Autophagy Intertwines with Different Diseases—Recent Strategies for Therapeutic Approaches


Abstract
1. Introduction
2. Overview of Autophagy for Intracellular Homeostasis
3. Role of Autophagy in Diseases
3.1. In Neurodegenerative Diseases
Autophagy in Neurodegenerative Protection
3.2. In Lysosomal Storage Diseases (LSDs)
3.2.1. In Mucolipidosis Type IV (MLIV)
3.2.2. Nephropathic Cystinosis
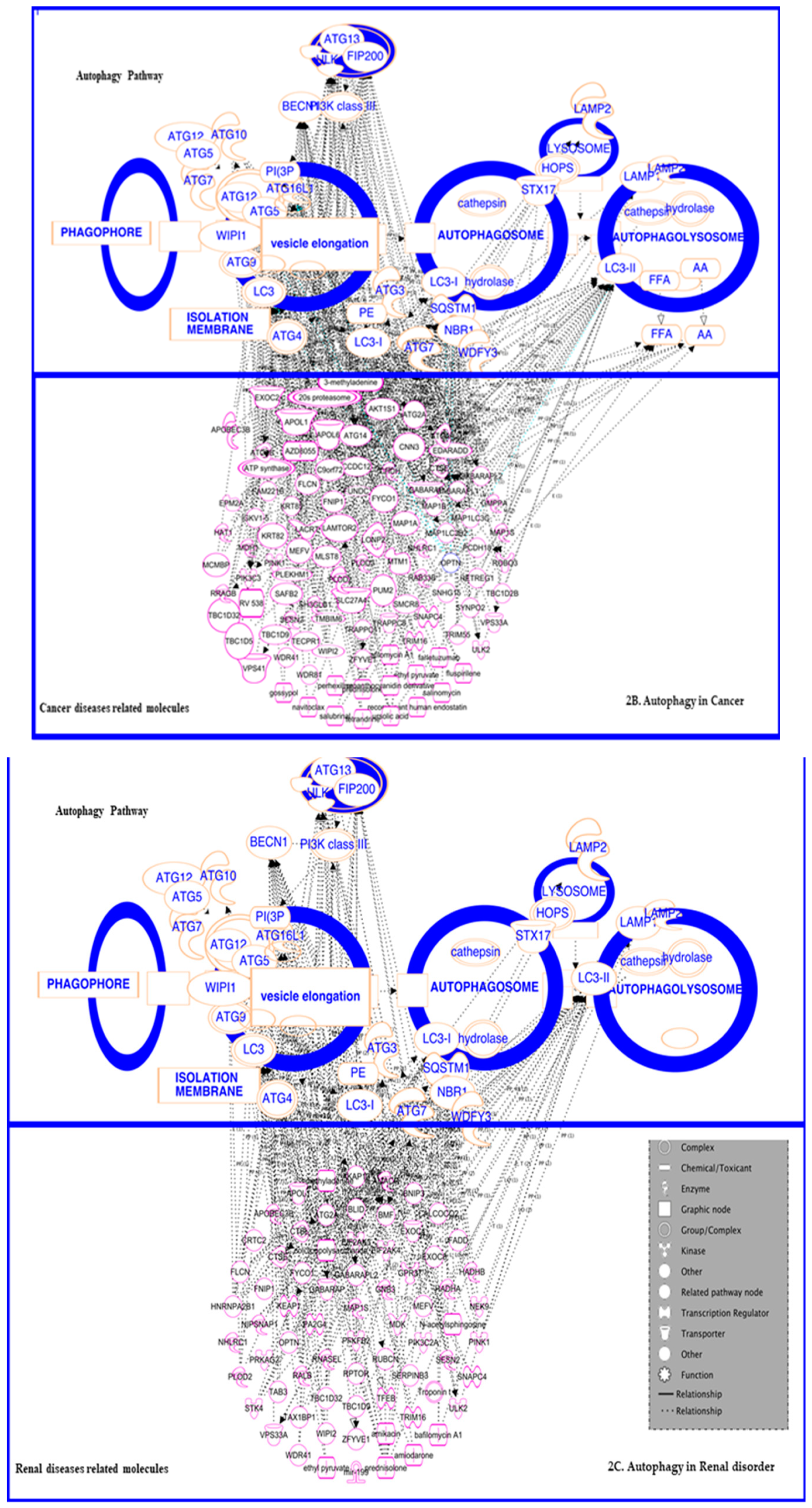
3.3. In Cancer: Biphasic Response
3.3.1. In Cancer Progression
3.3.2. In Suppression
3.4. In Renal Disorders
3.4.1. In Glomerulosclerosis
3.4.2. In Fibrosis
3.4.3. In Diabetic Nephropathy (DN)
3.4.4. In Transplantation/Renal Ischemic Injury (IR)
3.4.5. In Cystic Diseases
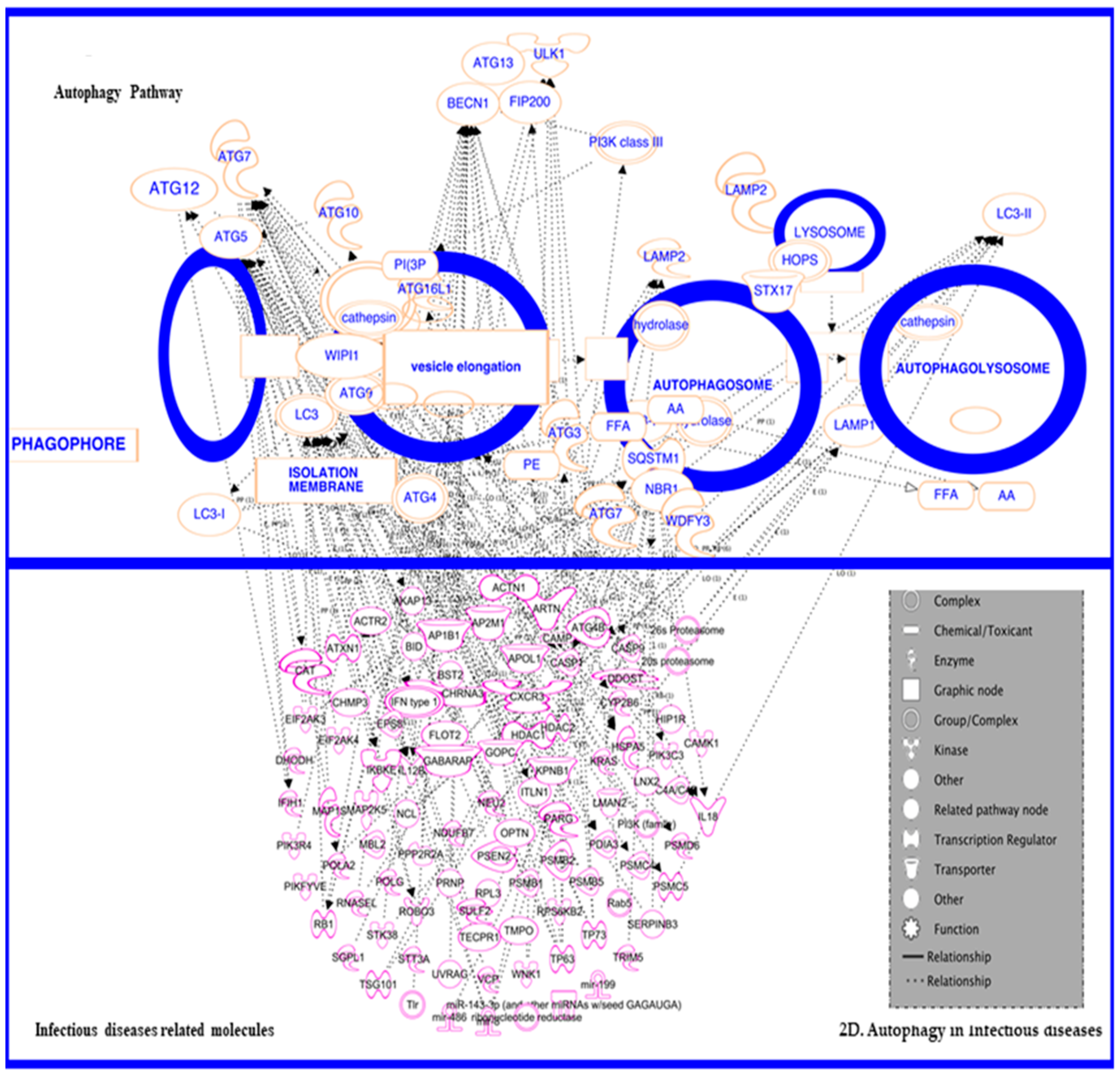
3.5. In Inflammatory and Infectious Disease
3.5.1. In Infectious Disease—HIV
3.5.2. Autophagy in HIV and HIV-Associated Neurological Disorders (HAND)
3.5.3. Endoplasmic Reticulum (ER) Stress-Mediated Autophagy
3.5.4. Autophagy and Drug Abuse-Mediated Cytotoxicity
4. Autophagy and Therapy
5. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Mizushima, N.; Yamamoto, A.; Matsui, M.; Yoshimori, T.; Ohsumi, Y. In vivo analysis of autophagy in response to nutrient starvation using transgenic mice expressing a fluorescent autophagosome marker. Mol. Biol. Cell 2004, 15, 1101–1111. [Google Scholar] [CrossRef] [PubMed]
- Eskelinen, E.L.; Saftig, P. Autophagy: A lysosomal degradation pathway with a central role in health and disease. Biochim. Biophys. Acta 2009, 1793, 664–673. [Google Scholar] [CrossRef] [PubMed]
- Eskelinen, E.L. New insights into the mechanisms of macroautophagy in mammalian cells. Int. Rev. Cell Mol. Biol. 2008, 266, 207–247. [Google Scholar] [PubMed]
- Kundu, M.; Thompson, C.B. Autophagy: Basic principles and relevance to disease. Annu. Rev. Pathol. 2008, 3, 427–455. [Google Scholar] [CrossRef] [PubMed]
- Bredesen, D.E.; Rao, R.V.; Mehlen, P. Cell death in the nervous system. Nature 2006, 443, 796–802. [Google Scholar] [CrossRef]
- De Duve, C.; Wattiaux, R. Functions of lysosomes. Annu. Rev. Physiol. 1966, 28, 435–492. [Google Scholar] [CrossRef]
- Feng, Y.; He, D.; Yao, Z.; Klionsky, D.J. The machinery of macroautophagy. Cell Res. 2014, 24, 24–41. [Google Scholar] [CrossRef]
- Klionsky, D.J. The molecular machinery of autophagy: Unanswered questions. J. Cell Sci. 2005, 118, 7–18. [Google Scholar] [CrossRef]
- Kroemer, G.; Levine, B. Autophagic cell death: The story of a misnomer. Nat. Rev. Mol. Cell Biol. 2008, 9, 1004–1010. [Google Scholar] [CrossRef]
- Wu, J.; Dang, Y.; Su, W.; Liu, C.; Ma, H.; Shan, Y.; Pei, Y.; Wan, B.; Guo, J.; Yu, L. Molecular cloning and characterization of rat lc3a and lc3b—Two novel markers of autophagosome. Biochem. Biophys. Res. Commun. 2006, 339, 437–442. [Google Scholar] [CrossRef]
- Cavallucci, V.; D’Amelio, M.; Cecconi, F. Abeta toxicity in alzheimer’s disease. Mol. Neurobiol. 2012, 45, 366–378. [Google Scholar] [CrossRef]
- Burchell, J.T.; Panegyres, P.K. Prion diseases: Immunotargets and therapy. Immunotargets Ther. 2016, 5, 57–68. [Google Scholar] [PubMed]
- Mullin, S.; Schapira, A. Alpha-synuclein and mitochondrial dysfunction in parkinson’s disease. Mol. Neurobiol. 2013, 47, 587–597. [Google Scholar] [CrossRef]
- Dehay, B.; Bourdenx, M.; Gorry, P.; Przedborski, S.; Vila, M.; Hunot, S.; Singleton, A.; Olanow, C.W.; Merchant, K.M.; Bezard, E.; et al. Targeting alpha-synuclein for treatment of parkinson’s disease: Mechanistic and therapeutic considerations. Lancet Neurol. 2015, 14, 855–866. [Google Scholar] [CrossRef]
- Sau, D.; De Biasi, S.; Vitellaro-Zuccarello, L.; Riso, P.; Guarnieri, S.; Porrini, M.; Simeoni, S.; Crippa, V.; Onesto, E.; Palazzolo, I.; et al. Mutation of sod1 in als: A gain of a loss of function. Hum. Mol. Genet. 2007, 16, 1604–1618. [Google Scholar] [CrossRef] [PubMed]
- Nagata, E.; Sawa, A.; Ross, C.A.; Snyder, S.H. Autophagosome-like vacuole formation in huntington’s disease lymphoblasts. Neuroreport 2004, 15, 1325–1328. [Google Scholar] [CrossRef]
- Komatsu, M.; Wang, Q.J.; Holstein, G.R.; Friedrich, V.L., Jr.; Iwata, J.; Kominami, E.; Chait, B.T.; Tanaka, K.; Yue, Z. Essential role for autophagy protein atg7 in the maintenance of axonal homeostasis and the prevention of axonal degeneration. Proc. Natl. Acad. Sci. USA 2007, 104, 14489–14494. [Google Scholar] [CrossRef] [PubMed]
- Jentsch, S. Ubiquitin-dependent protein degradation: A cellular perspective. Trends Cell Biol. 1992, 2, 98–103. [Google Scholar] [CrossRef]
- Chung, K.M.; Hernandez, N.; Sproul, A.A.; Yu, W.H. Alzheimer’s disease and the autophagic-lysosomal system. Neurosci. Lett. 2018. [Google Scholar] [CrossRef]
- Mishra, A.K.; ur Rasheed, M.S.; Shukla, S.; Tripathi, M.K.; Dixit, A.; Singh, M.P. Aberrant autophagy and parkinsonism: Does correction rescue from disease progression? Mol. Neurobiol. 2015, 51, 893–908. [Google Scholar] [CrossRef]
- Thomson, S.B.; Leavitt, B.R. Transcriptional regulation of the huntingtin gene. J. Huntingtons Dis. 2018. [Google Scholar] [CrossRef] [PubMed]
- Musi, N.; Valentine, J.M.; Sickora, K.R.; Baeuerle, E.; Thompson, C.S.; Shen, Q.; Orr, M.E. Tau protein aggregation is associated with cellular senescence in the brain. Aging Cell 2018, 17, e12840. [Google Scholar] [CrossRef] [PubMed]
- Batlevi, Y.; Martin, D.N.; Pandey, U.B.; Simon, C.R.; Powers, C.M.; Taylor, J.P.; Baehrecke, E.H. Dynein light chain 1 is required for autophagy, protein clearance, and cell death in drosophila. Proc. Natl. Acad. Sci. USA 2010, 107, 742–747. [Google Scholar] [CrossRef] [PubMed]
- Bjorkoy, G.; Lamark, T.; Brech, A.; Outzen, H.; Perander, M.; Overvatn, A.; Stenmark, H.; Johansen, T. P62/sqstm1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J. Cell Biol. 2005, 171, 603–614. [Google Scholar] [CrossRef] [PubMed]
- Rubinsztein, D.C.; Gestwicki, J.E.; Murphy, L.O.; Klionsky, D.J. Potential therapeutic applications of autophagy. Nat. Rev. Drug Discov. 2007, 6, 304–312. [Google Scholar] [CrossRef] [PubMed]
- Rubinsztein, D.C. The roles of intracellular protein-degradation pathways in neurodegeneration. Nature 2006, 443, 780–786. [Google Scholar] [CrossRef] [PubMed]
- Penna, F.; Costamagna, D.; Pin, F.; Camperi, A.; Fanzani, A.; Chiarpotto, E.M.; Cavallini, G.; Bonelli, G.; Baccino, F.M.; Costelli, P. Autophagic degradation contributes to muscle wasting in cancer cachexia. Am. J. Pathol. 2013, 182, 1367–1378. [Google Scholar] [CrossRef]
- Dobrowolny, G.; Aucello, M.; Rizzuto, E.; Beccafico, S.; Mammucari, C.; Boncompagni, S.; Belia, S.; Wannenes, F.; Nicoletti, C.; Del Prete, Z.; et al. Skeletal muscle is a primary target of sod1g93a-mediated toxicity. Cell Metab. 2008, 8, 425–436. [Google Scholar] [CrossRef]
- Masiero, E.; Agatea, L.; Mammucari, C.; Blaauw, B.; Loro, E.; Komatsu, M.; Metzger, D.; Reggiani, C.; Schiaffino, S.; Sandri, M. Autophagy is required to maintain muscle mass. Cell Metab. 2009, 10, 507–515. [Google Scholar] [CrossRef]
- Pigna, E.; Sanna, K.; Coletti, D.; Li, Z.; Parlakian, A.; Adamo, S.; Moresi, V. Increasing autophagy does not affect neurogenic muscle atrophy. Eur. J. Transl. Myol. 2018, 28, 7687. [Google Scholar] [CrossRef]
