Epigenetic Mechanisms of Tamoxifen Resistance in Luminal Breast Cancer

{kind=link}
Abstract
1. Introduction
2. Predisposition to Drug Resistance among Luminal Breast Cancer Patients
The Molecular Mechanisms behind Luminal Tumor-Cell Heterogeneity
3. Epigenetic Regulation of Gene Signaling in Breast Cancers
3.1. DNA Methylation
3.2. Histone Modifications
3.2.1. Histone Acetylation and HAT Inhibitors
3.2.2. Histone Deacetylation and HDAC Inhibitor
3.2.3. Histone Methylation and Demethylation
3.3. microRNAs
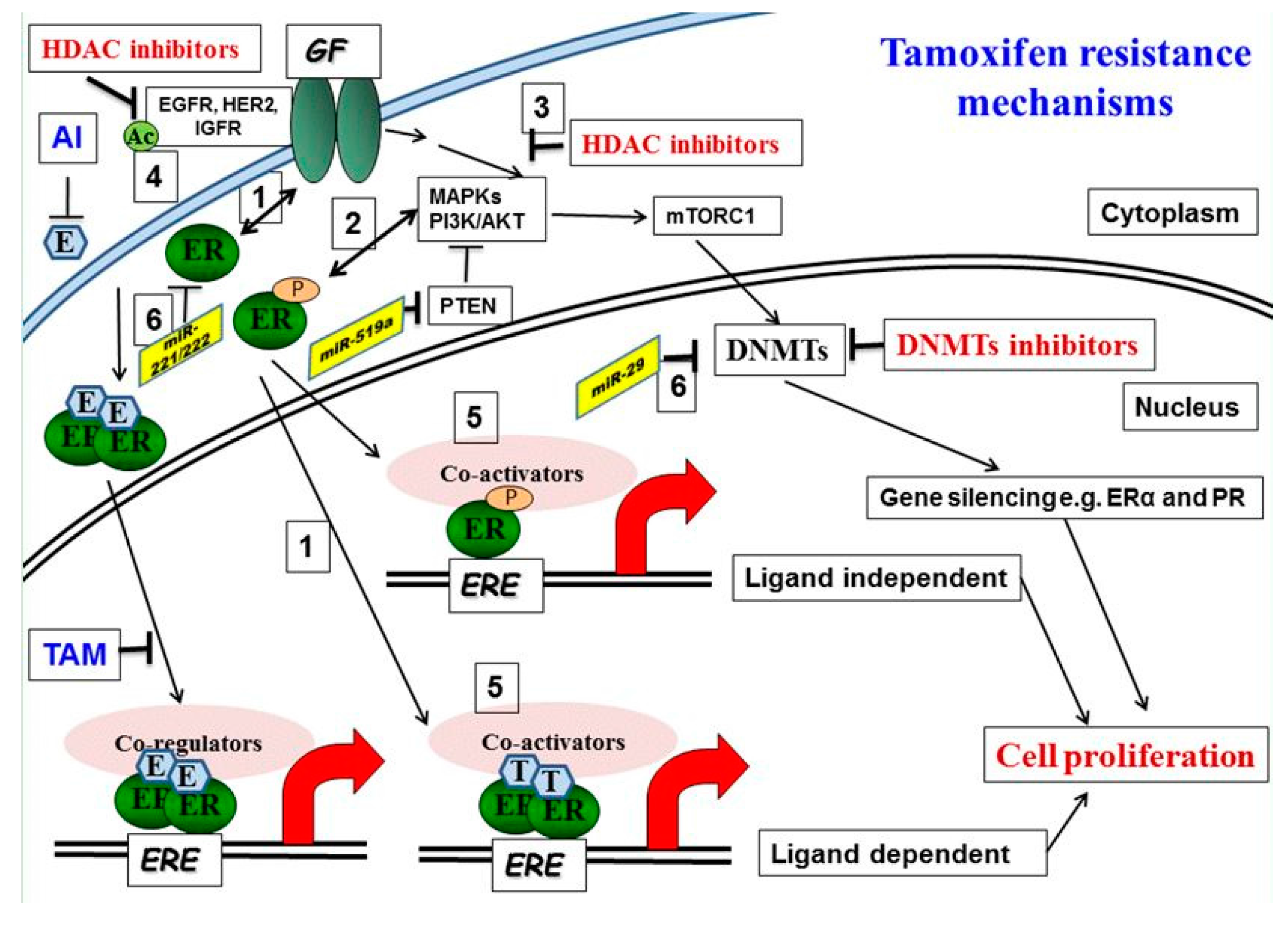
4. Epigenetic Modulation of Tamoxifen Resistance
Estrogen Receptor
5. Epigenetic Regulation of Breast Cancer Stem Cells
6. Summary and Future Perspectives
Acknowledgments
Conflicts of Interest
References
- Mendes, T.F.; Kluskens, L.D.; Rodrigues, L.R. Triple Negative Breast Cancer: Nanosolutions for a Big Challenge. Adv. Sci. 2015, 2, 1500053. [Google Scholar] [CrossRef] [PubMed]
- Olsen, S.N.; Wronski, A.; Castano, Z.; Dake, B.; Malone, C.; De Raedt, T.; Enos, M.; DeRose, Y.S.; Zhou, W.; Guerra, S.; et al. Loss of RasGAP Tumor Suppressors Underlie the Aggressive Nature of Luminal B Breast Cancers. Cancer Discov. 2016, 7, 202–217. [Google Scholar] [CrossRef] [PubMed]
- Tran, B; Bedard, P.L. Luminal-B breast cancer and novel therapeutic targets. Breast Cancer Res. 2011, 13, 221. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature 2012, 490, 61–70. [Google Scholar]
- Hammond, M.E.; Hayes, D.F.; Dowsett, M.; Allred, D.C.; Hagerty, K.L.; Badve, S.; Fitzgibbons, P.L.; Francis, G.; Goldstein, N.S.; Hayes, M.; et al. American Society of Clinical Oncology/College of American Pathologists guideline recommendations for immunohistochemical testing of estrogen and progesterone receptors in breast cancer (unabridged version). Arch. Pathol. Lab. Med. 2010, 134, e48–e72. [Google Scholar] [PubMed]
- Allred, D.C.; Brown, P.; Medina, D. The origins of estrogen receptor alpha-positive and estrogen receptor alpha-negative human breast cancer. Breast Cancer Res. 2004, 6, 240–245. [Google Scholar] [CrossRef] [PubMed]
- Osborne, C.K. Steroid hormone receptors in breast cancer management. Breast Cancer Res. Treat. 1998, 51, 227–238. [Google Scholar] [CrossRef] [PubMed]
- Badia, E.; Oliva, J.; Balaguer, P.; Cavailles, V. Tamoxifen resistance and epigenetic modifications in breast cancer cell lines. Curr. Med. Chem. 2007, 14, 3035–3045. [Google Scholar] [CrossRef] [PubMed]
- Hart, C.D.; Sanna, G.; Siclari, O.; Biganzoli, L.; Di Leo, A. Defining optimal duration and predicting benefit from chemotherapy in patients with luminal-like subtypes. Breast 2015, 24 (Suppl. 2), S136–S142. [Google Scholar] [CrossRef] [PubMed]
- Diessner, J.; Wischnewsky, M.; Blettner, M.; Hausler, S.; Janni, W.; Kreienberg, R.; Stein, R.; Stuber, T.; Schwentner, L.; Bartmann, C.; et al. Do Patients with Luminal A Breast Cancer Profit from Adjuvant Systemic Therapy? A Retrospective Multicenter Study. PLoS ONE 2016, 11, e0168730. [Google Scholar] [CrossRef] [PubMed]
- Cheang, M.C.; Chia, S.K.; Voduc, D.; Gao, D.; Leung, S.; Snider, J.; Watson, M.; Davies, S.; Bernard, P.S.; Parker, J.S.; et al. Ki67 index, HER2 status, and prognosis of patients with luminal B breast cancer. J. Natl. Cancer Inst. 2009, 101, 736–750. [Google Scholar] [CrossRef] [PubMed]
- Prat, A; Cheang, M.C.; Martin, M.; Parker, J.S.; Carrasco, E.; Caballero, R.; Tyldesley, S.; Gelmon, K.; Bernard, P.S.; Nielsen, T.O.; et al. Prognostic significance of progesterone receptor-positive tumor cells within immunohistochemically defined luminal A breast cancer. J. Clin. Oncol. 2013, 31, 203–209. [Google Scholar] [CrossRef] [PubMed]
