The Presence of Gut Microbial Genes Encoding Bacterial Genotoxins or Pro-Inflammatory Factors in Stool Samples from Individuals with Colorectal Neoplasia


Abstract
1. Introduction
2. Materials and Methods
2.1. Stool Sample Collection
2.2. Bacterial DNA Extraction
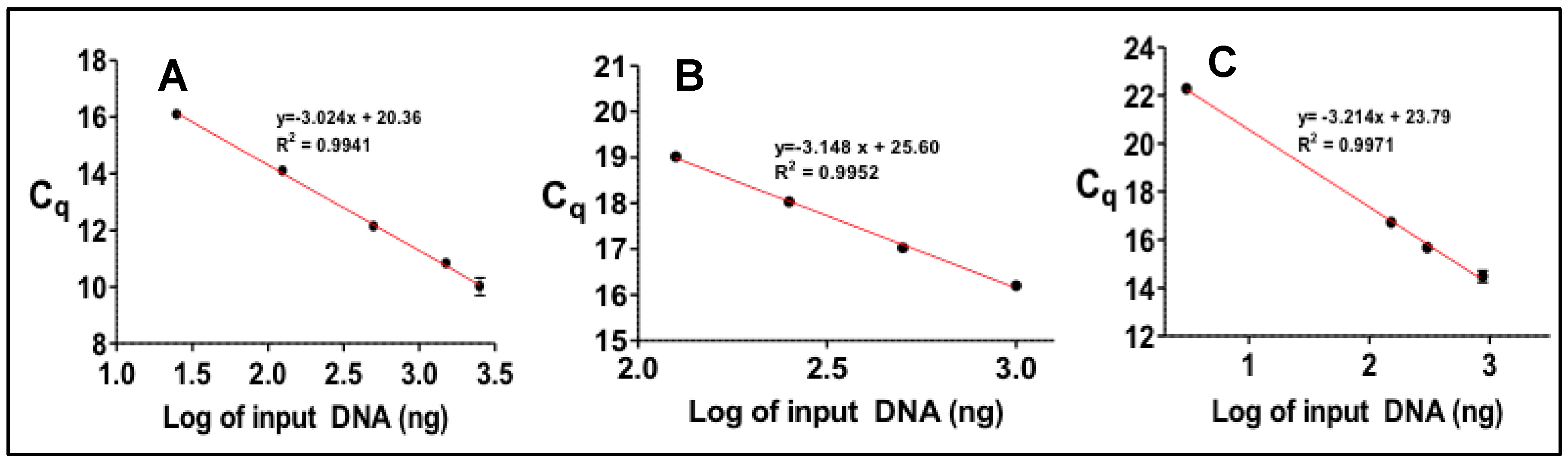
2.3. PCR Profiling of Specific Bacterial Toxin Genes
2.4. Statistical Analysis
3. Results
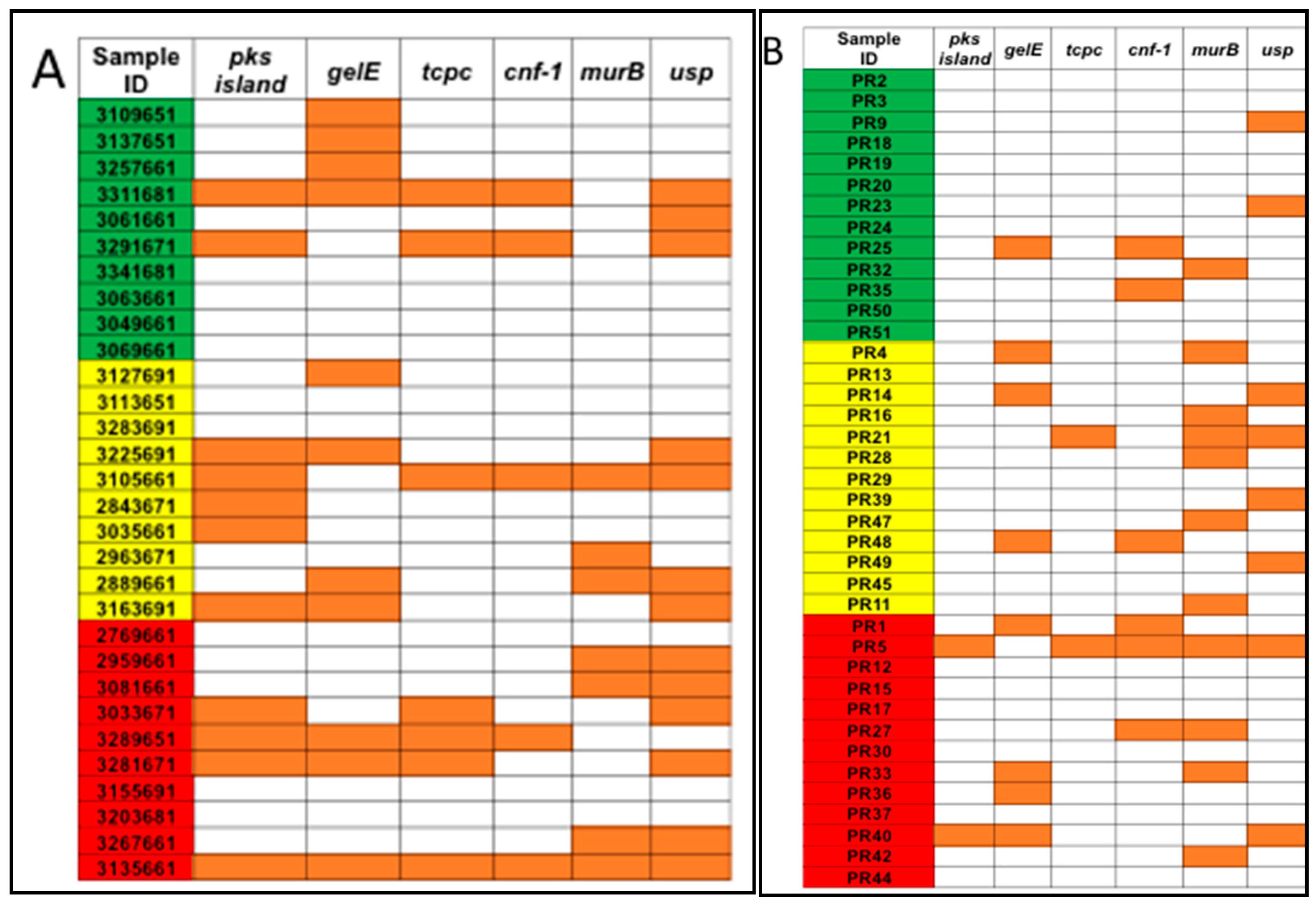
3.1. Detection of Genotoxic or Pro-Inflammatory Bacterial Genes by PCR
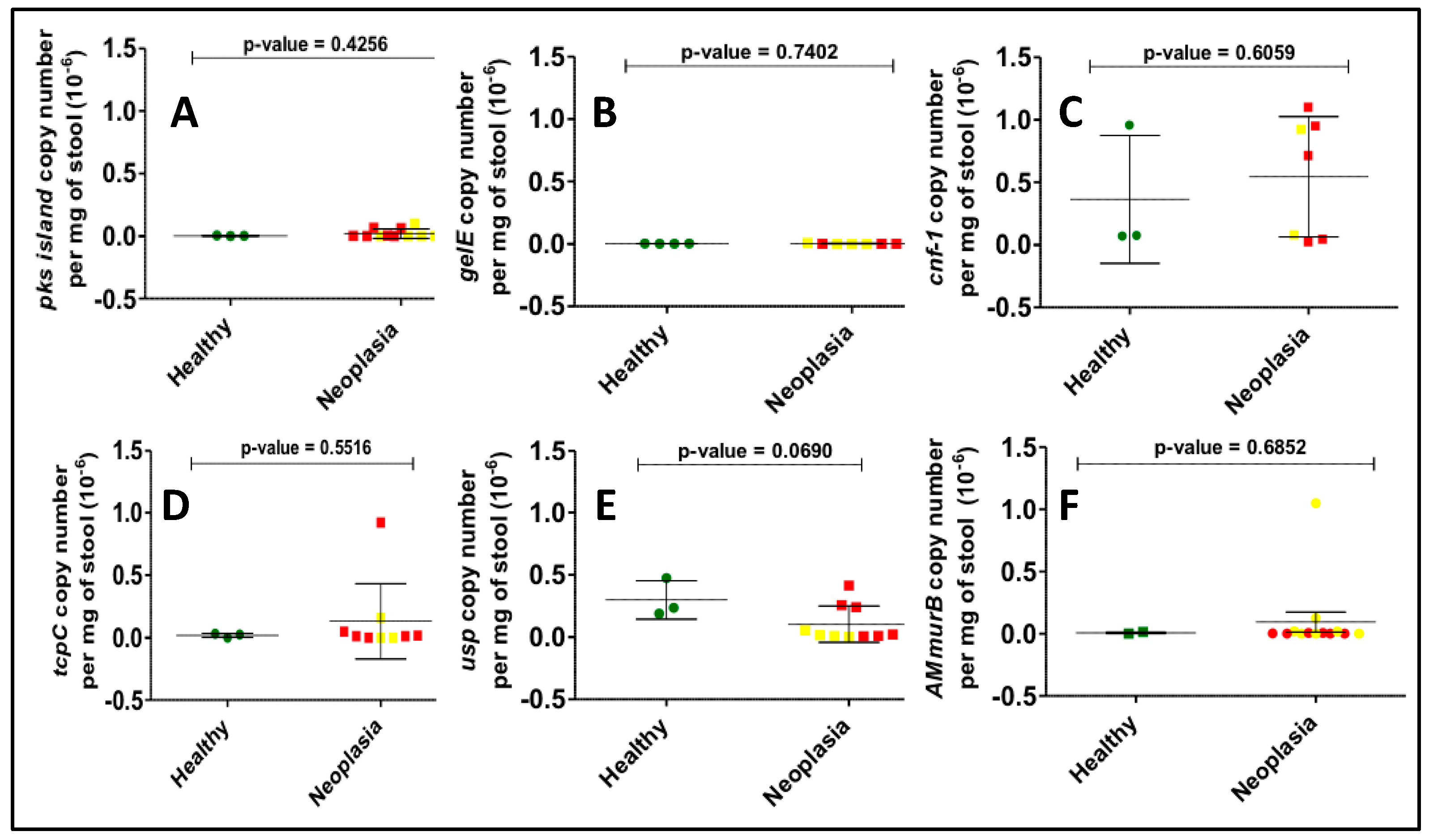
3.2. Gene Copy Number Measurements
4. Discussion
Author Contributions
Funding
Acknowledgments
Consent of Participants
Conflicts of Interest
Appendix A


References
- Smith, B.D.; Smith, G.L.; Hurria, A.; Hortobagyi, G.N.; Buchholz, T.A. Future of Cancer Incidence in the United States: Burdens Upon an Aging, Changing Nation. JCO 2009, 27, 2758–2765. [Google Scholar] [CrossRef] [PubMed]
- Colorectal Cancer Facts & Figures 2017–2019; American Cancer Society: Atlanta, GA, USA, 2017.
- Tjalsma, H.; Boleij, A.; Marchesi, J.R.; Dutilh, B.E. A bacterial driver-passenger model for colorectal cancer: Beyond the usual suspects. Nat. Rev. Microbiol. 2012, 10, 575–582. [Google Scholar] [CrossRef] [PubMed]
- Sears, C.L.; Garrett, W.S. Microbes, microbiota, and colon cancer. Cell Host Microbe 2014, 15, 317–328. [Google Scholar] [CrossRef] [PubMed]
- Konstantinov, S.R.; Kuipers, E.J.; Peppelenbosch, M.P. Functional genomic analyses of the gut microbiota for CRC screening. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 741–745. [Google Scholar] [CrossRef] [PubMed]
- Sobhani, I.; Tap, J.; Roudot-Thoraval, F.; Roperch, J.P.; Letulle, S.; Langella, P.; Corthier, G.; Van Nhieu, J.T.; Furet, J.P. Microbial Dysbiosis in Colorectal Cancer (CRC) Patients. PLoS ONE 2011, 6, e16393. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Cai, G.; Qiu, Y.; Fei, N.; Zhang, M.; Pang, X.; Jia, W.; Cai, S.; Zhao, L. Structural segregation of gut microbiota between colorectal cancer patients and healthy volunteers. ISME J. 2011, 6, 320–329. [Google Scholar] [CrossRef] [PubMed]