- Jia, K.; Hart, A.C.; Levine, B. Autophagy genes protect against disease caused by polyglutamine expansion proteins in caenorhabditis elegans. Autophagy 2007, 3, 21–25. [Google Scholar] [CrossRef] [PubMed]
- Schnoder, L.; Hao, W.; Qin, Y.; Liu, S.; Tomic, I.; Liu, X.; Fassbender, K.; Liu, Y. Deficiency of neuronal p38alpha mapk attenuates amyloid pathology in alzheimer disease mouse and cell models through facilitating lysosomal degradation of bace1. J. Biol. Chem. 2016, 291, 2067–2079. [Google Scholar] [CrossRef] [PubMed]
- Yoshimura, K.; Shibata, M.; Koike, M.; Gotoh, K.; Fukaya, M.; Watanabe, M.; Uchiyama, Y. Effects of rna interference of atg4b on the limited proteolysis of lc3 in pc12 cells and expression of atg4b in various rat tissues. Autophagy 2006, 2, 200–208. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Vicente, M.; Cuervo, A.M. Autophagy and neurodegeneration: When the cleaning crew goes on strike. Lancet Neurol. 2007, 6, 352–361. [Google Scholar] [CrossRef]
- Williams, A.; Jahreiss, L.; Sarkar, S.; Saiki, S.; Menzies, F.M.; Ravikumar, B.; Rubinsztein, D.C. Aggregate-prone proteins are cleared from the cytosol by autophagy: Therapeutic implications. Curr. Top Dev. Biol. 2006, 76, 89–101. [Google Scholar] [PubMed]
- Wong, E.S.; Tan, J.M.; Soong, W.E.; Hussein, K.; Nukina, N.; Dawson, V.L.; Dawson, T.M.; Cuervo, A.M.; Lim, K.L. Autophagy-mediated clearance of aggresomes is not a universal phenomenon. Hum. Mol. Genet. 2008, 17, 2570–2582. [Google Scholar] [CrossRef] [PubMed]
- Levine, B.; Kroemer, G. Autophagy in the pathogenesis of disease. Cell 2008, 132, 27–42. [Google Scholar] [CrossRef] [PubMed]
- Hara, T.; Mizushima, N. [Regulators of mammalian cellular autophagy]. Tanpakushitsu Kakusan Koso 2006, 51, 1484–1489. [Google Scholar] [PubMed]
- Lee, J.A.; Beigneux, A.; Ahmad, S.T.; Young, S.G.; Gao, F.B. Escrt-iii dysfunction causes autophagosome accumulation and neurodegeneration. Curr. Biol. 2007, 17, 1561–1567. [Google Scholar] [CrossRef]
- Azarnia Tehran, D.; Kuijpers, M.; Haucke, V. Presynaptic endocytic factors in autophagy and neurodegeneration. Curr. Opin. Neurobiol. 2018, 48, 153–159. [Google Scholar] [CrossRef]
- Vijayan, V.; Verstreken, P. Autophagy in the presynaptic compartment in health and disease. J. Cell. Biol. 2017, 216, 1895–1906. [Google Scholar] [CrossRef]
- Ryter, S.W.; Bhatia, D.; Choi, M.E. Autophagy: A lysosome-dependent process with implications in cellular redox homeostasis and human disease. Antioxid. Redox Signal. 2019, 30, 138–159. [Google Scholar] [CrossRef] [PubMed]
- Elrick, M.J.; Yu, T.; Chung, C.; Lieberman, A.P. Impaired proteolysis underlies autophagic dysfunction in niemann-pick type c disease. Hum. Mol. Genet. 2012, 21, 4876–4887. [Google Scholar]
- Lieberman, A.P.; Puertollano, R.; Raben, N.; Slaugenhaupt, S.; Walkley, S.U.; Ballabio, A. Autophagy in lysosomal storage disorders. Autophagy 2012, 8, 719–730. [Google Scholar] [CrossRef] [PubMed]
- Elrick, M.J.; Lieberman, A.P. Autophagic dysfunction in a lysosomal storage disorder due to impaired proteolysis. Autophagy 2013, 9, 234–235. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, Y.; Guhde, G.; Suter, A.; Eskelinen, E.L.; Hartmann, D.; Lullmann-Rauch, R.; Janssen, P.M.; Blanz, J.; von Figura, K.; Saftig, P. Accumulation of autophagic vacuoles and cardiomyopathy in lamp-2-deficient mice. Nature 2000, 406, 902–906. [Google Scholar] [PubMed]
- Venugopal, B.; Mesires, N.T.; Kennedy, J.C.; Curcio-Morelli, C.; Laplante, J.M.; Dice, J.F.; Slaugenhaupt, S.A. Chaperone-mediated autophagy is defective in mucolipidosis type iv. J. Cell Physiol. 2009, 219, 344–353. [Google Scholar] [CrossRef] [PubMed]
- Rajakumar, T.; Munkacsi, A.B.; Sturley, S.L. Exacerbating and reversing lysosomal storage diseases: From yeast to humans. Microb. Cell 2017, 4, 278–293. [Google Scholar] [CrossRef] [PubMed]
- Medic, G.; van der Weijden, M.; Karabis, A.; Hemels, M. A systematic literature review of cysteamine bitartrate in the treatment of nephropathic cystinosis. Curr. Med. Res. Opin. 2017, 33, 2065–2076. [Google Scholar] [CrossRef]
- Zhang, J.; Johnson, J.L.; He, J.; Napolitano, G.; Ramadass, M.; Rocca, C.; Kiosses, W.B.; Bucci, C.; Xin, Q.; Gavathiotis, E.; et al. Cystinosin, the small gtpase rab11, and the rab7 effector rilp regulate intracellular trafficking of the chaperone-mediated autophagy receptor lamp2a. J. Biol. Chem. 2017, 292, 10328–10346. [Google Scholar] [CrossRef]
- Napolitano, G.; Johnson, J.L.; He, J.; Rocca, C.J.; Monfregola, J.; Pestonjamasp, K.; Cherqui, S.; Catz, S.D. Impairment of chaperone-mediated autophagy leads to selective lysosomal degradation defects in the lysosomal storage disease cystinosis. EMBO Mol. Med. 2015, 7, 158–174. [Google Scholar] [CrossRef] [PubMed]
- Sansanwal, P.; Yen, B.; Gahl, W.A.; Ma, Y.; Ying, L.; Wong, L.J.; Sarwal, M.M. Mitochondrial autophagy promotes cellular injury in nephropathic cystinosis. J. Am. Soc. Nephrol. 2010, 21, 272–283. [Google Scholar] [CrossRef] [PubMed]
- Festa, B.P.; Chen, Z.; Berquez, M.; Debaix, H.; Tokonami, N.; Prange, J.A.; Hoek, G.V.; Alessio, C.; Raimondi, A.; Nevo, N.; et al. Impaired autophagy bridges lysosomal storage disease and epithelial dysfunction in the kidney. Nat. Commun. 2018, 9, 161. [Google Scholar] [CrossRef] [PubMed]
- Sansanwal, P.; Sarwal, M.M. Abnormal mitochondrial autophagy in nephropathic cystinosis. Autophagy 2010, 6, 971–973. [Google Scholar] [CrossRef]
- White, E. Deconvoluting the context-dependent role for autophagy in cancer. Nat. Rev. Cancer 2012, 12, 401–410. [Google Scholar] [CrossRef] [PubMed]
- Bialik, S.; Kimchi, A. Autophagy and tumor suppression: Recent advances in understanding the link between autophagic cell death pathways and tumor development. Adv. Exp. Med. Biol. 2008, 615, 177–200. [Google Scholar]
- Liang, X.H.; Jackson, S.; Seaman, M.; Brown, K.; Kempkes, B.; Hibshoosh, H.; Levine, B. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature 1999, 402, 672–676. [Google Scholar] [CrossRef]
- Yue, Z.; Jin, S.; Yang, C.; Levine, A.J.; Heintz, N. Beclin 1, an autophagy gene essential for early embryonic development, is a haploinsufficient tumor suppressor. Proc. Natl. Acad. Sci. USA 2003, 100, 15077–15082. [Google Scholar] [CrossRef]
- Salminen, A.; Kaarniranta, K.; Kauppinen, A.; Ojala, J.; Haapasalo, A.; Soininen, H.; Hiltunen, M. Impaired autophagy and app processing in alzheimer’s disease: The potential role of beclin 1 interactome. Prog. Neurobiol. 2013, 106–107, 33–54. [Google Scholar] [CrossRef]
- Qu, X.; Yu, J.; Bhagat, G.; Furuya, N.; Hibshoosh, H.; Troxel, A.; Rosen, J.; Eskelinen, E.L.; Mizushima, N.; Ohsumi, Y.; et al. Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J. Clin. Investig. 2003, 112, 1809–1820. [Google Scholar] [CrossRef]
- Fimia, G.M.; Stoykova, A.; Romagnoli, A.; Giunta, L.; Di Bartolomeo, S.; Nardacci, R.; Corazzari, M.; Fuoco, C.; Ucar, A.; Schwartz, P.; et al. Ambra1 regulates autophagy and development of the nervous system. Nature 2007, 447, 1121–1125. [Google Scholar] [PubMed]
- Liang, C.; Feng, P.; Ku, B.; Dotan, I.; Canaani, D.; Oh, B.H.; Jung, J.U. Autophagic and tumour suppressor activity of a novel beclin1-binding protein uvrag. Nat. Cell Biol. 2006, 8, 688–699. [Google Scholar] [CrossRef] [PubMed]