- Cancello, G.; Maisonneuve, P.; Rotmensz, N.; Viale, G.; Mastropasqua, M.G.; Pruneri, G.; Montagna, E.; Iorfida, M.; Mazza, M.; Balduzzi, A.; et al. Progesterone receptor loss identifies Luminal B breast cancer subgroups at higher risk of relapse. Ann. Oncol. 2013, 24, 661–668. [Google Scholar] [CrossRef] [PubMed]
- Sorlie, T.; Perou, C.M.; Tibshirani, R.; Aas, T.; Geisler, S.; Johnsen, H.; Hastie, T.; Eisen, M.B.; van de Rijn, M.; Jeffrey, S.S.; et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc. Natl. Acad. Sci. USA 2001, 98, 10869–10874. [Google Scholar] [CrossRef] [PubMed]
- Sotiriou, C.; Neo, S.Y.; McShane, L.M.; Korn, E.L.; Long, P.M.; Jazaeri, A.; Martiat, P.; Fox, S.B.; Harris, A.L.; Liu, E.T. Breast cancer classification and prognosis based on gene expression profiles from a population-based study. Proc. Natl. Acad. Sci. USA 2003, 100, 10393–10398. [Google Scholar] [CrossRef] [PubMed]
- Weigelt, B.; Mackay, A.; A’Hern, R.; Natrajan, R.; Tan, D.S.; Dowsett, M.; Ashworth, A.; Reis-Filho, J.S. Breast cancer molecular profiling with single sample predictors: A retrospective analysis. Lancet Oncol. 2010, 11, 339–349. [Google Scholar] [CrossRef]
- Dowsett, M.; Sestak, I.; Lopez-Knowles, E.; Sidhu, K.; Dunbier, A.K.; Cowens, J.W.; Ferree, S.; Storhoff, J.; Schaper, C.; Cuzick, J. Comparison of PAM50 risk of recurrence score with oncotype DX and IHC4 for predicting risk of distant recurrence after endocrine therapy. J. Clin. Oncol. 2013, 31, 2783–2790. [Google Scholar] [CrossRef] [PubMed]
- Wirapati, P.; Sotiriou, C.; Kunkel, S.; Farmer, P.; Pradervand, S.; Haibe-Kains, B.; Desmedt, C.; Ignatiadis, M.; Sengstag, T.; Schutz, F.; et al. Meta-analysis of gene expression profiles in breast cancer: Toward a unified understanding of breast cancer subtyping and prognosis signatures. Breast Cancer Res. 2008, 10, R65. [Google Scholar] [CrossRef] [PubMed]
- Ali, S; Coombes, R.C. Endocrine-responsive breast cancer and strategies for combating resistance. Nat. Rev. Cancer 2002, 2, 101–112. [Google Scholar] [CrossRef] [PubMed]
- Smith, I.E.; Walsh, G.; Skene, A.; Llombart, A.; Mayordomo, J.I.; Detre, S.; Salter, J.; Clark, E.; Magill, P.; Dowsett, M. A phase II placebo-controlled trial of neoadjuvant anastrozole alone or with gefitinib in early breast cancer. J. Clin. Oncol. 2007, 25, 3816–3822. [Google Scholar] [CrossRef] [PubMed]
- Johnston, S.R.; Martin, L.A.; Leary, A.; Head, J.; Dowsett, M. Clinical strategies for rationale combinations of aromatase inhibitors with novel therapies for breast cancer. J. Steroid Biochem. Mol. Biol. 2007, 106, 180–186. [Google Scholar] [CrossRef] [PubMed]
- Campbell, L.L.; Polyak, K. Breast tumor heterogeneity: Cancer stem cells or clonal evolution? Cell Cycle 2007, 6, 2332–2338. [Google Scholar] [CrossRef] [PubMed]
- Marjanovic, N.D.; Weinberg, R.A.; Chaffer, C.L. Cell plasticity and heterogeneity in cancer. Clin. Chem. 2013, 59, 168–179. [Google Scholar] [CrossRef] [PubMed]
- Meacham, C.E.; Morrison, S.J. Tumour heterogeneity and cancer cell plasticity. Nature 2013, 501, 328–337. [Google Scholar] [CrossRef] [PubMed]
- Mazor, T.; Pankov, A.; Song, J.S.; Costello, J.F. Intratumoral Heterogeneity of the Epigenome. Cancer Cell 2016, 29, 440–451. [Google Scholar] [CrossRef] [PubMed]
- Hammond, M.E.; Hayes, D.F.; Dowsett, M.; Allred, D.C.; Hagerty, K.L.; Badve, S.; Fitzgibbons, P.L.; Francis, G.; Goldstein, N.S.; Hayes, M.; et al. American Society of Clinical Oncology/College Of American Pathologists guideline recommendations for immunohistochemical testing of estrogen and progesterone receptors in breast cancer. J. Clin. Oncol. 2010, 28, 2784–2795. [Google Scholar] [CrossRef] [PubMed]