- Arthur, J.C.; Perez-Chanona, E.; Mühlbauer, M.; Tomkovich, S.; Uronis, J.M.; Fan, T.; Campbell, B.J.; Abujamel, T.; Dogan, B.; Rogers, A.B.; et al. Intestinal Inflammation Targets Cancer-Inducing Activity of the Microbiota. Science 2012, 338, 120–123. [Google Scholar] [CrossRef]
- Bezine, E.; Vignard, J.; Mirey, G. The Cytolethal Distending Toxin Effects on Mammalian Cells: A DNA Damage Perspective. Cells 2014, 3, 592–615. [Google Scholar] [CrossRef] [PubMed]
- Putze, J.; Hennequin, C.; Nougayrede, J.; Zhang, W.; Homburg, S.; Karch, H.; Bringer, M.; Fayolle, C.; Carniel, E.; Rabsch, W.; et al. Genetic Structure and Distribution of the Colibactin Genomic Island among Members of the Family Enterobacteriaceae. Infect. Immun. 2009, 77, 4696–4703. [Google Scholar] [CrossRef]
- Dejea, C.M.; Fathi, P.; Craig, J.M.; Boleij, A.; Taddese, R.; Geis, A.L.; Wu, X.; DeStefano Shields, C.E.; Hechenbleikner, E.M.; Huso, D.L.; et al. Patients with familial adenomatous polyposis harbor colonic biofilms containing tumorigenic bacteria. Science 2018, 359, 592. [Google Scholar] [CrossRef]
- Brennan, C.A.; Garrett, W.S. Gut Microbiota, Inflammation, and Colorectal Cancer. Annu. Rev. Microbiol. 2016, 70, 395–411. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Kundu, P.; Seow, S.W.; Texeira de Matos, C.; Aronsson, L.; Chin, K.C.; Kärre, K.; Pettersson, S.; Greicius, G. Gut microbiota accelerate tumor growth via c-jun and STAT3 phosphorylation in APC Min/+ mice. Carcinogenesis 2012, 33, 1231–1238. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Rhee, K.; Albesiano, E.; Rabizadeh, S.; Wu, X.; Yen, H.; Huso, D.L.; Brancati, F.L.; Wick, E.; McAllister, F.; et al. A human colonic commensal promotes colon tumorigenesis via activation of T helper type 17 T cell responses. Nat. Med. 2009, 15, 1016–1022. [Google Scholar] [CrossRef] [PubMed]
- Arthur, J.C.; Gharaibeh, R.Z.; Mühlbauer, M.; Perez-Chanona, E.; Uronis, J.M.; McCafferty, J.; Fodor, A.A.; Jobin, C. Microbial genomic analysis reveals the essential role of inflammation in bacteria-induced colorectal cancer. Nat. Commun. 2014, 5, 4724. [Google Scholar] [CrossRef] [PubMed]
- Schwabe, R.F.; Jobin, C. The microbiome and cancer. Nat. Rev. Cancer 2013, 13, 800–812. [Google Scholar] [CrossRef] [PubMed]