- Gentile, M.; Ahnstrom, M.; Schon, F.; Wingren, S. Candidate tumour suppressor genes at 11q23-q24 in breast cancer: Evidence of alterations in pig8, a gene involved in p53-induced apoptosis. Oncogene 2001, 20, 7753–7760. [Google Scholar] [CrossRef] [PubMed]
- Tian, S.; Lin, J.; Jun Zhou, J.; Wang, X.; Li, Y.; Ren, X.; Yu, W.; Zhong, W.; Xiao, J.; Sheng, F.; et al. Beclin 1-independent autophagy induced by a bcl-xl/bcl-2 targeting compound, z18. Autophagy 2010, 6, 1032–1041. [Google Scholar] [CrossRef]
- Lee, S.J.; Kim, H.P.; Jin, Y.; Choi, A.M.; Ryter, S.W. Beclin 1 deficiency is associated with increased hypoxia-induced angiogenesis. Autophagy 2011, 7, 829–839. [Google Scholar] [CrossRef]
- Liu, H.; He, Z.; von Rutte, T.; Yousefi, S.; Hunger, R.E.; Simon, H.U. Down-regulation of autophagy-related protein 5 (atg5) contributes to the pathogenesis of early-stage cutaneous melanoma. Sci. Transl. Med. 2013, 5, 202ra123. [Google Scholar] [CrossRef]
- Cheng, S.; Wu, Y.; Lu, Q.; Yan, J.; Zhang, H.; Wang, X. Autophagy genes coordinate with the class ii pi/ptdins 3-kinase piki-1 to regulate apoptotic cell clearance in c. Elegans. Autophagy 2013, 9, 2022–2032. [Google Scholar] [CrossRef] [PubMed]
- Kisen, G.O.; Tessitore, L.; Costelli, P.; Gordon, P.B.; Schwarze, P.E.; Baccino, F.M.; Seglen, P.O. Reduced autophagic activity in primary rat hepatocellular carcinoma and ascites hepatoma cells. Carcinogenesis 1993, 14, 2501–2505. [Google Scholar] [CrossRef]
- Mathew, R.; Karp, C.M.; Beaudoin, B.; Vuong, N.; Chen, G.; Chen, H.Y.; Bray, K.; Reddy, A.; Bhanot, G.; Gelinas, C.; et al. Autophagy suppresses tumorigenesis through elimination of p62. Cell 2009, 137, 1062–1075. [Google Scholar] [CrossRef]
- Boya, P.; Gonzalez-Polo, R.A.; Casares, N.; Perfettini, J.L.; Dessen, P.; Larochette, N.; Metivier, D.; Meley, D.; Souquere, S.; Yoshimori, T.; et al. Inhibition of macroautophagy triggers apoptosis. Mol. Cell. Biol. 2005, 25, 1025–1040. [Google Scholar] [CrossRef]
- Guo, J.Y.; Teng, X.; Laddha, S.V.; Ma, S.; Van Nostrand, S.C.; Yang, Y.; Khor, S.; Chan, C.S.; Rabinowitz, J.D.; White, E. Autophagy provides metabolic substrates to maintain energy charge and nucleotide pools in ras-driven lung cancer cells. Genes Dev. 2016, 30, 1704–1717. [Google Scholar] [PubMed]
- Mancias, J.D.; Kimmelman, A.C. Targeting autophagy addiction in cancer. Oncotarget 2011, 2, 1302–1306. [Google Scholar] [CrossRef]
- Mancias, J.D.; Kimmelman, A.C. Mechanisms of selective autophagy in normal physiology and cancer. J. Mol. Biol. 2016, 428, 1659–1680. [Google Scholar] [CrossRef]
- Yang, S.; Wang, X.; Contino, G.; Liesa, M.; Sahin, E.; Ying, H.; Bause, A.; Li, Y.; Stommel, J.M.; Dell’antonio, G.; et al. Pancreatic cancers require autophagy for tumor growth. Genes Dev. 2011, 25, 717–729. [Google Scholar] [CrossRef] [PubMed]
- Lock, R.; Roy, S.; Kenific, C.M.; Su, J.S.; Salas, E.; Ronen, S.M.; Debnath, J. Autophagy facilitates glycolysis during ras-mediated oncogenic transformation. Mol. Biol. Cell 2011, 22, 165–178. [Google Scholar] [CrossRef] [PubMed]
- Gewirtz, D.A. Autophagy, senescence and tumor dormancy in cancer therapy. Autophagy 2009, 5, 1232–1234. [Google Scholar] [CrossRef]
- Gewirtz, D.A.; Hilliker, M.L.; Wilson, E.N. Promotion of autophagy as a mechanism for radiation sensitization of breast tumor cells. Radiother Oncol. 2009, 92, 323–328. [Google Scholar] [CrossRef]
- Young, J.E.; La Spada, A.R. Development of selective nutrient deprivation as a system to study autophagy induction and regulation in neurons. Autophagy 2009, 5, 555–557. [Google Scholar] [CrossRef] [PubMed]
- Narita, M.; Young, A.R.; Narita, M. Autophagy facilitates oncogene-induced senescence. Autophagy 2009, 5, 1046–1047. [Google Scholar] [CrossRef] [PubMed]
- Janku, F.; McConkey, D.J.; Hong, D.S.; Kurzrock, R. Autophagy as a target for anticancer therapy. Nat. Rev. Clin. Oncol. 2011, 8, 528–539. [Google Scholar] [CrossRef] [PubMed]
- Takamura, A.; Komatsu, M.; Hara, T.; Sakamoto, A.; Kishi, C.; Waguri, S.; Eishi, Y.; Hino, O.; Tanaka, K.; Mizushima, N. Autophagy-deficient mice develop multiple liver tumors. Genes Dev. 2011, 25, 795–800. [Google Scholar] [CrossRef] [PubMed]
- Marino, G.; Salvador-Montoliu, N.; Fueyo, A.; Knecht, E.; Mizushima, N.; Lopez-Otin, C. Tissue-specific autophagy alterations and increased tumorigenesis in mice deficient in atg4c/autophagin-3. J. Biol. Chem. 2007, 282, 18573–18583. [Google Scholar] [CrossRef] [PubMed]
- Jung, H.S.; Chung, K.W.; Won Kim, J.; Kim, J.; Komatsu, M.; Tanaka, K.; Nguyen, Y.H.; Kang, T.M.; Yoon, K.H.; Kim, J.W.; et al. Loss of autophagy diminishes pancreatic beta cell mass and function with resultant hyperglycemia. Cell Metab. 2008, 8, 318–324. [Google Scholar] [CrossRef] [PubMed]
- Nakai, A.; Yamaguchi, O.; Takeda, T.; Higuchi, Y.; Hikoso, S.; Taniike, M.; Omiya, S.; Mizote, I.; Matsumura, Y.; Asahi, M.; et al. The role of autophagy in cardiomyocytes in the basal state and in response to hemodynamic stress. Nat. Med. 2007, 13, 619–624. [Google Scholar] [CrossRef] [PubMed]
- Nishino, I.; Fu, J.; Tanji, K.; Yamada, T.; Shimojo, S.; Koori, T.; Mora, M.; Riggs, J.E.; Oh, S.J.; Koga, Y.; et al. Primary lamp-2 deficiency causes x-linked vacuolar cardiomyopathy and myopathy (danon disease). Nature 2000, 406, 906–910. [Google Scholar] [CrossRef] [PubMed]
- Pickford, F.; Masliah, E.; Britschgi, M.; Lucin, K.; Narasimhan, R.; Jaeger, P.A.; Small, S.; Spencer, B.; Rockenstein, E.; Levine, B.; et al. The autophagy-related protein beclin 1 shows reduced expression in early alzheimer disease and regulates amyloid beta accumulation in mice. J. Clin. Investig. 2008, 118, 2190–2199. [Google Scholar] [PubMed]
- Spencer, B.; Potkar, R.; Trejo, M.; Rockenstein, E.; Patrick, C.; Gindi, R.; Adame, A.; Wyss-Coray, T.; Masliah, E. Beclin 1 gene transfer activates autophagy and ameliorates the neurodegenerative pathology in alpha-synuclein models of parkinson’s and lewy body diseases. J. Neurosci. 2009, 29, 13578–13588. [Google Scholar] [CrossRef] [PubMed]
- Uchiyama, Y.; Shibata, M.; Koike, M.; Yoshimura, K.; Sasaki, M. Autophagy-physiology and pathophysiology. Histochem. Cell Biol. 2008, 129, 407–420. [Google Scholar] [CrossRef] [PubMed]
- Shacka, J.J.; Klocke, B.J.; Young, C.; Shibata, M.; Olney, J.W.; Uchiyama, Y.; Saftig, P.; Roth, K.A. Cathepsin d deficiency induces persistent neurodegeneration in the absence of bax-dependent apoptosis. J. Neurosci. 2007, 27, 2081–2090. [Google Scholar] [CrossRef] [PubMed]
- Felbor, U.; Kessler, B.; Mothes, W.; Goebel, H.H.; Ploegh, H.L.; Bronson, R.T.; Olsen, B.R. Neuronal loss and brain atrophy in mice lacking cathepsins b and l. Proc. Natl. Acad Sci. USA 2002, 99, 7883–7888. [Google Scholar] [CrossRef] [PubMed]
- Cao, C.; Subhawong, T.; Albert, J.M.; Kim, K.W.; Geng, L.; Sekhar, K.R.; Gi, Y.J.; Lu, B. Inhibition of mammalian target of rapamycin or apoptotic pathway induces autophagy and radiosensitizes pten null prostate cancer cells. Cancer Res. 2006, 66, 10040–10047. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, Y.; Meyerkord, C.L.; Wang, H.G. Bargaining membranes for autophagosome formation: Regulation of autophagy and tumorigenesis by bif-1/endophilin b1. Autophagy 2008, 4, 121–124. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Liao, W.; Yang, J.; Ma, K.; Li, X.; Wang, Y.; Wang, D.; Wang, L.; Zhang, Y.; Yin, Y.; et al. Foxo3 induces foxo1-dependent autophagy by activating the akt1 signaling pathway. Autophagy 2012, 8, 1712–1723. [Google Scholar] [CrossRef] [PubMed]