- Maruyama, R.; Choudhury, S.; Kowalczyk, A.; Bessarabova, M.; Beresford-Smith, B.; Conway, T.; Kaspi, A.; Wu, Z.; Nikolskaya, T.; Merino, V.F.; et al. Epigenetic regulation of cell type-specific expression patterns in the human mammary epithelium. PLoS Genet. 2011, 7, e1001369. [Google Scholar] [CrossRef] [PubMed]
- Lustberg, M.B.; Ramaswamy, B. Epigenetic targeting in breast cancer: Therapeutic impact and future direction. Drug News Perspect. 2009, 22, 369–381. [Google Scholar] [CrossRef] [PubMed]
- Issa, J.P.; Kantarjian, H.M. Targeting DNA methylation. Clin. Cancer Res. 2009, 15, 3938–3946. [Google Scholar] [CrossRef] [PubMed]
- Jelinic, P.; Shaw, P. Loss of imprinting and cancer. J. Pathol. 2007, 211, 261–268. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.A. DNA methylation and cancer. Oncogene 2002, 21, 5358–5360. [Google Scholar] [CrossRef] [PubMed]
- Colacino, J.A.; Arthur, A.E.; Dolinoy, D.C.; Sartor, M.A.; Duffy, S.A.; Chepeha, D.B.; Bradford, C.R.; Walline, H.M.; McHugh, J.B.; D’Silva, N.; et al. Pretreatment dietary intake is associated with tumor suppressor DNA methylation in head and neck squamous cell carcinomas. Epigenetics 2012, 7, 883–891. [Google Scholar] [CrossRef] [PubMed]
- Portela, A.; Esteller, M. Epigenetic modifications and human disease. Nat. Biotechnol. 2010, 28, 1057–1068. [Google Scholar] [CrossRef] [PubMed]
- Duthie, S.J. Epigenetic modifications and human pathologies: Cancer and CVD. Proc. Nutr. Soc. 2011, 70, 47–56. [Google Scholar] [CrossRef] [PubMed]
- Duthie, S.J. Folate and cancer: How DNA damage, repair and methylation impact on colon carcinogenesis. J. Inherit. Metab. Dis. 2011, 34, 101–109. [Google Scholar] [CrossRef] [PubMed]
- Lan, J.; Hua, S.; He, X.; Zhang, Y. DNA methyltransferases and methyl-binding proteins of mammals. Acta Biochim. Biophys. Sin. (Shanghai) 2010, 42, 243–252. [Google Scholar] [CrossRef] [PubMed]
- Szyf, M. DNA methylation signatures for breast cancer classification and prognosis. Genome Med. 2012, 4, 26. [Google Scholar] [CrossRef] [PubMed]
- Lubecka, K.; Kurzava, L.; Flower, K.; Buvala, H.; Zhang, H.; Teegarden, D.; Camarillo, I.; Suderman, M.; Kuang, S.; Andrisani, O.; et al. Stilbenoids remodel the DNA methylation patterns in breast cancer cells and inhibit oncogenic NOTCH signaling through epigenetic regulation of MAML2 transcriptional activity. Carcinogenesis 2016, 37, 656–668. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.I.; Aumsuwan, P.; Khan, I.A.; Walker, L.A.; Dasmahapatra, A.K. Epigenetic events associated with breast cancer and their prevention by dietary components targeting the epigenome. Chem. Res. Toxicol. 2012, 25, 61–73. [Google Scholar] [CrossRef] [PubMed]
- Sunami, E.; Shinozaki, M.; Sim, M.S.; Nguyen, S.L.; Vu, A.T.; Giuliano, A.E.; Hoon, D.S. Estrogen receptor and HER2/neu status affect epigenetic differences of tumor-related genes in primary breast tumors. Breast Cancer Res. 2008, 10, R46. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Nayak, S.; Jankowitz, R.; Davidson, N.E. Epigenetics in breast cancer: What’s new? Breast Cancer Res. 2011, 13, 225. [Google Scholar] [CrossRef] [PubMed]
- Huang, T.H.; Esteller, M. Chromatin remodeling in mammary gland differentiation and breast tumorigenesis. Cold Spring Harb. Perspect. Biol. 2010, 2, a004515. [Google Scholar] [CrossRef] [PubMed]
- Jovanovic, J.; Ronneberg, J.A.; Tost, J.; Kristensen, V. The epigenetics of breast cancer. Mol. Oncol. 2010, 4, 242–254. [Google Scholar] [CrossRef] [PubMed]
