kuttenn, F.; Misrahi, M. Delayed puberty and primary amenorrhea associated with a novel mutation of the human follicle-stimulating hormone receptor: Clinical, histological, and molecular studies. J. Clin. Endocrinol. Metab. 2003, 88, 3491–3498. [Google Scholar] [CrossRef]
- Nenonen, H.A.; Lindgren, I.A.; Prahl, A.S.; Trzybulska, D.; Kharraziha, I.; Hultén, M.; Giwercman, Y.L.; Henic, E. The N680S variant in the follicle-stimulating hormone receptor gene identifies hyperresponders to controlled ovarian stimulation. Pharm. Genom. 2019, 29, 114. [Google Scholar] [CrossRef]
- Vezzoli, V.; Duminuco, P.; Vottero, A.; Kleinau, G.; Schülein, R.; Minari, R.; Bassi, I.; Bernasconi, S.; Persani, L.; Bonomi, M. A new variant in signal peptide of the human luteinizing hormone receptor (LHCGR) affects receptor biogenesis causing leydig cell hypoplasia. Hum. Mol. Genet. 2015, 24, 6003–6012. [Google Scholar] [CrossRef]
- Charmandari, E.; Guan, R.; Zhang, M.; Silveira, L.; Fan, Q.; Chrousos, G.; Sertedaki, A.; Latronico, A.; Segaloff, D. Misfolding ectodomain mutations of the lutropin receptor increase efficacy of hormone stimulation. Mol. Endocrinol. 2016, 30, 62–76. [Google Scholar] [CrossRef][Green Version]
- Leung, M.Y.-K.; Steinbach, P.J.; Bear, D.; Baxendale, V.; Fechner, P.Y.; Rennert, O.M.; Chan, W.-Y. Biological effect of a novel mutation in the third leucine-rich repeat of human luteinizing hormone receptor. Mol. Endocrinol. 2006, 20, 2493–2503. [Google Scholar] [CrossRef][Green Version]
- Martens, J.W.M.; Verhoef-Post, M.; Abelin, N.; Ezabella, M.; Toledo, S.P.A.; Brunner, H.G.; Themmen, A.P.N. A homozygous mutation in the luteinizing hormone receptor causes partial Leydig cell hypoplasia: Correlation between receptor activity and phenotype. Mol. Endocrinol. 1998, 12, 775–784. [Google Scholar] [CrossRef] [PubMed]
- Richter-Unruh, A.; Verhoef-Post, M.; Malak, S.; Homoki, J.; Hauffa, B.; Themmen, A. Leydig cell hypoplasia: Absent luteinizing hormone receptor cell surface expression caused by a novel homozygous mutation in the extracellular domain. J. Clin. Endocrinol. Metab. 2004, 89, 5161–5167. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Qiao, J.; Han, B.; Liu, B.L.; Chen, X.; Ru, Y.; Cheng, K.X.; Chen, F.G.; Zhao, S.-X.; Liang, J.; Lu, Y.L. A splice site mutation combined with a novel missense mutation of LHCGR cause male pseudohermaphroditism. Hum. Mutat. 2009, 30, E855–E865. [Google Scholar] [CrossRef] [PubMed]
- Hmida, I.B.H.; Mougou-Zerelli, S.; Hadded, A.; Dimassi, S.; Kammoun, M.; Bignon-Topalovic, J.; Bibi, M.; Saad, A.; Bashamboo, A.; McElreavey, K. Novel homozygous nonsense mutations in the luteinizing hormone receptor (LHCGR) gene associated with 46, XY primary amenorrhea. Fertil. Steril. 2016, 106, 225–229.e211. [Google Scholar] [CrossRef]
- Gromoll, J.; Schulz, A.; Borta, H.; Gudermann, T.; Teerds, K.J.; Greschniok, A.; Nieschlag, E.; Seif, F.J. Homozygous mutation within the conserved Ala-Phe-Asn-Glu-Thr motif of exon 7 of the LH receptor causes male pseudohermaphroditism. Eur. J. Endocrinol 2002, 147, 597–608. [Google Scholar] [CrossRef][Green Version]