- Grivennikov, S.I.; Wang, K.; Mucida, D.; Stewart, C.A.; Schnabl, B.; Jauch, D.; Taniguchi, K.; Yu, G.; Osterreicher, C.H.; Hung, K.E.; et al. Adenoma-linked barrier defects and microbial products drive IL-23/IL-17-mediated tumour growth. Nature 2012, 491, 254–258. [Google Scholar] [CrossRef] [PubMed]
- Vannucci, L.; Stepankova, R.; Kozakova, H.; Fiserova, A.; Rossmann, P.; Tlaskalova-Hogenova, H. Colorectal carcinogenesis in germ-free and conventionally reared rats: Different intestinal environments affect the systemic immunity. Int. J. Oncol. 2008, 32, 609–617. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.Y.; Shaw, M.H.; Redondo, G.; Núñez, G. The Innate Immune Receptor Nod1 Protects the Intestine from Inflammation-Induced Tumorigenesis. Cancer Res. 2008, 68, 10060. [Google Scholar] [CrossRef]
- Dove, W.F.; Clipson, L.; Gould, K.A.; Luongo, C.; Marshall, D.J.; Moser, A.R.; Newton, M.A.; Jacoby, R.F. Intestinal Neoplasia in the ApcMin Mouse: Independence from the Microbial and Natural Killer (beige Locus) Status. Cancer Res. 1997, 57, 812. [Google Scholar] [PubMed]
- Kostic, A.D.; Gevers, D.; Pedamallu, C.S.; Michaud, M.; Duke, F.; Earl, A.M.; Ojesina, A.I.; Jung, J.; Bass, A.J.; Tabernero, J.; et al. Genomic analysis identifies association of Fusobacterium with colorectal carcinoma. Genome Res. 2011, 22, 292–298. [Google Scholar] [CrossRef]
- Kostic, A.D.; Chun, E.; Robertson, L.; Glickman, J.N.; Gallini, C.A.; Michaud, M.; Clancy, T.E.; Chung, D.C.; Lochhead, P.; Hold, G.L.; et al. Fusobacterium nucleatum potentiates intestinal tumorigenesis and modulates the tumor immune microenvironment. Cell Host Microbe 2013, 14, 207–215. [Google Scholar] [CrossRef] [PubMed]
- Buc, E.; Dubois, D.; Sauvanet, P.; Raisch, J.; Delmas, J.; Darfeuille-Michaud, A.; Pezet, D.; Bonnet, R. High Prevalence of Mucosa-Associated E. coli Producing Cyclomodulin and Genotoxin in Colon Cancer. PLoS ONE 2013, 8, e56964. [Google Scholar] [CrossRef] [PubMed]
- Prorok-Hamon, M.; Friswell, M.K.; Alswied, A.; Roberts, C.L.; Song, F.; Flanagan, P.K.; Knight, P.; Codling, C.; Marchesi, J.R.; Winstanley, C.; et al. Colonic mucosa-associated diffusely adherent afaC+ Escherichia coli expressing lpfA and pks are increased in inflammatory bowel disease and colon cancer. Gut 2013, 63, 761–770. [Google Scholar] [CrossRef] [PubMed]