- Lo, P.K.; Sukumar, S. Epigenomics and breast cancer. Pharmacogenomics 2008, 9, 1879–1902. [Google Scholar] [CrossRef] [PubMed]
- Pathiraja, T.N.; Stearns, V.; Oesterreich, S. Epigenetic regulation in estrogen receptor positive breast cancer--role in treatment response. J. Mammary Gland Biol. Neoplasia 2010, 15, 35–47. [Google Scholar] [CrossRef] [PubMed]
- Shinozaki, M.; Hoon, D.S.; Giuliano, A.E.; Hansen, N.M.; Wang, H.J.; Turner, R.; Taback, B. Distinct hypermethylation profile of primary breast cancer is associated with sentinel lymph node metastasis. Clin. Cancer Res. 2005, 11, 2156–2162. [Google Scholar] [CrossRef] [PubMed]
- Ward, A.K.; Mellor, P.; Smith, S.E.; Kendall, S.; Just, N.A.; Vizeacoumar, F.S.; Sarker, S.; Phillips, Z.; Alvi, R.; Saxena, A.; et al. Epigenetic silencing of CREB3L1 by DNA methylation is associated with high-grade metastatic breast cancers with poor prognosis and is prevalent in triple negative breast cancers. Breast Cancer Res. 2016, 18, 12. [Google Scholar] [CrossRef] [PubMed]
- Menschikowski, M.; Hagelgans, A.; Nacke, B.; Jandeck, C.; Sukocheva, O.; Siegert, G. Epigenetic control of phospholipase A2 receptor expression in mammary cancer cells. BMC Cancer 2015, 15, 971. [Google Scholar] [CrossRef] [PubMed]
- Ordway, J.M.; Budiman, M.A.; Korshunova, Y.; Maloney, R.K.; Bedell, J.A.; Citek, R.W.; Bacher, B.; Peterson, S.; Rohlfing, T.; Hall, J.; et al. Identification of novel high-frequency DNA methylation changes in breast cancer. PLoS ONE 2007, 2, e1314. [Google Scholar] [CrossRef] [PubMed]
- Fang, F.; Turcan, S.; Rimner, A.; Kaufman, A.; Giri, D.; Morris, L.G.; Shen, R.; Seshan, V.; Mo, Q.; Heguy, A.; et al. Breast cancer methylomes establish an epigenomic foundation for metastasis. Sci. Transl. Med. 2011, 3, 75ra25. [Google Scholar] [CrossRef] [PubMed]
- Bediaga, N.G.; Acha-Sagredo, A.; Guerra, I.; Viguri, A.; Albaina, C.; Ruiz Diaz, I.; Rezola, R.; Alberdi, M.J.; Dopazo, J.; Montaner, D.; et al. DNA methylation epigenotypes in breast cancer molecular subtypes. Breast Cancer Res. 2010, 12, R77. [Google Scholar] [CrossRef] [PubMed]
- Feng, W.; Shen, L.; Wen, S.; Rosen, D.G.; Jelinek, J.; Hu, X.; Huan, S.; Huang, M.; Liu, J.; Sahin, A.A.; et al. Correlation between CpG methylation profiles and hormone receptor status in breast cancers. Breast Cancer Res. 2007, 9, R57. [Google Scholar] [CrossRef] [PubMed]
- Bertolo, C.; Guerrero, D.; Vicente, F.; Cordoba, A.; Esteller, M.; Ropero, S.; Guillen-Grima, F.; Martinez-Penuela, J.M.; Lera, J.M. Differences and molecular immunohistochemical parameters in the subtypes of infiltrating ductal breast cancer. Am. J. Clin. Pathol. 2008, 130, 414–424. [Google Scholar] [CrossRef] [PubMed]
- Fiegl, H.; Millinger, S.; Goebel, G.; Muller-Holzner, E.; Marth, C.; Laird, P.W. Breast cancer DNA methylation profiles in cancer cells and tumor stroma: association with HER-2/neu status in primary breast cancer. Cancer Res. 2006, 66, 29–33. [Google Scholar] [CrossRef] [PubMed]
- Holm, K.; Hegardt, C.; Staaf, J.; Vallon-Christersson, J.; Jonsson, G.; Olsson, H.; Borg, A.; Ringner, M. Molecular subtypes of breast cancer are associated with characteristic DNA methylation patterns. Breast Cancer Res. 2010, 12, R36. [Google Scholar] [CrossRef] [PubMed]
- Holm, K.; Staaf, J.; Lauss, M.; Aine, M.; Lindgren, D.; Bendahl, P.O.; Vallon-Christersson, J.; Barkardottir, R.B.; Hoglund, M.; Borg, A.; et al. An integrated genomics analysis of epigenetic subtypes in human breast tumors links DNA methylation patterns to chromatin states in normal mammary cells. Breast Cancer Res. 2016, 18, 27. [Google Scholar] [CrossRef] [PubMed]