- Lindgren, I.; Nenonen, H.; Henic, E.; Bungum, L.; Prahl, A.; Bungum, M.; Leijonhufvud, I.; Huhtaniemi, I.; Andersen, C.Y.; Giwercman, Y.L. Gonadotropin receptor variants are linked to cumulative live birth rate after in vitro fertilization. J. Assist. ReproGenet. 2019, 36, 29–38. [Google Scholar] [CrossRef]
- Bruysters, M.; Christin-Maitre, S.; Verhoef-Post, M.; Sultan, C.; Auger, J.; Faugeron, I.; Larue, L.; Lumbroso, S.; Themmen, A.; Bouchard, P. A new LH receptor splice mutation responsible for male hypogonadism with subnormal sperm production in the propositus, and infertility with regular cycles in an affected sister. Hum. Repro. 2008, 23, 1917–1923. [Google Scholar] [CrossRef]
- Martens, J.; Lumbroso, S.; Verhoef-Post, M.; Georget, V.; Richter-Unruh, A.; Szarras-Czapnik, M.; Romer, T.; Brunner, H.; Themmen, A.; Sultan, C. Mutant luteinizing hormone receptors in a compound heterozygous patient with complete Leydig cell hypoplasia: Abnormal processing causes signaling deficiency. J. Clin. Endocrinol. Metab. 2002, 87, 2506–2513. [Google Scholar] [CrossRef]
- Brüser, A.; Schulz, A.; Rothemund, S.; Ricken, A.; Calebiro, D.; Kleinau, G.; Schöneberg, T. The activation mechanism of glycoprotein hormone receptors with implications in the cause and therapy of endocrine diseases. J. Biol. Chem. 2016, 291, 508–520. [Google Scholar] [CrossRef]
- Latronico, A.C.; Shinozaki, H.; Guerra, G., Jr.; Pereira, M.A.A.; Lemos Marini, S.H.V.; Baptista, M.T.M.; Arnhold, I.J.P.; Fanelli, F.; Mendonca, B.B.; Segaloff, D.L. Gonadotropin-independent precocious puberty due to luteinizing hormone receptor mutations in Brazilian boys: A novel constitutively activating mutation in the first transmembrane helix. J. Clin. Endocrinol. Metab. 2000, 85, 4799–4805. [Google Scholar]
- Pals-Rylaarsdam, R.; Liu, G.; Brickman, W.; Duranteau, L.; Monroe, J.; El-Awady, M.K.; Gad, Y.Z.; Shenker, A. A novel double mutation in the luteinizing hormone receptor in a kindred with familial Leydig cell hypoplasia and male pseudohermaphroditism. Endocr. Res. 2005, 31, 307–323. [Google Scholar] [CrossRef]
- Leschek, E.W.; Flor, A.C.; Bryant, J.C.; Jones, J.V.; Barnes, K.M.; Cutler, G.B., Jr. Effect of antiandrogen, aromatase inhibitor, and gonadotropin-releasing hormone analog on adult height in familial male precocious puberty. J. Pediatrics 2017, 190, 229–235. [Google Scholar] [CrossRef]
- Yariz, K.O.; Walsh, T.; Uzak, A.; Spiliopoulos, M.; Duman, D.; Onalan, G.; King, M.-C.; Tekin, M. Inherited mutation of the luteinizing hormone/choriogonadotropin receptor (LHCGR) in empty follicle syndrome. Fertil. Steril. 2011, 96, e125–e130. [Google Scholar] [CrossRef] [PubMed]
- Kossack, N.; Troppmann, B.; Richter-Unruh, A.; Kleinau, G.; Gromoll, J. Aberrant transcription of the LHCGR gene caused by a mutation in exon 6A leads to Leydig cell hypoplasia type II. Mol. Cell. Endocrinol. 2013, 366, 59–67. [Google Scholar] [CrossRef] [PubMed]
- Kossack, N.; Simoni, M.; Richter-Unruh, A.; Themmen, A.P.N.; Gromoll, J. Mutations in a novel, cryptic exon of the luteinizing hormone/chorionic gonadotropin receptor gene cause male pseudohermaphroditism. PLoS Med. 2008, 5, e88. [Google Scholar] [CrossRef]