- Rubinsztein, D.C.; Codogno, P.; Levine, B. Autophagy modulation as a potential therapeutic target for diverse diseases. Nat. Rev. Drug Discov. 2012, 11, 709–730. [Google Scholar] [CrossRef] [PubMed]
- Jin, S. Autophagy, mitochondrial quality control, and oncogenesis. Autophagy 2006, 2, 80–84. [Google Scholar] [CrossRef]
- Mathew, R.; Karantza-Wadsworth, V.; White, E. Role of autophagy in cancer. Nat. Rev. Cancer 2007, 7, 961–967. [Google Scholar] [CrossRef]
- Degenhardt, K.; Mathew, R.; Beaudoin, B.; Bray, K.; Anderson, D.; Chen, G.; Mukherjee, C.; Shi, Y.; Gelinas, C.; Fan, Y.; et al. Autophagy promotes tumor cell survival and restricts necrosis, inflammation, and tumorigenesis. Cancer Cell 2006, 10, 51–64. [Google Scholar] [CrossRef]
- White, E.J.; Martin, V.; Liu, J.L.; Klein, S.R.; Piya, S.; Gomez-Manzano, C.; Fueyo, J.; Jiang, H. Autophagy regulation in cancer development and therapy. Am. J. Cancer Res. 2011, 1, 362–372. [Google Scholar]
- Maiuri, M.C.; Zalckvar, E.; Kimchi, A.; Kroemer, G. Self-eating and self-killing: Crosstalk between autophagy and apoptosis. Nat. Rev. Mol. Cell Biol. 2007, 8, 741–752. [Google Scholar] [CrossRef]
- Huber, T.B.; Edelstein, C.L.; Hartleben, B.; Inoki, K.; Jiang, M.; Koya, D.; Kume, S.; Lieberthal, W.; Pallet, N.; Quiroga, A.; et al. Emerging role of autophagy in kidney function, diseases and aging. Autophagy 2012, 8, 1009–1031. [Google Scholar] [CrossRef]
- Livingston, M.J.; Ding, H.F.; Huang, S.; Hill, J.A.; Yin, X.M.; Dong, Z. Persistent activation of autophagy in kidney tubular cells promotes renal interstitial fibrosis during unilateral ureteral obstruction. Autophagy 2016, 12, 976–998. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Livingston, M.J.; Su, Y.; Dong, Z. Reciprocal regulation of cilia and autophagy via the mtor and proteasome pathways. Autophagy 2015, 11, 607–616. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Peng, X.; Wang, Y.; Cao, S.; Xiong, L.; Fan, J.; Wang, Y.; Zhuang, S.; Yu, X.; Mao, H. Atg5-mediated autophagy deficiency in proximal tubules promotes cell cycle g2/m arrest and renal fibrosis. Autophagy 2016, 12, 1472–1486. [Google Scholar] [CrossRef] [PubMed]
- Inoki, K. Mtor signaling in autophagy regulation in the kidney. Semin. Nephrol. 2014, 34, 2–8. [Google Scholar] [CrossRef] [PubMed]
- Hartleben, B.; Godel, M.; Meyer-Schwesinger, C.; Liu, S.; Ulrich, T.; Kobler, S.; Wiech, T.; Grahammer, F.; Arnold, S.J.; Lindenmeyer, M.T.; et al. Autophagy influences glomerular disease susceptibility and maintains podocyte homeostasis in aging mice. J. Clin. Investig. 2010, 120, 1084–1096. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.L.; Saleem, M.A.; Chan, K.W.; Yung, B.Y.; Law, H.K. The cytoprotective role of autophagy in puromycin aminonucleoside treated human podocytes. Biochem. Biophys Res. Commun. 2014, 443, 628–634. [Google Scholar] [CrossRef]
- Asanuma, K.; Tanida, I.; Shirato, I.; Ueno, T.; Takahara, H.; Nishitani, T.; Kominami, E.; Tomino, Y. Map-lc3, a promising autophagosomal marker, is processed during the differentiation and recovery of podocytes from pan nephrosis. FASEB J. 2003, 17, 1165–1167. [Google Scholar] [CrossRef] [PubMed]
- Liang, S.; Jin, J.; Lin, B.; Gong, J.; Li, Y.; He, Q. Rapamycin induces autophagy and reduces the apoptosis of podocytes under a stimulated condition of immunoglobulin a nephropathy. Kidney Blood Press Res. 2017, 42, 177–187. [Google Scholar] [CrossRef] [PubMed]
- Fang, L.; Li, X.; Luo, Y.; He, W.; Dai, C.; Yang, J. Autophagy inhibition induces podocyte apoptosis by activating the pro-apoptotic pathway of endoplasmic reticulum stress. Exp. Cell Res. 2014, 322, 290–301. [Google Scholar] [CrossRef] [PubMed]
- Zeng, C.; Fan, Y.; Wu, J.; Shi, S.; Chen, Z.; Zhong, Y.; Zhang, C.; Zen, K.; Liu, Z. Podocyte autophagic activity plays a protective role in renal injury and delays the progression of podocytopathies. J. Pathol. 2014, 234, 203–213. [Google Scholar] [CrossRef] [PubMed]
- Liebau, M.C.; Braun, F.; Hopker, K.; Weitbrecht, C.; Bartels, V.; Muller, R.U.; Brodesser, S.; Saleem, M.A.; Benzing, T.; Schermer, B.; et al. Dysregulated autophagy contributes to podocyte damage in fabry’s disease. PLoS ONE 2013, 8, e63506. [Google Scholar] [CrossRef] [PubMed]
- Kawakami, T.; Gomez, I.G.; Ren, S.; Hudkins, K.; Roach, A.; Alpers, C.E.; Shankland, S.J.; D’Agati, V.D.; Duffield, J.S. Deficient autophagy results in mitochondrial dysfunction and fsgs. J. Am. Soc. Nephrol. 2015, 26, 1040–1052. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Kim, S.; Lee, S.Y.; Koo, J.K.; Wang, Z.; Choi, M.E. Autophagy regulates tgf-beta expression and suppresses kidney fibrosis induced by unilateral ureteral obstruction. J. Am. Soc. Nephrol. 2014, 25, 2835–2846. [Google Scholar] [CrossRef] [PubMed]
- Ju, K.D.; Kim, H.J.; Tsogbadrakh, B.; Lee, J.; Ryu, H.; Cho, E.J.; Hwang, Y.H.; Kim, K.; Yang, J.; Ahn, C.; et al. Hl156a, a novel amp-activated protein kinase activator, is protective against peritoneal fibrosis in an in vivo and in vitro model of peritoneal fibrosis. Am. J. Physiol. Renal. Physiol. 2016, 310, F342–F350. [Google Scholar] [CrossRef] [PubMed]
- Grahammer, F.; Haenisch, N.; Steinhardt, F.; Sandner, L.; Roerden, M.; Arnold, F.; Cordts, T.; Wanner, N.; Reichardt, W.; Kerjaschki, D.; et al. Mtorc1 maintains renal tubular homeostasis and is essential in response to ischemic stress. Proc. Natl. Acad. Sci. USA 2014, 111, E2817–E2826. [Google Scholar] [CrossRef] [PubMed]
- Godel, M.; Hartleben, B.; Herbach, N.; Liu, S.; Zschiedrich, S.; Lu, S.; Debreczeni-Mor, A.; Lindenmeyer, M.T.; Rastaldi, M.P.; Hartleben, G.; et al. Role of mtor in podocyte function and diabetic nephropathy in humans and mice. J. Clin. Investig. 2011, 121, 2197–2209. [Google Scholar] [CrossRef] [PubMed]
- Kume, S.; Koya, D.; Uzu, T.; Maegawa, H. Role of nutrient-sensing signals in the pathogenesis of diabetic nephropathy. Biomed Res. Int. 2014, 2014, 315494. [Google Scholar] [CrossRef]
- Klionsky, D.J.; Abdalla, F.C.; Abeliovich, H.; Abraham, R.T.; Acevedo-Arozena, A.; Adeli, K.; Agholme, L.; Agnello, M.; Agostinis, P.; Aguirre-Ghiso, J.A.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 2012, 8, 445–544. [Google Scholar] [CrossRef] [PubMed]
- Yeh, C.H.; Hsu, S.P.; Yang, C.C.; Chien, C.T.; Wang, N.P. Hypoxic preconditioning reinforces hif-alpha-dependent hsp70 signaling to reduce ischemic renal failure-induced renal tubular apoptosis and autophagy. Life Sci. 2010, 86, 115–123. [Google Scholar] [CrossRef]
- Wu, H.H.; Hsiao, T.Y.; Chien, C.T.; Lai, M.K. Ischemic conditioning by short periods of reperfusion attenuates renal ischemia/reperfusion induced apoptosis and autophagy in the rat. J. Biomed. Sci. 2009, 16, 19. [Google Scholar] [CrossRef]