- Jenuwein, T; Allis, C.D. Translating the histone code. Science 2001, 293, 1074–1080. [Google Scholar] [CrossRef] [PubMed]
- Sawan, C.; Herceg, Z. Histone modifications and cancer. Adv. Genet. 2010, 70, 57–85. [Google Scholar] [PubMed]
- Zhao, Q.Y.; Lei, P.J.; Zhang, X.; Zheng, J.Y.; Wang, H.Y.; Zhao, J.; Li, Y.M.; Ye, M.; Li, L.; Wei, G.; et al. Global histone modification profiling reveals the epigenomic dynamics during malignant transformation in a four-stage breast cancer model. Clin. Epigenetics 2016, 8, 34. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Chen, H. Silencing of Wnt5a during colon cancer metastasis involves histone modifications. Epigenetics 2012, 7, 551–558. [Google Scholar] [CrossRef] [PubMed]
- Wapenaar, H.; Dekker, F.J. Histone acetyltransferases: Challenges in targeting bi-substrate enzymes. Clin. Epigenetics 2016, 8, 59. [Google Scholar] [CrossRef] [PubMed]
- Manzo, F.; Tambaro, F.P.; Mai, A.; Altucci, L. Histone acetyltransferase inhibitors and preclinical studies. Expert Opin. Ther. Pat. 2009, 19, 761–774. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.K.; Workman, J.L. Histone acetyltransferase complexes: One size doesn’t fit all. Nat. Rev. Mol. Cell Biol. 2007, 8, 284–295. [Google Scholar] [CrossRef] [PubMed]
- Huang, F.; Abmayr, S.M.; Workman, J.L. Regulation of KAT6 Acetyltransferases and Their Roles in Cell Cycle Progression, Stem Cell Maintenance, and Human Disease. Mol. Cell Biol. 2016, 36, 1900–1907. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Liang, Y.; Cao, X.; Wang, X.; Gao, H.; Lin, S.Y.; Schiff, R.; Wang, X.S.; Li, K. Identification of MYST3 as a novel epigenetic activator of ERalpha frequently amplified in breast cancer. Oncogene 2016, 36, 2910–2918. [Google Scholar] [CrossRef] [PubMed]
- Gao, C.; Bourke, E.; Scobie, M.; Famme, M.A.; Koolmeister, T.; Helleday, T.; Eriksson, L.A.; Lowndes, N.F.; Brown, J.A. Rational design and validation of a Tip60 histone acetyltransferase inhibitor. Sci. Rep. 2014, 4, 5372. [Google Scholar] [CrossRef] [PubMed]
- Simon, R.P.; Robaa, D.; Alhalabi, Z.; Sippl, W.; Jung, M. KATching-Up on Small Molecule Modulators of Lysine Acetyltransferases. J. Med. Chem. 2016, 59, 1249–1270. [Google Scholar] [CrossRef] [PubMed]
- Stimson, L.; Rowlands, M.G.; Newbatt, Y.M.; Smith, N.F.; Raynaud, F.I.; Rogers, P.; Bavetsias, V.; Gorsuch, S.; Jarman, M.; Bannister, A.; et al. Isothiazolones as inhibitors of PCAF and p300 histone acetyltransferase activity. Mol. Cancer Ther. 2005, 4, 1521–1532. [Google Scholar] [CrossRef] [PubMed]
- Gajer, J.M.; Furdas, S.D.; Grunder, A.; Gothwal, M.; Heinicke, U.; Keller, K.; Colland, F.; Fulda, S.; Pahl, H.L.; Fichtner, I.; et al. Histone acetyltransferase inhibitors block neuroblastoma cell growth in vivo. Oncogenesis 2015, 4, e137. [Google Scholar] [CrossRef] [PubMed]
- Balasubramanyam, K.; Swaminathan, V.; Ranganathan, A.; Kundu, T.K. Small molecule modulators of histone acetyltransferase p300. J. Biol. Chem. 2003, 278, 19134–19140. [Google Scholar] [CrossRef] [PubMed]
- Eliseeva, E.D.; Valkov, V.; Jung, M.; Jung, M.O. Characterization of novel inhibitors of histone acetyltransferases. Mol. Cancer Ther. 2007, 6, 2391–2398. [Google Scholar] [CrossRef] [PubMed]
- Minucci, S.; Pelicci, P.G. Histone deacetylase inhibitors and the promise of epigenetic (and more) treatments for cancer. Nat. Rev. Cancer 2006, 6, 38–51. [Google Scholar] [CrossRef] [PubMed]