- Martin, H.M.; Campbell, B.J.; Hart, C.A.; Mpofu, C.; Nayar, M.; Singh, R.; Englyst, H.; Williams, H.F.; Rhodes, J.M. Enhanced Escherichia coli adherence and invasion in Crohn’s disease and colon cancer 1. Gastroenterology 2004, 127, 80–93. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Moreno, R.; Robledo, I.E.; Baerga-Ortiz, A. Direct Detection and Quantification of Bacterial Genes Associated with Inflammation in DNA Isolated from Stool. Adv. Microbiol. 2014, 4, 1065–1075. [Google Scholar] [CrossRef]
- Nougayrede, J.P.; Homburg, S.; Taieb, F.; Boury, M.; Brzuszkiewicz, E.; Gottschalk, G.; Buchrieser, C.; Hacker, J.; Dobrindt, U.; Oswald, E. Escherichia coli induces DNA double-strand breaks in eukaryotic cells. Science 2006, 313, 848–851. [Google Scholar] [CrossRef] [PubMed]
- Cuevas-Ramos, G.; Petit, C.R.; Marcq, I.; Boury, M.; Oswald, E.; Nougayrède, J. Escherichia coli induces DNA damage in vivo and triggers genomic instability in mammalian cells. Proc. Natl. Acad. Sci. USA 2010, 107, 11537–11542. [Google Scholar] [CrossRef]
- Snyder, G.A.; Cirl, C.; Jiang, J.; Chen, K.; Waldhuber, A.; Smith, P.; Römmler, F.; Snyder, N.; Fresquez, T.; Dürr, S.; et al. Molecular mechanisms for the subversion of MyD88 signaling by TcpC from virulent uropathogenic Escherichia coli. Proc. Natl. Acad. Sci. USA 2013, 110, 6985–6990. [Google Scholar] [CrossRef]
- Sifri, C.D.; Mylonakis, E.; Singh, K.V.; Qin, X.; Garsin, D.A.; Murray, B.E.; Ausubel, F.M.; Calderwood, S.B. Virulence Effect of Enterococcus faecalis Protease Genes and the Quorum-Sensing Locus fsr in Caenorhabditis elegans and Mice. Infect. Immun. 2002, 70, 5647–5650. [Google Scholar] [CrossRef]
- Nougayrède, J.; Taieb, F.; Rycke, J.D.; Oswald, E. Cyclomodulins: Bacterial effectors that modulate the eukaryotic cell cycle. Trends Microbiol. 2005, 13, 103–110. [Google Scholar] [CrossRef]
- Nipic, D.; Podlesek, Z.; Budic, M.; Crnigoj, M.; Zgur-Bertok, D. Escherichia coli uropathogenic-specific protein, Usp, is a bacteriocin-like genotoxin. J. Infect. Dis. 2013, 208, 1545–1552. [Google Scholar] [CrossRef] [PubMed]