- Leung, M.Y.K.; Al-Muslim, O.; Wu, S.M.; Aziz, A.; Inam, S.l.; Awadh, M.; Rennert, O.M.; Chan, W.Y. A novel missense homozygous inactivating mutation in the fourth transmembrane helix of the luteinizing hormone receptor in leydig cell hypoplasia. Am. J. Med. Genet. Part A 2004, 130, 146–153. [Google Scholar] [CrossRef]
- Xu, Y.; Chen, Y.; Li, N.; Hu, X.; Li, G.; Ding, Y.; Li, J.; Shen, Y.; Wang, X.; Wang, J. Novel compound heterozygous variants in the LHCGR gene identified in a subject with Leydig cell hypoplasia type 1. J. Pediatric Endocrinol. Metab. 2018, 31, 239–245. [Google Scholar] [CrossRef]
- Laue, L.; Wu, S.-M.; Kudo, M.; Hsueh, A.J.W.; Cutler, G.B., Jr.; Griffin, J.E.; Wilson, J.D.; Brain, C.; Berry, A.C.; Grant, D.B. A nonsense mutation of the human luteinizing hormone receptor gene in Leydig cell hypoplasia. Hum. Mol. Genet. 1995, 4, 1429–1433. [Google Scholar] [CrossRef]
- Latronico, A.C.; Anasti, J.; Arnhold, I.J.P.; Rapaport, R.; Mendonca, B.B.; Bloise, W.; Castro, M.; Tsigos, C.; Chrousos, G.P. Brief Report: Testicular and Ovarian Resistance to Luteinizing Hormone Caused by Inactivating Mutations of the Luteinizing Hormone-Receptor Gene. Obstet. Gynecol. Surv. 1996, 51, 416–419. [Google Scholar] [CrossRef]
- Leschek, E.W.; Chan, W.-Y.; Diamond, D.A.; Kaefer, M.; Jones, J.; Barnes, K.M.; Cutler, G.B., Jr. Nodular Leydig cell hyperplasia in a boy with familial male-limited precocious puberty. J. Pediatrics 2001, 138, 949–951. [Google Scholar] [CrossRef] [PubMed]
- Boot, A.M.; Lumbroso, S.; Verhoef-Post, M.; Richter-Unruh, A.; Looijenga, L.H.J.; Funaro, A.; Beishuizen, A.; van Marle, A.; Drop, S.L.S.; Themmen, A.P.N. Mutation Analysis of the LH Receptor Gene in Leydig Cell Adenoma and Hyperplasia and Functional and Biochemical Studies of Activating Mutations of the LH Receptor Gene. J. Clin. Endocrinol Metab. 2011, 96, E1197–E1205. [Google Scholar] [CrossRef] [PubMed]
- Müller, J.; Gondos, B.; Kosugi, S.; Mori, T.; Shenker, A. Severe testotoxicosis phenotype associated with Asp578--> Tyr mutation of the lutrophin/choriogonadotrophin receptor gene. J. Med. Genet. 1998, 35, 340–341. [Google Scholar] [CrossRef] [PubMed]
- Yano, K.; Hidaka, A.; Saji, M.; Polymeropoulos, M.H.; Okuno, A.; Kohn, L.D.; Cutler, G.B., Jr. A sporadic case of male-limited precocious puberty has the same constitutively activating point mutation in luteinizing hormone/choriogonadotropin receptor gene as familial cases. J. Clin. Endocrinol. Metab. 1994, 79, 1818–1823. [Google Scholar] [PubMed]
- Wu, S.-M.; Leschek, E.W.; Brain, C.; Chan, W.-Y. A novel luteinizing hormone receptor mutation in a patient with familial male-limited precocious puberty: Effect of the size of a critical amino acid on receptor activity. Mol. Genet. Metab. 1999, 66, 68–73. [Google Scholar] [CrossRef]
- Laue, L.L.; Wu, S.-M.; Kudo, M.; Bourdony, C.J.; Cutler, G.B., Jr.; Hsueh, A.J.; Chan, W.-Y. Compound heterozygous mutations of the luteinizing hormone receptor gene in Leydig cell hypoplasia. Mol. Endocrinol. 1996, 10, 987–997. [Google Scholar]