- Olzscha, H.; Sheikh, S.; La Thangue, N.B. Deacetylation of chromatin and gene expression regulation: A new target for epigenetic therapy. Crit. Rev. Oncog. 2015, 20, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Damaskos, C.; Valsami, S.; Kontos, M.; Spartalis, E.; Kalampokas, T.; Kalampokas, E.; Athanasiou, A.; Moris, D.; Daskalopoulou, A.; Davakis, S.; et al. Histone Deacetylase Inhibitors: An Attractive Therapeutic Strategy Against Breast Cancer. Anticancer Res. 2017, 37, 35–46. [Google Scholar] [CrossRef] [PubMed]
- Dokmanovic, M.; Marks, P.A. Prospects: Histone deacetylase inhibitors. J. Cell Biochem. 2005, 96, 293–304. [Google Scholar] [CrossRef] [PubMed]
- Marson, C.M. Histone deacetylase inhibitors: Design, structure-activity relationships and therapeutic implications for cancer. Anticancer Agents Med. Chem. 2009, 9, 661–692. [Google Scholar] [CrossRef] [PubMed]
- Khan, O.; La Thangue, N.B. Drug Insight: Histone deacetylase inhibitor-based therapies for cutaneous T-cell lymphomas. Nat. Clin. Pract. Oncol. 2008, 5, 714–726. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Atadja, P.; Davidson, N.E. Histone deacetylase inhibitor LBH589 reactivates silenced estrogen receptor alpha (ER) gene expression without loss of DNA hypermethylation. Cancer Biol. Ther. 2007, 6, 64–69. [Google Scholar] [CrossRef] [PubMed]
- Stearns, V.; Zhou, Q.; Davidson, N.E. Epigenetic regulation as a new target for breast cancer therapy. Cancer Invest. 2007, 25, 659–665. [Google Scholar] [CrossRef] [PubMed]
- Chiu, H.W.; Yeh, Y.L.; Wang, Y.C.; Huang, W.J.; Ho, S.Y.; Lin, P.; Wang, Y.J. Combination of the novel histone deacetylase inhibitor YCW1 and radiation induces autophagic cell death through the downregulation of BNIP3 in triple-negative breast cancer cells in vitro and in an orthotopic mouse model. Mol. Cancer 2016, 15, 46. [Google Scholar] [CrossRef] [PubMed]
- Huang, T.; Lin, C.; Zhong, L.L.; Zhao, L.; Zhang, G.; Lu, A.; Wu, J.; Bian, Z. Targeting histone methylation for colorectal cancer. Ther. Adv. Gastroenterol. 2017, 10, 114–131. [Google Scholar] [CrossRef] [PubMed]
- Cohen, I.; Poreba, E.; Kamieniarz, K.; Schneider, R. Histone modifiers in cancer: Friends or foes? Genes Cancer 2011, 2, 631–647. [Google Scholar] [CrossRef] [PubMed]
- Biswas, S.; Rao, C.M. Epigenetics in cancer: Fundamentals and Beyond. Pharmacol. Ther. 2017, 173, 118–134. [Google Scholar] [CrossRef] [PubMed]
- van der Vlag, J; Otte, A.P. Transcriptional repression mediated by the human polycomb-group protein EED involves histone deacetylation. Nat. Genet. 1999, 23, 474–478. [Google Scholar] [PubMed]
- Kleer, C.G.; Cao, Q.; Varambally, S.; Shen, R.; Ota, I.; Tomlins, S.A.; Ghosh, D.; Sewalt, R.G.; Otte, A.P.; Hayes, D.F.; et al. EZH2 is a marker of aggressive breast cancer and promotes neoplastic transformation of breast epithelial cells. Proc. Natl. Acad. Sci. USA 2003, 100, 11606–11611. [Google Scholar] [CrossRef] [PubMed]
- Collett, K.; Eide, G.E.; Arnes, J.; Stefansson, I.M.; Eide, J.; Braaten, A.; Aas, T.; Otte, A.P.; Akslen, L.A. Expression of enhancer of zeste homologue 2 is significantly associated with increased tumor cell proliferation and is a marker of aggressive breast cancer. Clin. Cancer Res. 2006, 12, 1168–1174. [Google Scholar] [CrossRef] [PubMed]
- Bachmann, I.M.; Halvorsen, O.J.; Collett, K.; Stefansson, I.M.; Straume, O.; Haukaas, S.A.; Salvesen, H.B.; Otte, A.P.; Akslen, L.A. EZH2 expression is associated with high proliferation rate and aggressive tumor subgroups in cutaneous melanoma and cancers of the endometrium, prostate, and breast. J. Clin. Oncol. 2006, 24, 268–273. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.H.; Roberts, C.W. Targeting EZH2 in cancer. Nat. Med. 2016, 22, 128–134. [Google Scholar] [CrossRef] [PubMed]