- Suzuki, C.; Isaka, Y.; Takabatake, Y.; Tanaka, H.; Koike, M.; Shibata, M.; Uchiyama, Y.; Takahara, S.; Imai, E. Participation of autophagy in renal ischemia/reperfusion injury. Biochem. Biophys Res. Commun. 2008, 368, 100–106. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, S.; Nishihara, K.; Inui, K.; Masuda, S. Involvement of autophagy in the pharmacological effects of the mtor inhibitor everolimus in acute kidney injury. Eur. J. Pharmacol. 2012, 696, 143–154. [Google Scholar] [CrossRef]
- Isaka, Y.; Suzuki, C.; Abe, T.; Okumi, M.; Ichimaru, N.; Imamura, R.; Kakuta, Y.; Matsui, I.; Takabatake, Y.; Rakugi, H.; et al. Bcl-2 protects tubular epithelial cells from ischemia/reperfusion injury by dual mechanisms. Transplant. Proc. 2009, 41, 52–54. [Google Scholar] [CrossRef] [PubMed]
- Kimura, T.; Takabatake, Y.; Takahashi, A.; Kaimori, J.Y.; Matsui, I.; Namba, T.; Kitamura, H.; Niimura, F.; Matsusaka, T.; Soga, T.; et al. Autophagy protects the proximal tubule from degeneration and acute ischemic injury. J. Am. Soc. Nephrol. 2011, 22, 902–913. [Google Scholar] [CrossRef] [PubMed]
- Jiang, M.; Wei, Q.; Dong, G.; Komatsu, M.; Su, Y.; Dong, Z. Autophagy in proximal tubules protects against acute kidney injury. Kidney Int. 2012, 82, 1271–1283. [Google Scholar] [CrossRef] [PubMed]
- Strand, D.W.; Jiang, M.; Murphy, T.A.; Yi, Y.; Konvinse, K.C.; Franco, O.E.; Wang, Y.; Young, J.D.; Hayward, S.W. Ppargamma isoforms differentially regulate metabolic networks to mediate mouse prostatic epithelial differentiation. Cell Death Dis. 2012, 3, e361. [Google Scholar] [CrossRef]
- Isaka, Y.; Kimura, T.; Takabatake, Y. The protective role of autophagy against aging and acute ischemic injury in kidney proximal tubular cells. Autophagy 2011, 7, 1085–1087. [Google Scholar] [CrossRef] [PubMed]
- Decuypere, J.P.; Pirenne, J.; Jochmans, I. Autophagy in renal ischemia-reperfusion injury: Friend or foe? Am. J. Transplant. 2014, 14, 1464–1465. [Google Scholar] [CrossRef] [PubMed]
- Cebotaru, V.; Cebotaru, L.; Kim, H.; Chiaravalli, M.; Boletta, A.; Qian, F.; Guggino, W.B. Polycystin-1 negatively regulates polycystin-2 expression via the aggresome/autophagosome pathway. J. Biol. Chem. 2014, 289, 6404–6414. [Google Scholar] [CrossRef] [PubMed]
- Harris, J. Autophagy and cytokines. Cytokine 2011, 56, 140–144. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, V.; Jaiswal, M.K.; Mallers, T.; Katara, G.K.; Gilman-Sachs, A.; Beaman, K.D.; Hirsch, E. Altered autophagic flux enhances inflammatory responses during inflammation-induced preterm labor. Sci. Rep. 2015, 5, 9410. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Fux, B.; Goodwin, M.; Dunay, I.R.; Strong, D.; Miller, B.C.; Cadwell, K.; Delgado, M.A.; Ponpuak, M.; Green, K.G.; et al. Autophagosome-independent essential function for the autophagy protein atg5 in cellular immunity to intracellular pathogens. Cell Host Microbe 2008, 4, 458–469. [Google Scholar] [CrossRef] [PubMed]
- Lai, Y.C.; Chuang, Y.C.; Chang, C.P.; Yeh, T.M. Macrophage migration inhibitory factor has a permissive role in concanavalin a-induced cell death of human hepatoma cells through autophagy. Cell Death Dis. 2015, 6, e2008. [Google Scholar] [CrossRef] [PubMed]
- Matsuo, H.; Itoh, H.; Kitamura, N.; Kamikubo, Y.; Higuchi, T.; Shiga, S.; Ichiyama, S.; Kondo, T.; Takaori-Kondo, A.; Adachi, S. Intravenous immunoglobulin enhances the killing activity and autophagy of neutrophils isolated from immunocompromised patients against multidrug-resistant bacteria. Biochem. Biophys. Res. Commun. 2015, 464, 94–99. [Google Scholar] [CrossRef] [PubMed]
- Mihalache, C.C.; Yousefi, S.; Conus, S.; Villiger, P.M.; Schneider, E.M.; Simon, H.U. Inflammation-associated autophagy-related programmed necrotic death of human neutrophils characterized by organelle fusion events. J. Immunol. 2011, 186, 6532–6542. [Google Scholar] [CrossRef] [PubMed]
- Tang, S.; Zhang, Y.; Yin, S.W.; Gao, X.J.; Shi, W.W.; Wang, Y.; Huang, X.; Wang, L.; Zou, L.Y.; Zhao, J.H.; et al. Neutrophil extracellular trap formation is associated with autophagy-related signalling in anca-associated vasculitis. Clin. Exp. Immunol. 2015, 180, 408–418. [Google Scholar] [CrossRef] [PubMed]
- Joska, J.A.; Gouse, H.; Paul, R.H.; Stein, D.J.; Flisher, A.J. Does highly active antiretroviral therapy improve neurocognitive function? A systematic review. J. Neurovirol. 2010, 16, 101–114. [Google Scholar] [CrossRef] [PubMed]
- Ronsard, L.; Lata, S.; Singh, J.; Ramachandran, V.G.; Das, S.; Banerjea, A.C. Molecular and genetic characterization of natural hiv-1 tat exon-1 variants from north india and their functional implications. PLoS ONE 2014, 9, e85452. [Google Scholar] [CrossRef] [PubMed]
- Ronsard, L.; Rai, T.; Rai, D.; Ramachandran, V.G.; Banerjea, A.C. In silico analyses of subtype specific hiv-1 tat-tar rna interaction reveals the structural determinants for viral activity. Front. Microbiol. 2017, 8, 1467. [Google Scholar] [CrossRef] [PubMed]
- Ronsard, L.; Raja, R.; Panwar, V.; Saini, S.; Mohankumar, K.; Sridharan, S.; Padmapriya, R.; Chaudhuri, S.; Ramachandran, V.G.; Banerjea, A.C. Genetic and functional characterization of hiv-1 vif on apobec3g degradation: First report of emergence of b/c recombinants from north india. Sci. Rep. 2015, 5, 15438. [Google Scholar] [CrossRef]
- Ronsard, L.; Ganguli, N.; Singh, V.K.; Mohankumar, K.; Rai, T.; Sridharan, S.; Pajaniradje, S.; Kumar, B.; Rai, D.; Chaudhuri, S.; et al. Impact of genetic variations in hiv-1 tat on ltr-mediated transcription via tar rna interaction. Front. Microbiol. 2017, 8, 706. [Google Scholar] [CrossRef] [PubMed]
- Bartz, S.R.; Emerman, M. Human immunodeficiency virus type 1 tat induces apoptosis and increases sensitivity to apoptotic signals by up-regulating flice/caspase-8. J. Virol. 1999, 73, 1956–1963. [Google Scholar] [PubMed]
- Li, J.C.; Au, K.Y.; Fang, J.W.; Yim, H.C.; Chow, K.H.; Ho, P.L.; Lau, A.S. Hiv-1 trans-activator protein dysregulates ifn-gamma signaling and contributes to the suppression of autophagy induction. AIDS 2011, 25, 15–25. [Google Scholar] [CrossRef] [PubMed]
- Brass, A.L.; Dykxhoorn, D.M.; Benita, Y.; Yan, N.; Engelman, A.; Xavier, R.J.; Lieberman, J.; Elledge, S.J. Identification of host proteins required for hiv infection through a functional genomic screen. Science 2008, 319, 921–926. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, M.N.; Ng, A.; Sukumaran, B.; Gilfoy, F.D.; Uchil, P.D.; Sultana, H.; Brass, A.L.; Adametz, R.; Tsui, M.; Qian, F.; et al. Rna interference screen for human genes associated with west nile virus infection. Nature 2008, 455, 242–245. [Google Scholar] [CrossRef] [PubMed]
- Eekels, J.J.; Sagnier, S.; Geerts, D.; Jeeninga, R.E.; Biard-Piechaczyk, M.; Berkhout, B. Inhibition of hiv-1 replication with stable rnai-mediated knockdown of autophagy factors. Virol J. 2012, 9, 69. [Google Scholar] [CrossRef] [PubMed]
- Kyei, G.B.; Dinkins, C.; Davis, A.S.; Roberts, E.; Singh, S.B.; Dong, C.; Wu, L.; Kominami, E.; Ueno, T.; Yamamoto, A.; et al. Autophagy pathway intersects with hiv-1 biosynthesis and regulates viral yields in macrophages. J. Cell Biol. 2009, 186, 255–268. [Google Scholar] [CrossRef] [PubMed]
- Killian, M.S. Dual role of autophagy in hiv-1 replication and pathogenesis. AIDS Res. Ther. 2012, 9, 16. [Google Scholar] [CrossRef] [PubMed]
- Daussy, C.F.; Beaumelle, B.; Espert, L. Autophagy restricts hiv-1 infection. Oncotarget 2015, 6, 20752–20753. [Google Scholar] [CrossRef]