- Gromoll, J.; Eiholzer, U.; Nieschlag, E.; Simoni, M. Male hypogonadism caused by homozygous deletion of exon 10 of the luteinizing hormone (LH) receptor: Differential action of human chorionic gonadotropin and LH. J. Clin. Endocrinol. Metab. 2000, 85, 2281–2286. [Google Scholar] [CrossRef]
- Althumairy, D.; Murakami, H.A.; Zhang, D.; Barisas, B.G.; Roess, D.A.; Crans, D.C. Effects of vanadium (IV) compounds on plasma membrane lipids lead to G protein-coupled receptor signal transduction. J. Inorg. Biochem. 2020, 203, 110873. [Google Scholar] [CrossRef]
- Gründker, C.; Völker, P.; Emons, G. Antiproliferative signaling of luteinizing hormone-releasing hormone in human endometrial and ovarian cancer cells through G proteinα I-mediated activation of phosphotyrosine phosphatase. Endocrinology 2001, 142, 2369–2380. [Google Scholar] [CrossRef]
- Kan, W.-C.; Lu, T.-L.; Ling, P.; Lee, T.-H.; Cho, C.-Y.; Huang, C.-Y.F.; Jeng, W.-Y.; Weng, Y.-P.; Chiang, C.-Y.; Wu, J.B. Pervanadate induces Mammalian Ste20 Kinase 3 (MST3) tyrosine phosphorylation but not activation. J. Inorg. Biochem. 2016, 160, 33–39. [Google Scholar] [CrossRef]
- Althumairy, D.; Postal, K.; Barisas, B.; Nunes, G.; Roess, D.; Crans, D. Polyoxometalates Function as Indirect Activators of a G Protein-Coupled Receptor. Metallomics 2020, 12, 1044–1061. [Google Scholar] [CrossRef]
- Crans, D.; Brown, M.; Roess, D. Vanadium compounds promote biocatalysis in cells through actions on cell membranes. Catal. Today 2020. under revision. [Google Scholar] [CrossRef]
- Alhumairy, D.; Murakami, H.; Colclough, R.; Barisas, B.; Roess, D.; Crans, D. Vanadium Compounds as Indirect Activators of a G Protein-Coupled Receptor. In Vanadium Catalysis; Chapter 21; ASAP Royal Society of Chemistry: London, UK, 2020. [Google Scholar]
- Samart, N.; Althumairy, D.; Zhang, D.; Roess, D.A.; Crans, D.C. Initiation of a novel mode of membrane signaling: Vanadium facilitated signal transduction. Coord. Chem. Rev. 2020, 416, 213286. [Google Scholar] [CrossRef]
- Crans, D.C.; Rithner, C.D.; Baruah, B.; Gourley, B.L.; Levinger, N.E. Molecular probe location in reverse micelles determined by NMR dipolar interactions. J. Am. Chem. Soc. 2006, 128, 4437–4445. [Google Scholar] [CrossRef] [PubMed]
- Crans, D.C.; Schoeberl, S.; Gaidamauskas, E.; Baruah, B.; Roess, D.A. Antidiabetic vanadium compound and membrane interfaces: Interface-facilitated metal complex hydrolysis. J. Biol. Inorg. Chem. 2011, 16, 961–972. [Google Scholar] [CrossRef]
- Winter, P.W.; Al-Qatati, A.; Wolf-Ringwall, A.L.; Schoeberl, S.; Chatterjee, P.B.; Barisas, B.G.; Roess, D.A.; Crans, D.C. The anti-diabetic bis (maltolato) oxovanadium (IV) decreases lipid order while increasing insulin receptor localization in membrane microdomains. Dalton Trans. 2012, 41, 6419–6430. [Google Scholar] [CrossRef]
- Roess, D.A.; Smith, S.M.; Holder, A.A.; Baruah, B.; Trujillo, A.M.; Gilsdorf, D.; Stahla, M.L.; Crans, D.C. Do Vanadium Compounds Drive Reorganization of the Plasma Membrane and Activation of Insulin Receptors with Lipid Rafts? In Vanadium: The Versatile Metal; ACS Publications: Washington, DC, USA, 2007. [Google Scholar]