- Weir, T.L.; Manter, D.K.; Sheflin, A.M.; Barnett, B.A.; Heuberger, A.L.; Ryan, E.P. Stool Microbiome and Metabolome Differences between Colorectal Cancer Patients and Healthy Adults. PLoS ONE 2013, 8, e70803. [Google Scholar] [CrossRef] [PubMed]
- Robledo, I.E.; Aquino, E.E.; Vazquez, G.J. Detection of the KPC gene in Escherichia coli, Klebsiella pneumoniae, Pseudomonas aeruginosa, and Acinetobacter baumannii during a PCR-based nosocomial surveillance study in Puerto Rico. Antimicrob. Agents Chemother. 2011, 55, 2968–2970. [Google Scholar] [CrossRef] [PubMed]
- Ferlay, J.; Soerjomataram, I.; Ervik, M.; Dikshit, R.; Eser, S.; Mathers, C.; Rebelo, M.; Parkin, D.M.; Forman, D.; Bray, F. GLOBOCAN 2012 v1.0, Cancer Incidence and Mortality Worldwide: IARC CancerBase No. 11 [Internet]; International Agency for Research on Cancer: Lyon, France, 2013. [Google Scholar]
- Guinane, C.M.; Cotter, P.D. Role of the gut microbiota in health and chronic gastrointestinal disease: Understanding a hidden metabolic organ. Ther. Adv. Gastroenterol. 2013, 6, 295–308. [Google Scholar] [CrossRef] [PubMed]
- Plottel, C.S.; Blaser, M.J. Microbiome and Malignancy. Cell Host Microbe 2011, 10, 324–335. [Google Scholar] [CrossRef] [PubMed]
- Stary, L.; Mezerova, K.; Skalicky, P.; Zboril, P.; Raclavsky, V. Are we any closer to screening for colorectal cancer using microbial markers? A critical review. Biomed. Pap. Med. Fac. Univ. Palacky Olomouc Czech Repub. 2017, 161, 333–338. [Google Scholar] [CrossRef] [PubMed]
- Walker, A.W.; Sanderson, J.D.; Churcher, C.; Parkes, G.C.; Hudspith, B.N.; Rayment, N.; Brostoff, J.; Parkhill, J.; Dougan, G.; Petrovska, L. High-throughput clone library analysis of the mucosa-associated microbiota reveals dysbiosis and differences between inflamed and non-inflamed regions of the intestine in inflammatory bowel disease. BMC Microbiol. 2011, 11, 7. [Google Scholar] [CrossRef]
- Wu, G.D.; Chen, J.; Hoffmann, C.; Bittinger, K.; Chen, Y.; Keilbaugh, S.A.; Bewtra, M.; Knights, D.; Walters, W.A.; Knight, R.; et al. Linking Long-Term Dietary Patterns with Gut Microbial Enterotypes. Science 2011, 334, 105–108. [Google Scholar] [CrossRef]
- Duncan, S.H.; Louis, P.; Flint, H.J. Cultivable bacterial diversity from the human colon. Lett. Appl. Microbiol. 2007, 44, 343–350. [Google Scholar] [CrossRef]