- Tan, J.; Yang, X.; Zhuang, L.; Jiang, X.; Chen, W.; Lee, P.L.; Karuturi, R.K.; Tan, P.B.; Liu, E.T.; Yu, Q. Pharmacologic disruption of Polycomb-repressive complex 2-mediated gene repression selectively induces apoptosis in cancer cells. Genes Dev. 2007, 21, 1050–1063. [Google Scholar] [CrossRef] [PubMed]
- Woo, J.; Kim, H.Y.; Byun, B.J.; Chae, C.H.; Lee, J.Y.; Ryu, S.Y.; Park, W.K.; Cho, H.; Choi, G. Biological evaluation of tanshindiols as EZH2 histone methyltransferase inhibitors. Bioorg. Med. Chem. Lett. 2014, 24, 2486–2492. [Google Scholar] [CrossRef] [PubMed]
- Smadbeck, J.; Peterson, M.B.; Zee, B.M.; Garapaty, S.; Mago, A.; Lee, C.; Giannis, A.; Trojer, P.; Garcia, B.A.; Floudas, C.A. De novo peptide design and experimental validation of histone methyltransferase inhibitors. PLoS ONE 2014, 9, e95535. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Xu, A.M. SET and MYND domain containing protein 3 in cancer. Am. J. Transl. Res. 2017, 9, 1–14. [Google Scholar] [PubMed]
- Hamamoto, R.; Silva, F.P.; Tsuge, M.; Nishidate, T.; Katagiri, T.; Nakamura, Y.; Furukawa, Y. Enhanced SMYD3 expression is essential for the growth of breast cancer cells. Cancer Sci. 2006, 97, 113–118. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.G.; Zou, J.N.; Wang, S.Z.; Zhang, T.C.; Xi, T. Novobiocin decreases SMYD3 expression and inhibits the migration of MDA-MB-231 human breast cancer cells. IUBMB Life 2010, 62, 194–199. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.G.; Wynder, C.; Schmidt, D.M.; McCafferty, D.G.; Shiekhattar, R. Histone H3 lysine 4 demethylation is a target of nonselective antidepressive medications. Chem. Biol. 2006, 13, 563–567. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Lan, F.; Matson, C.; Mulligan, P.; Whetstine, J.R.; Cole, P.A.; Casero, R.A. Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell 2004, 119, 941–953. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.G.; Wynder, C.; Cooch, N.; Shiekhattar, R. An essential role for CoREST in nucleosomal histone 3 lysine 4 demethylation. Nature 2005, 437, 432–435. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Sengupta, R.; Espejo, A.B.; Lee, M.G.; Dorsey, J.A.; Richter, M.; Opravil, S.; Shiekhattar, R.; Bedford, M.T.; Jenuwein, T.; et al. p53 is regulated by the lysine demethylase LSD1. Nature 2007, 449, 105–108. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Hevi, S.; Kurash, J.K.; Lei, H.; Gay, F.; Bajko, J.; Su, H.; Sun, W.; Chang, H.; Xu, G.; et al. The lysine demethylase LSD1 (KDM1) is required for maintenance of global DNA methylation. Nat. Genet. 2009, 41, 125–129. [Google Scholar] [CrossRef] [PubMed]
- Hino, S.; Kohrogi, K.; Nakao, M. Histone demethylase LSD1 controls the phenotypic plasticity of cancer cells. Cancer Sci. 2016, 107, 1187–1192. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.; Janzer, A.; Becker, A.; Zimmer, A.; Schule, R.; Buettner, R.; Kirfel, J. Lysine-specific demethylase 1 (LSD1) is highly expressed in ER-negative breast cancers and a biomarker predicting aggressive biology. Carcinogenesis 2010, 31, 512–520. [Google Scholar] [CrossRef] [PubMed]
- Nagasawa, S.; Sedukhina, A.S.; Nakagawa, Y.; Maeda, I.; Kubota, M.; Ohnuma, S.; Tsugawa, K.; Ohta, T.; Roche-Molina, M.; Bernal, J.A.; et al. LSD1 overexpression is associated with poor prognosis in basal-like breast cancer, and sensitivity to PARP inhibition. PLoS ONE 2015, 10, e0118002. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Ma, S.; Zhao, G.; Yang, L.; Zhang, P.; Yi, Q.; Cheng, S. miR-708/LSD1 axis regulates the proliferation and invasion of breast cancer cells. Cancer Med. 2016, 5, 684–692. [Google Scholar] [CrossRef] [PubMed]