- Al-Qatati, A.; Fontes, F.L.; Barisas, B.G.; Zhang, D.; Roess, D.A.; Crans, D.C. Raft localization of Type I Fcε receptor and degranulation of RBL-2H3 cells exposed to decavanadate, a structural model for V2O5. Dalton Trans. 2013, 42, 11912–11920. [Google Scholar] [CrossRef]
- Chattopadhyay, A. GPCRs: Lipid-dependent membrane receptors that act as drug targets. Adv. Biol. 2014, 2014, 1–12. [Google Scholar] [CrossRef]
- Newton, C.L.; Anderson, R.C.; Kreuchwig, A.; Krause, G.; Katz, A.A.; Millar, R.P. Rescue of function of mutant luteinising hormone receptors with deficiencies in cell surface expression, hormone binding and hormone signalling. Neuroendocrinology 2020. [Google Scholar] [CrossRef]
- Lei, Y.; Hagen, G.M.; Smith, S.M.; Liu, J.; Barisas, G.; Roess, D.A. Constitutively-active human LH receptors are self-associated and located in rafts. Mol. Cell. Endocrinol. 2007, 260, 65–72. [Google Scholar] [CrossRef][Green Version]
- Edge, A.S.; Faltynek, C.R.; Hof, L.; Reichert, L.E., Jr.; Weber, P. Deglycosylation of glycoproteins by trifluoromethanesulfonic acid. Anal. Biochem. 1981, 118, 131–137. [Google Scholar] [CrossRef]
- Kalyan, N.K.; Bahl, O.P. Effect of deglycosylation on the subunit interactions and receptor binding activity of human chorionic gonadotropin. Biochem. Biophys. Res. Commun. 1981, 102, 1246–1253. [Google Scholar] [CrossRef]
- Keutmann, H.T.; Johnson, L.; Ryan, R.J. Evidence for a conformational change in deglycosylated glycoprotein hormones. FEBS Lett. 1985, 185, 333–338. [Google Scholar] [CrossRef]
- Sairam, M. Role of carbohydrates in glycoprotein hormone signal transduction. Faseb J. 1989, 3, 1915–1926. [Google Scholar] [CrossRef] [PubMed]
- Weinberg, Z.Y.; Puthenveedu, M.A. Regulation of G protein-coupled receptor signaling by plasma membrane organization and endocytosis. Traffic 2019, 20, 121–129. [Google Scholar] [CrossRef]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Althumairy, D.; Zhang, X.; Baez, N.; Barisas, G.; Roess, D.A.; Bousfield, G.R.; Crans, D.C. Glycoprotein G-protein Coupled Receptors in Disease: Luteinizing Hormone Receptors and Follicle Stimulating Hormone Receptors. Diseases 2020, 8, 35. https://doi.org/10.3390/diseases8030035
Althumairy D, Zhang X, Baez N, Barisas G, Roess DA, Bousfield GR, Crans DC. Glycoprotein G-protein Coupled Receptors in Disease: Luteinizing Hormone Receptors and Follicle Stimulating Hormone Receptors. Diseases. 2020; 8(3):35. https://doi.org/10.3390/diseases8030035
Chicago/Turabian StyleAlthumairy, Duaa, Xiaoping Zhang, Nicholas Baez, George Barisas, Deborah A. Roess, George R. Bousfield, and Debbie C. Crans. 2020. "Glycoprotein G-protein Coupled Receptors in Disease: Luteinizing Hormone Receptors and Follicle Stimulating Hormone Receptors" Diseases 8, no. 3: 35. https://doi.org/10.3390/diseases8030035
APA StyleAlthumairy, D., Zhang, X., Baez, N., Barisas, G., Roess, D. A., Bousfield, G. R., & Crans, D. C. (2020). Glycoprotein G-protein Coupled Receptors in Disease: Luteinizing Hormone Receptors and Follicle Stimulating Hormone Receptors. Diseases, 8(3), 35. https://doi.org/10.3390/diseases8030035