- Tucker, K.L.; Bianchi, L.A.; Maras, J.; Bermudez, O.I. Adaptation of a food frequency questionnaire to assess diets of Puerto Rican and non-Hispanic adults. Am. J. Epidemiol. 1998, 148, 507–518. [Google Scholar] [CrossRef]
- Goodrich, J.; Waters, J.; Poole, A.; Sutter, J.; Koren, O.; Blekhman, R.; Beaumont, M.; Van Treuren, W.; Knight, R.; Bell, J.; et al. Human Genetics Shape the Gut Microbiome. Cell 2014, 159, 789–799. [Google Scholar] [CrossRef] [PubMed]
- Blekhman, R.; Goodrich, J.K.; Huang, K.; Sun, Q.; Bukowski, R.; Bell, J.T.; Spector, T.D.; Keinan, A.; Ley, R.E.; Gevers, D.; et al. Host genetic variation impacts microbiome composition across human body sites. Genome Biol. 2015, 16, 191. [Google Scholar] [CrossRef] [PubMed]
- Spor, A.; Koren, O.; Ley, R. Unravelling the effects of the environment and host genotype on the gut microbiome. Nat. Rev. Microbiol. 2011, 9, 279. [Google Scholar] [CrossRef] [PubMed]
- Via, M.; Gignoux, C.R.; Roth, L.A.; Fejerman, L.; Galanter, J.; Choudhry, S.; Toro-Labrador, G.; Viera-Vera, J.; Oleksyk, T.K.; Beckman, K.; et al. History Shaped the Geographic Distribution of Genomic Admixture on the Island of Puerto Rico. PLoS ONE 2011, 6, e16513. [Google Scholar] [CrossRef] [PubMed]
- Dingemanse, C.; Belzer, C.; van Hijum, S.A.F.T.; Günthel, M.; Salvatori, D.; Dunnen, J.T.D.; Kuijper, E.J.; Devilee, P.; de Vos, W.M.; van Ommen, G.B.; et al. Akkermansia muciniphila and Helicobacter typhlonius modulate intestinal tumor development in mice. Carcinogenesis 2015, 36, 1388–1396. [Google Scholar] [CrossRef] [PubMed]
- Byrd, J.C.; Bresalier, R.S. Mucins and mucin binding proteins in colorectal cancer. Cancer Metastasis Rev. 2004, 23, 77–99. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Name | Pathogenic Mechanism |
|---|---|
| pks island (pks) | Encodes colibactin, a genotoxin that induces double-strand DNA breaks and genome instability [27,28] |
| TIR domain-containing protein (tcpC) | Toxin modulates host immune response [29] |
| gelatinase-E (gelE) | Pro-inflammatory toxin [30] |
| cytotoxic necrotizing factor (CNF) | Cyclomodulin that promotes proliferation [31] |