- Leymarie, O.; Lepont, L.; Berlioz-Torrent, C. Canonical and non-canonical autophagy in hiv-1 replication cycle. Viruses 2017, 9. [Google Scholar] [CrossRef] [PubMed]
- Gupta, M.K.; Kaminski, R.; Mullen, B.; Gordon, J.; Burdo, T.H.; Cheung, J.Y.; Feldman, A.M.; Madesh, M.; Khalili, K. Hiv-1 nef-induced cardiotoxicity through dysregulation of autophagy. Sci. Rep. 2017, 7, 8572. [Google Scholar] [CrossRef] [PubMed]
- Spector, S.A.; Zhou, D. Autophagy: An overlooked mechanism of hiv-1 pathogenesis and neuroaids? Autophagy 2008, 4, 704–706. [Google Scholar] [CrossRef] [PubMed]
- Fields, J.; Dumaop, W.; Eleuteri, S.; Campos, S.; Serger, E.; Trejo, M.; Kosberg, K.; Adame, A.; Spencer, B.; Rockenstein, E.; et al. Hiv-1 tat alters neuronal autophagy by modulating autophagosome fusion to the lysosome: Implications for hiv-associated neurocognitive disorders. J. Neurosci. 2015, 35, 1921–1938. [Google Scholar] [CrossRef] [PubMed]
- Menzies, F.M.; Fleming, A.; Caricasole, A.; Bento, C.F.; Andrews, S.P.; Ashkenazi, A.; Fullgrabe, J.; Jackson, A.; Jimenez Sanchez, M.; Karabiyik, C.; et al. Autophagy and neurodegeneration: Pathogenic mechanisms and therapeutic opportunities. Neuron 2017, 93, 1015–1034. [Google Scholar] [CrossRef] [PubMed]
- Van Grol, J.; Subauste, C.; Andrade, R.M.; Fujinaga, K.; Nelson, J.; Subauste, C.S. Hiv-1 inhibits autophagy in bystander macrophage/monocytic cells through src-akt and stat3. PLoS ONE 2010, 5, e11733. [Google Scholar] [CrossRef] [PubMed]
- He, C.; Levine, B. The beclin 1 interactome. Curr. Opin. Cell Biol. 2010, 22, 140–149. [Google Scholar] [CrossRef] [PubMed]
- Saribas, A.S.; Khalili, K.; Sariyer, I.K. Dysregulation of autophagy by hiv-1 nef in human astrocytes. Cell Cycle 2015, 14, 2899–2904. [Google Scholar] [CrossRef] [PubMed]
- Silverstein, P.S.; Shah, A.; Weemhoff, J.; Kumar, S.; Singh, D.P.; Kumar, A. Hiv-1 gp120 and drugs of abuse: Interactions in the central nervous system. Curr. HIV Res. 2012, 10, 369–383. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Qiao, L.; Zhang, Y.; Zang, Y.; Shi, Y.; Liu, K.; Zhang, X.; Lu, X.; Yuan, L.; Su, B.; et al. Aspp2 plays a dual role in gp120-induced autophagy and apoptosis of neuroblastoma cells. Front. Neurosci. 2017, 11, 150. [Google Scholar] [CrossRef] [PubMed]
- Manches, O.; Frleta, D.; Bhardwaj, N. Dendritic cells in progression and pathology of hiv infection. Trends Immunol. 2014, 35, 114–122. [Google Scholar] [CrossRef] [PubMed]
- Cinti, A.; Le Sage, V.; Milev, M.P.; Valiente-Echeverria, F.; Crossie, C.; Miron, M.J.; Pante, N.; Olivier, M.; Mouland, A.J. Hiv-1 enhances mtorc1 activity and repositions lysosomes to the periphery by co-opting rag gtpases. Sci. Rep. 2017, 7, 5515. [Google Scholar] [CrossRef] [PubMed]
- Pleet, M.L.; Branscome, H.; DeMarino, C.; Pinto, D.O.; Zadeh, M.A.; Rodriguez, M.; Sariyer, I.K.; El-Hage, N.; Kashanchi, F. Autophagy, evs, and infections: A perfect question for a perfect time. Front. Cell. Infect. Microbiol. 2018, 8, 362. [Google Scholar] [CrossRef] [PubMed]
- Espert, L.; Denizot, M.; Grimaldi, M.; Robert-Hebmann, V.; Gay, B.; Varbanov, M.; Codogno, P.; Biard-Piechaczyk, M. Autophagy is involved in t cell death after binding of hiv-1 envelope proteins to cxcr4. J. Clin. Investig. 2006, 116, 2161–2172. [Google Scholar] [CrossRef] [PubMed]
- Zhou, D.; Masliah, E.; Spector, S.A. Autophagy is increased in postmortem brains of persons with hiv-1-associated encephalitis. J. Infect. Dis. 2011, 203, 1647–1657. [Google Scholar] [CrossRef] [PubMed]
- Nardacci, R.; Amendola, A.; Ciccosanti, F.; Corazzari, M.; Esposito, V.; Vlassi, C.; Taibi, C.; Fimia, G.M.; Del Nonno, F.; Ippolito, G.; et al. Autophagy plays an important role in the containment of hiv-1 in nonprogressor-infected patients. Autophagy 2014, 10, 1167–1178. [Google Scholar] [CrossRef] [PubMed]
- Maday, S. Mechanisms of neuronal homeostasis: Autophagy in the axon. Brain Res. 2016, 1649, 143–150. [Google Scholar] [CrossRef] [PubMed]
- Ojha, C.R.; Lapierre, J.; Rodriguez, M.; Dever, S.M.; Zadeh, M.A.; DeMarino, C.; Pleet, M.L.; Kashanchi, F.; El-Hage, N. Interplay between autophagy, exosomes and hiv-1 associated neurological disorders: New insights for diagnosis and therapeutic applications. Viruses 2017, 9. [Google Scholar] [CrossRef] [PubMed]
- Nooka, S.; Ghorpade, A. Hiv-1-associated inflammation and antiretroviral therapy regulate astrocyte endoplasmic reticulum stress responses. Cell Death Discov. 2017, 3, 17061. [Google Scholar] [CrossRef] [PubMed]
- Espert, L.; Varbanov, M.; Robert-Hebmann, V.; Sagnier, S.; Robbins, I.; Sanchez, F.; Lafont, V.; Biard-Piechaczyk, M. Differential role of autophagy in cd4 t cells and macrophages during x4 and r5 hiv-1 infection. PLoS ONE 2009, 4, e5787. [Google Scholar] [CrossRef] [PubMed]
- Meng, L.; Zhang, Z.; Xu, K.; Qi, G. Hiv-1 gp120 induces autophagy in cardiomyocytes via the nmda receptor. Int. J. Cardiol. 2013, 167, 2517–2523. [Google Scholar] [CrossRef]
- Sagnier, S.; Daussy, C.F.; Borel, S.; Robert-Hebmann, V.; Faure, M.; Blanchet, F.P.; Beaumelle, B.; Biard-Piechaczyk, M.; Espert, L. Autophagy restricts hiv-1 infection by selectively degrading tat in CD4+ t lymphocytes. J. Virol. 2015, 89, 615–625. [Google Scholar] [CrossRef] [PubMed]
- Bruno, A.P.; De Simone, F.I.; Iorio, V.; De Marco, M.; Khalili, K.; Sariyer, I.K.; Capunzo, M.; Nori, S.L.; Rosati, A. Hiv-1 tat protein induces glial cell autophagy through enhancement of bag3 protein levels. Cell Cycle 2014, 13, 3640–3644. [Google Scholar] [CrossRef] [PubMed]
- Campbell, G.R.; Rawat, P.; Bruckman, R.S.; Spector, S.A. Human immunodeficiency virus type 1 nef inhibits autophagy through transcription factor eb sequestration. PLoS Pathog. 2015, 11, e1005018. [Google Scholar] [CrossRef] [PubMed]
- Sardo, L.; Iordanskiy, S.; Klase, Z.; Kashanchi, F. Hiv-1 nef blocks autophagy in human astrocytes. Cell Cycle 2015, 14, 3781–3782. [Google Scholar] [CrossRef] [PubMed]
- Gregoire, I.P.; Richetta, C.; Meyniel-Schicklin, L.; Borel, S.; Pradezynski, F.; Diaz, O.; Deloire, A.; Azocar, O.; Baguet, J.; Le Breton, M.; et al. Irgm is a common target of rna viruses that subvert the autophagy network. PLoS Pathog. 2011, 7, e1002422. [Google Scholar] [CrossRef] [PubMed]
- Beaupere, C.; Garcia, M.; Larghero, J.; Feve, B.; Capeau, J.; Lagathu, C. The hiv proteins tat and nef promote human bone marrow mesenchymal stem cell senescence and alter osteoblastic differentiation. Aging Cell 2015, 14, 534–546. [Google Scholar] [CrossRef] [PubMed]
- Bravo, R.; Parra, V.; Gatica, D.; Rodriguez, A.E.; Torrealba, N.; Paredes, F.; Wang, Z.V.; Zorzano, A.; Hill, J.A.; Jaimovich, E.; et al. Endoplasmic reticulum and the unfolded protein response: Dynamics and metabolic integration. Int. Rev. Cell Mol. Biol. 2013, 301, 215–290. [Google Scholar]
- Kolattukudy, P.E.; Niu, J. Inflammation, endoplasmic reticulum stress, autophagy, and the monocyte chemoattractant protein-1/ccr2 pathway. Circ. Res. 2012, 110, 174–189. [Google Scholar] [CrossRef]
- Garg, A.D.; Kaczmarek, A.; Krysko, O.; Vandenabeele, P.; Krysko, D.V.; Agostinis, P. Er stress-induced inflammation: Does it aid or impede disease progression? Trends Mol. Med. 2012, 18, 589–598. [Google Scholar] [CrossRef]