| uropathogenic specific protein (USP) | Genotoxin that induces DNA damage [32] |
| UDP-N-acetylenolpyruvylglucosamine reductase (murB) | Nonpathogenic; surrogate marker for Akkermansia muciniphila, a mucolytic bacterium associated with CRC [33] |
| Bacterial Gene | Primer Sequence | Annealing Temp (°C) | Size (bp) | Reference |
|---|---|---|---|---|
| pks island | F: GTTTTGCTCGCCAGATAGTCATTC | 63 | 733 | Ref. [26] |
| R: CAGTTCGGGTATGTGTGGAAGG | ||||
| tcpC | F: TCGGCGATAGCTTAAGGAGA | 56 | 216 | Ref. [26] |
| R: CCGCCAAATAATGGCTGTAT | ||||
| gelE | F: TATGACAATGCTTTTTGGGAT | 49 | 213 | Ref. [26] |
| R: AGATGCACCCGAAATAATATA | ||||
| cnf-1 | F: AGCGTGCAATCTATCCGTATTT | 56 | 173 | Ref. [26] |
| R: TGGAATTTCCCCAGTATAGGTG | ||||
| usp | F: GGTGTTCATACGGGTGAAGG | 63 | 618 | This study |
| R: CTCAGGGACATAGGGGGAAT | ||||
| AMmurB | F: GAAATCCGCAGCCATACAAG | 57.3 | 135 | This study |
| R: CTCCAGAAGACGCTCCATTT |
| Bacterial Gene | Primer Sequence | Annealing Temp (°C) | Size (bp) | Reference |
|---|---|---|---|---|
| pks island | F: TCGATATAGTCACGCCACCA | 63 | 137 | This study |
| R: GTCAAGCGAGCATACGAACA | ||||
| tcpC | F: AGATGGGAGTGGAAGGAGGT | 61 | 144 | This study |
| R: TGCTTGTAATTTTGCCCAGTC | ||||
| gelE | F: GGTACAGGCATCTTTGTTGGA | 61 | 131 | This study |
| R: GCCTCAGAAATTGCCTCCTT | ||||
| cnf-1 | F: AGCGTGCAATCTATCCGTATTT | 56 | 173 | Ref. [26] |
| R: TGGAATTTCCCCAGTATAGGTG | ||||
| usp | F: GGTGTTCATACGGGTGAAGG | 63 | 618 | This study |
| R: CTCAGGGACATAGGGGGAAT | ||||
| AMmurB | F: GAAATCCGCAGCCATACAAG | 57.3 | 135 | This study |
| R: CTCCAGAAGACGCTCCATTT |
| Model 1 | Model 2 | |||||
|---|---|---|---|---|---|---|
| Bacterial Gene | Adenoma | CRC | Neoplasia | |||
| OR (% CI) | p-Value | OR (% CI) | p-Value | OR (% CI) | p-Value | |
| pks | 2.7 (0.4–29.1) | 0.17 | 2.7 (0.4–19.7) | 0.34 | 3.3 (0.6–19.4) | 0.19 |
| tcpC | 0.5 (0.03–5.9) | 0.53 | 2.7 (0.4–19.7) | 0.33 | 1.3 (0.21–8.5) | 0.76 |
| cnf | 0.5 (0.03–5.9) | 0.53 | 1 (0.1–8.9) | 0.99 | 0.7 (0.1–5.1) | 0.73 |
| usp | 1.6 (0.2–9.9) | 0.64 | 3.5 (0.5–22.3) | 0.18 | 2.3 (0.5–11.7) | 0.30 |
| gelE | 1 (0.2–6.0) | 0.99 | 0.64 (0.1–4.1) | 0.64 | 0.8 (0.2–3.9) | 0.79 |
| AMmurB * | 9.8 (0.4–219.2) | 0.15 | 14.5 (0.7–316.7) | 0.09 | 11.7 (0.6–228.4) | 0.11 |
| ≥2 genes | 3.2 (0.4–28.8) | 0.29 | 3.7 (0.5–28.5) | 0.19 | 4.4 (0.6–32.5) | 0.15 |
| Model 1 | Model 2 | |||||
|---|---|---|---|---|---|---|
| Bacterial Gene | Adenoma | CRC | Neoplasia | |||
| OR (% CI) | p-Value | OR (% CI) | p-Value | OR (% CI) | p-Value | |
| Pks * | 1.6 (0.02–87.8) | 0.82 | 10.0 (0.5–215.8) | 0.82 | 5.4 (0.3–113.7) | 0.28 |
| tcpC * | 10.4 (0.4–249.0) | 0.15 | 3.5 (0.1–95.1) | 0.46 | 5.4 (0.3–113.7) | 0.28 |
| cnf * | 0.3 (0.01–6.4) | 0.41 | 1.7 (0.3–10.6) | 0.57 | 1.0 (0.1–13.4) | 0.99 |
| usp | 3.3 (0.4–26.4) | 0.26 | 1.1 (0.1–9.3) | 0.93 | 1.8 (0.2–22.3) | 0.84 |
| gelE | 4.0 (0.3–53.5) | 0.30 | 8.6 (0.8–89.0) | 0.07 | 6.2 (0.6–315.0) | 0.16 |
| AMmurB | 5.5 (0.7–42.6) | 0.10 | 2.8 (0.4–18.9) | 0.30 | 3.5 (0.5–41.3) | 0.26 |
| ≥2 genes | 24.0 (1.1–518.6) | 0.04 | 10.0 (0.9–117.0) | 0.07 | 11.3 (1.0–637.1) | 0.05 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gómez-Moreno, R.; González-Pons, M.; Soto-Salgado, M.; Cruz-Correa, M.; Baerga-Ortiz, A. The Presence of Gut Microbial Genes Encoding Bacterial Genotoxins or Pro-Inflammatory Factors in Stool Samples from Individuals with Colorectal Neoplasia. Diseases 2019, 7, 16. https://doi.org/10.3390/diseases7010016
Gómez-Moreno R, González-Pons M, Soto-Salgado M, Cruz-Correa M, Baerga-Ortiz A. The Presence of Gut Microbial Genes Encoding Bacterial Genotoxins or Pro-Inflammatory Factors in Stool Samples from Individuals with Colorectal Neoplasia. Diseases. 2019; 7(1):16. https://doi.org/10.3390/diseases7010016
Chicago/Turabian StyleGómez-Moreno, Ramón, María González-Pons, Marievelisse Soto-Salgado, Marcia Cruz-Correa, and Abel Baerga-Ortiz. 2019. "The Presence of Gut Microbial Genes Encoding Bacterial Genotoxins or Pro-Inflammatory Factors in Stool Samples from Individuals with Colorectal Neoplasia" Diseases 7, no. 1: 16. https://doi.org/10.3390/diseases7010016
APA StyleGómez-Moreno, R., González-Pons, M., Soto-Salgado, M., Cruz-Correa, M., & Baerga-Ortiz, A. (2019). The Presence of Gut Microbial Genes Encoding Bacterial Genotoxins or Pro-Inflammatory Factors in Stool Samples from Individuals with Colorectal Neoplasia. Diseases, 7(1), 16. https://doi.org/10.3390/diseases7010016