- Song, S.; Tan, J.; Miao, Y.; Zhang, Q. Crosstalk of er stress-mediated autophagy and er-phagy: Involvement of upr and the core autophagy machinery. J. Cell Physiol. 2018, 233, 3867–3874. [Google Scholar] [CrossRef]
- Kadowaki, H.; Nishitoh, H. Signaling pathways from the endoplasmic reticulum and their roles in disease. Genes 2013, 4, 306–333. [Google Scholar] [CrossRef] [PubMed]
- Rao, R.V.; Bredesen, D.E. Misfolded proteins, endoplasmic reticulum stress and neurodegeneration. Curr. Opin. Cell Biol. 2004, 16, 653–662. [Google Scholar] [CrossRef]
- Lindholm, D.; Korhonen, L.; Eriksson, O.; Koks, S. Recent insights into the role of unfolded protein response in er stress in health and disease. Front. Cell Dev. Biol. 2017, 5, 48. [Google Scholar] [CrossRef] [PubMed]
- Santos, L.E.; Ferreira, S.T. Crosstalk between endoplasmic reticulum stress and brain inflammation in alzheimer’s disease. Neuropharmacology 2018, 136, 350–360. [Google Scholar] [CrossRef] [PubMed]
- Mochida, K.; Oikawa, Y.; Kimura, Y.; Kirisako, H.; Hirano, H.; Ohsumi, Y.; Nakatogawa, H. Receptor-mediated selective autophagy degrades the endoplasmic reticulum and the nucleus. Nature 2015, 522, 359–362. [Google Scholar] [CrossRef] [PubMed]
- Mekahli, D.; Bultynck, G.; Parys, J.B.; De Smedt, H.; Missiaen, L. Endoplasmic-reticulum calcium depletion and disease. Cold Spring Harb. Perspect. Biol. 2011, 3. [Google Scholar] [CrossRef]
- Rebbeck, R.T.; Essawy, M.M.; Nitu, F.R.; Grant, B.D.; Gillispie, G.D.; Thomas, D.D.; Bers, D.M.; Cornea, R.L. High-throughput screens to discover small-molecule modulators of ryanodine receptor calcium release channels. SLAS Discov. 2017, 22, 176–186. [Google Scholar] [CrossRef] [PubMed]
- Senft, D.; Ronai, Z.A. Upr, autophagy, and mitochondria crosstalk underlies the er stress response. Trends Biochem. Sci. 2015, 40, 141–148. [Google Scholar] [CrossRef]
- Cai, Y.; Arikkath, J.; Yang, L.; Guo, M.L.; Periyasamy, P.; Buch, S. Interplay of endoplasmic reticulum stress and autophagy in neurodegenerative disorders. Autophagy 2016, 12, 225–244. [Google Scholar] [CrossRef]
- Buch, S.; Yao, H.; Guo, M.; Mori, T.; Su, T.P.; Wang, J. Cocaine and hiv-1 interplay: Molecular mechanisms of action and addiction. J. Neuroimmune Pharmacol. 2011, 6, 503–515. [Google Scholar] [CrossRef]
- Scheckel, C.; Aguzzi, A. Prions, prionoids and protein misfolding disorders. Nat. Rev. Genet. 2018, 19, 405–418. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, S. Endoplasmic reticulum stress in motor neurons of the spinal cord in sporadic amyotrophic lateral sclerosis. J. Neuropathol. Exp. Neurol. 2010, 69, 346–355. [Google Scholar] [CrossRef] [PubMed]
- Norman, J.P.; Perry, S.W.; Reynolds, H.M.; Kiebala, M.; De Mesy Bentley, K.L.; Trejo, M.; Volsky, D.J.; Maggirwar, S.B.; Dewhurst, S.; Masliah, E.; et al. Hiv-1 tat activates neuronal ryanodine receptors with rapid induction of the unfolded protein response and mitochondrial hyperpolarization. PLoS ONE 2008, 3, e3731. [Google Scholar] [CrossRef] [PubMed]
- Ilieva, E.V.; Ayala, V.; Jove, M.; Dalfo, E.; Cacabelos, D.; Povedano, M.; Bellmunt, M.J.; Ferrer, I.; Pamplona, R.; Portero-Otin, M. Oxidative and endoplasmic reticulum stress interplay in sporadic amyotrophic lateral sclerosis. Brain 2007, 130, 3111–3123. [Google Scholar] [CrossRef] [PubMed]
- Vidal, R.L.; Figueroa, A.; Court, F.A.; Thielen, P.; Molina, C.; Wirth, C.; Caballero, B.; Kiffin, R.; Segura-Aguilar, J.; Cuervo, A.M.; et al. Targeting the upr transcription factor xbp1 protects against huntington’s disease through the regulation of foxo1 and autophagy. Hum. Mol. Genet. 2012, 21, 2245–2262. [Google Scholar] [CrossRef] [PubMed]
- Matus, S.; Lisbona, F.; Torres, M.; Leon, C.; Thielen, P.; Hetz, C. The stress rheostat: An interplay between the unfolded protein response (upr) and autophagy in neurodegeneration. Curr. Mol. Med. 2008, 8, 157–172. [Google Scholar] [CrossRef] [PubMed]
- Rouschop, K.M.; van den Beucken, T.; Dubois, L.; Niessen, H.; Bussink, J.; Savelkouls, K.; Keulers, T.; Mujcic, H.; Landuyt, W.; Voncken, J.W.; et al. The unfolded protein response protects human tumor cells during hypoxia through regulation of the autophagy genes map1lc3b and atg5. J. Clin. Investig. 2010, 120, 127–141. [Google Scholar] [CrossRef] [PubMed]
- Rashid, H.O.; Yadav, R.K.; Kim, H.R.; Chae, H.J. Er stress: Autophagy induction, inhibition and selection. Autophagy 2015, 11, 1956–1977. [Google Scholar] [CrossRef]
- Lonskaya, I.; Desforges, N.M.; Hebron, M.L.; Moussa, C.E. Ubiquitination increases parkin activity to promote autophagic alpha-synuclein clearance. PLoS ONE 2013, 8, e83914. [Google Scholar] [CrossRef]
- Plaza-Zabala, A.; Sierra-Torre, V.; Sierra, A. Autophagy and microglia: Novel partners in neurodegeneration and aging. Int. J. Mol. Sci. 2017, 18. [Google Scholar] [CrossRef]
- Von Bernhardi, R.; Eugenin-von Bernhardi, L.; Eugenin, J. Microglial cell dysregulation in brain aging and neurodegeneration. Front. Aging Neurosci. 2015, 7, 124. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Zhang, J.; Jiang, W.; Cao, Z.; Zhao, F.; Cai, T.; Aschner, M.; Luo, W. The role of nlrp3-casp1 in inflammasome-mediated neuroinflammation and autophagy dysfunction in manganese-induced, hippocampal-dependent impairment of learning and memory ability. Autophagy 2017, 13, 914–927. [Google Scholar] [CrossRef] [PubMed]
- Caldeira, C.; Cunha, C.; Vaz, A.R.; Falcao, A.S.; Barateiro, A.; Seixas, E.; Fernandes, A.; Brites, D. Key aging-associated alterations in primary microglia response to beta-amyloid stimulation. Front. Aging Neurosci. 2017, 9, 277. [Google Scholar] [CrossRef]
- Verfaillie, T.; Salazar, M.; Velasco, G.; Agostinis, P. Linking er stress to autophagy: Potential implications for cancer therapy. Int. J. Cell Biol. 2010, 2010, 930509. [Google Scholar] [CrossRef] [PubMed]
- Rozpedek, W.; Nowak, A.; Pytel, D.; Diehl, J.A.; Majsterek, I. Molecular basis of human diseases and targeted therapy based on small-molecule inhibitors of er stress-induced signaling pathways. Curr. Mol. Med. 2017, 17, 118–132. [Google Scholar]
- Ma, R.; Yang, L.; Niu, F.; Buch, S. Hiv tat-mediated induction of human brain microvascular endothelial cell apoptosis involves endoplasmic reticulum stress and mitochondrial dysfunction. Mol. Neurobiol. 2016, 53, 132–142. [Google Scholar] [CrossRef]
- Chang, J.R.; Mukerjee, R.; Bagashev, A.; Del Valle, L.; Chabrashvili, T.; Hawkins, B.J.; He, J.J.; Sawaya, B.E. Hiv-1 tat protein promotes neuronal dysfunction through disruption of micrornas. J. Biol. Chem. 2011, 286, 41125–41134. [Google Scholar] [CrossRef]
- Kim, J.E.; Ko, A.R.; Hyun, H.W.; Min, S.J.; Kang, T.C. P2rx7-mapk1/2-sp1 axis inhibits mtor independent hspb1-mediated astroglial autophagy. Cell Death Dis. 2018, 9, 546. [Google Scholar] [CrossRef] [PubMed]
- Morrow, B.A.; Roth, R.H.; Redmond, D.E.; Elsworth, J.D. Impact of methamphetamine on dopamine neurons in primates is dependent on age: Implications for development of parkinson’s disease. Neuroscience 2011, 189, 277–285. [Google Scholar] [CrossRef]
- Larsen, K.E.; Fon, E.A.; Hastings, T.G.; Edwards, R.H.; Sulzer, D. Methamphetamine-induced degeneration of dopaminergic neurons involves autophagy and upregulation of dopamine synthesis. J. Neurosci. 2002, 22, 8951–8960. [Google Scholar] [CrossRef]
- Ma, J.; Wan, J.; Meng, J.; Banerjee, S.; Ramakrishnan, S.; Roy, S. Methamphetamine induces autophagy as a pro-survival response against apoptotic endothelial cell death through the kappa opioid receptor. Cell Death Dis. 2014, 5, e1099. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.; Chandramani-Shivalingappa, P.; Jin, H.; Ghosh, A.; Anantharam, V.; Ali, S.; Kanthasamy, A.G.; Kanthasamy, A. Methamphetamine-induced neurotoxicity linked to ubiquitin-proteasome system dysfunction and autophagy-related changes that can be modulated by protein kinase c delta in dopaminergic neuronal cells. Neuroscience 2012, 210, 308–332. [Google Scholar] [CrossRef] [PubMed]
- Kongsuphol, P.; Mukda, S.; Nopparat, C.; Villarroel, A.; Govitrapong, P. Melatonin attenuates methamphetamine-induced deactivation of the mammalian target of rapamycin signaling to induce autophagy in sk-n-sh cells. J. Pineal. Res. 2009, 46, 199–206. [Google Scholar] [CrossRef] [PubMed]
- Kesby, J.P.; Hubbard, D.T.; Markou, A.; Semenova, S. Expression of hiv gp120 protein increases sensitivity to the rewarding properties of methamphetamine in mice. Addict. Biol. 2014, 19, 593–605. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, R.; Hoshijima, H.; Nagasaka, H.; Chowdhury, S.A.; Kikuchi, H.; Kanda, Y.; Kunii, S.; Kawase, M.; Sakagami, H. Induction of non-apoptotic cell death by morphinone in human promyelocytic leukemia hl-60 cells. Anticancer Res. 2006, 26, 3343–3348. [Google Scholar] [PubMed]
- Zhao, L.; Zhu, Y.; Wang, D.; Chen, M.; Gao, P.; Xiao, W.; Rao, G.; Wang, X.; Jin, H.; Xu, L.; et al. Morphine induces beclin 1- and atg5-dependent autophagy in human neuroblastoma sh-sy5y cells and in the rat hippocampus. Autophagy 2010, 6, 386–394. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, M.; Lapierre, J.; Ojha, C.R.; Estrada-Bueno, H.; Dever, S.M.; Gewirtz, D.A.; Kashanchi, F.; El-Hage, N. Importance of autophagy in mediating human immunodeficiency virus (hiv) and morphine-induced metabolic dysfunction and inflammation in human astrocytes. Viruses 2017, 9. [Google Scholar] [CrossRef]
- El-Hage, N.; Rodriguez, M.; Dever, S.M.; Masvekar, R.R.; Gewirtz, D.A.; Shacka, J.J. Hiv-1 and morphine regulation of autophagy in microglia: Limited interactions in the context of hiv-1 infection and opioid abuse. J. Virol. 2015, 89, 1024–1035. [Google Scholar] [CrossRef]
- Feng, Y.M.; Jia, Y.F.; Su, L.Y.; Wang, D.; Lv, L.; Xu, L.; Yao, Y.G. Decreased mitochondrial DNA copy number in the hippocampus and peripheral blood during opiate addiction is mediated by autophagy and can be salvaged by melatonin. Autophagy 2013, 9, 1395–1406. [Google Scholar] [CrossRef]
- Guha, P.; Harraz, M.M.; Snyder, S.H. Cocaine elicits autophagic cytotoxicity via a nitric oxide-gapdh signaling cascade. Proc. Natl. Acad. Sci. USA 2016, 113, 1417–1422. [Google Scholar] [CrossRef]
- Von Haefen, C.; Sifringer, M.; Menk, M.; Spies, C.D. Ethanol enhances susceptibility to apoptotic cell death via down-regulation of autophagy-related proteins. Alcohol. Clin. Exp. Res. 2011, 35, 1381–1391. [Google Scholar] [CrossRef] [PubMed]
- Prock, T.L.; Miranda, R.C. Embryonic cerebral cortical progenitors are resistant to apoptosis, but increase expression of suicide receptor disc-complex genes and suppress autophagy following ethanol exposure. Alcohol. Clin. Exp. Res. 2007, 31, 694–703. [Google Scholar] [CrossRef] [PubMed]
- You, M.; Matsumoto, M.; Pacold, C.M.; Cho, W.K.; Crabb, D.W. The role of amp-activated protein kinase in the action of ethanol in the liver. Gastroenterology 2004, 127, 1798–1808. [Google Scholar] [CrossRef] [PubMed]
- Donohue, T.M., Jr. Autophagy and ethanol-induced liver injury. World J. Gastroenterol. 2009, 15, 1178–1185. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.; White, E. Role of autophagy in cancer: Management of metabolic stress. Autophagy 2007, 3, 28–31. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genotype of the Autophagy-Related Genes | Phenotype | Relevant Diseases | Reference |
|---|---|---|---|
| Atg5 deletion, liver-specific Atg7 deficiency and Atg4C deficiency | Mice develop benign tumors in the liver. | Liver cancer | [81] |
| Atg7 mutation | Mice show impaired glucose tolerance and a decreased level of serum insulin: protective autophagy. | Diabetes | [83] |
| Atg5 deficiency | Mice show cardiac hypertrophy and contractile dysfunction. | Cardiomyopathy | [84] |
| Beclin1 deletion | Mice show an increased frequency of spontaneous tumors: protective autophagy. | Breast, ovarian, and prostate cancer | [58,60,84] |
| Beclin1 overexpression | Decreases in MCF-7, cellular proliferation, in vitro clonogenicity, and tumorigenesis in nude mice. | Breast cancer | [84] |
| LAMP2 deficiency | Ultrastructural defects in cardiac myocytes and severely reduced cardiac contractibility. | Danon disease | [85] |
| Ambra1 deficiency | Severe neural tube defects in mice. | Neurodegenerative disease | [61] |
| Beclin 1 deficiency | Accelerated amyloid-b accumulation: protective autophagy. | Alzheimer’s disease | [86] |
| Bec-1, Ce-Atg7, or Ce-Atg18 knockdown | Increase in aggregate formation and toxicity of PolyQ expansion proteins in Caenorhabditis elegans. | Huntington’s disease | [31] |
| Beclin1 overexpression | Reduced accumulation of a-syn and the associated neuronal pathology in mouse. | Parkinson’s disease | [87] |
| cathepsin D−/− (all tissues) | Neuronal ceroid lipofuscinosis with autophagic vacuolization and LC3-I to LC3-II conversion. Bax knockout reduced enhanced apoptosis but not autophagic degeneration and neuronal loss. | Neurodegenerative disease | [88,89] |
| cathepsin B−/−L−/− (all tissues) | Severe brain atrophy with enhanced apoptosis, autophagic vacuolization, and LC3-I to LC3-II conversion. | Neurodegenerative disease | [88,89,90] |
| cln3 (all tissues) | Juvenile neuronal ceroid lipofuscinosis, with autophagic vacuolization and LC3-I to LC3-II conversion. | Neurodegenerative disease | [91] |
| ambra1−/− (all tissues) | Decreased autophagy, increased apoptosis, and increased cell proliferation in fetal brain. Neural tube defects and embryonic death. | Neurodegenerative disease | [61] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ramesh, J.; Ronsard, L.; Gao, A.; Venugopal, B. Autophagy Intertwines with Different Diseases—Recent Strategies for Therapeutic Approaches. Diseases 2019, 7, 15. https://doi.org/10.3390/diseases7010015
Ramesh J, Ronsard L, Gao A, Venugopal B. Autophagy Intertwines with Different Diseases—Recent Strategies for Therapeutic Approaches. Diseases. 2019; 7(1):15. https://doi.org/10.3390/diseases7010015
Chicago/Turabian StyleRamesh, Janani, Larance Ronsard, Anthony Gao, and Bhuvarahamurthy Venugopal. 2019. "Autophagy Intertwines with Different Diseases—Recent Strategies for Therapeutic Approaches" Diseases 7, no. 1: 15. https://doi.org/10.3390/diseases7010015
APA StyleRamesh, J., Ronsard, L., Gao, A., & Venugopal, B. (2019). Autophagy Intertwines with Different Diseases—Recent Strategies for Therapeutic Approaches. Diseases, 7(1), 15. https://doi.org/10.3390/diseases7010015