- Perillo, B.; Ombra, M.N.; Bertoni, A.; Cuozzo, C.; Sacchetti, S.; Sasso, A.; Chiariotti, L.; Malorni, A.; Abbondanza, C.; Avvedimento, E.V. DNA oxidation as triggered by H3K9me2 demethylation drives estrogen-induced gene expression. Science 2008, 319, 202–206. [Google Scholar] [CrossRef] [PubMed]
- Hayward, D.; Cole, P.A. LSD1 Histone Demethylase Assays and Inhibition. Methods Enzymol. 2016, 573, 261–278. [Google Scholar] [PubMed]
- Morera, L.; Lubbert, M.; Jung, M. Targeting histone methyltransferases and demethylases in clinical trials for cancer therapy. Clin. Epigenetics 2016, 8, 57. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Greene, E.; Murray Stewart, T.; Goodwin, A.C.; Baylin, S.B.; Woster, P.M.; Casero, R.A., Jr. Inhibition of lysine-specific demethylase 1 by polyamine analogues results in reexpression of aberrantly silenced genes. Proc. Natl. Acad. Sci. USA 2007, 104, 8023–8028. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Stewart, T.M.; Wu, Y.; Baylin, S.B.; Marton, L.J.; Perkins, B.; Jones, R.J.; Woster, P.M.; Casero, R.A., Jr. Novel oligoamine analogues inhibit lysine-specific demethylase 1 and induce reexpression of epigenetically silenced genes. Clin. Cancer Res. 2009, 15, 7217–7228. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Q.; Huang, Y.; Marton, L.J.; Woster, P.M.; Davidson, N.E.; Casero, R.A., Jr. Polyamine analogs modulate gene expression by inhibiting lysine-specific demethylase 1 (LSD1) and altering chromatin structure in human breast cancer cells. Amino Acids 2011, 42, 887–898. [Google Scholar] [CrossRef] [PubMed]
- Vasilatos, S.N.; Katz, T.A.; Oesterreich, S.; Wan, Y.; Davidson, N.E.; Huang, Y. Crosstalk between lysine-specific demethylase 1 (LSD1) and histone deacetylases mediates antineoplastic efficacy of HDAC inhibitors in human breast cancer cells. Carcinogenesis 2013, 34, 1196–1207. [Google Scholar] [CrossRef] [PubMed]
- Cao, C.; Vasilatos, S.N.; Bhargava, R.; Fine, J.L.; Oesterreich, S.; Davidson, N.E.; Huang, Y. Functional interaction of histone deacetylase 5 (HDAC5) and lysine-specific demethylase 1 (LSD1) promotes breast cancer progression. Oncogene 2017, 36, 133–145. [Google Scholar] [CrossRef] [PubMed]
- Tachibana, M.; Sugimoto, K.; Nozaki, M.; Ueda, J.; Ohta, T.; Ohki, M.; Fukuda, M.; Takeda, N.; Niida, H.; Kato, H.; et al. G9a histone methyltransferase plays a dominant role in euchromatic histone H3 lysine 9 methylation and is essential for early embryogenesis. Genes Dev. 2002, 16, 1779–1791. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Dorsey, J.; Chuikov, S.; Perez-Burgos, L.; Zhang, X.; Jenuwein, T.; Reinberg, D.; Berger, S.L. G9a and Glp methylate lysine 373 in the tumor suppressor p53. J. Biol. Chem. 2010, 285, 9636–9641. [Google Scholar] [CrossRef] [PubMed]
- Kondo, Y.; Shen, L.; Suzuki, S.; Kurokawa, T.; Masuko, K.; Tanaka, Y.; Kato, H.; Mizuno, Y.; Yokoe, M.; Sugauchi, F.; et al. Alterations of DNA methylation and histone modifications contribute to gene silencing in hepatocellular carcinomas. Hepatol. Res. 2007, 37, 974–983. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, C.T.; Weisenberger, D.J.; Velicescu, M.; Gonzales, F.A.; Lin, J.C.; Liang, G.; Jones, P.A. Histone H3-lysine 9 methylation is associated with aberrant gene silencing in cancer cells and is rapidly reversed by 5-aza-2′-deoxycytidine. Cancer Res. 2002, 62, 6456–6461. [Google Scholar] [PubMed]
- Casciello, F.; Windloch, K.; Gannon, F.; Lee, J.S. Functional Role of G9a Histone Methyltransferase in Cancer. Front. Immunol. 2015, 6, 487. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.W.; Hua, K.T.; Kao, H.J.; Chi, C.C.; Wei, L.H.; Johansson, G.; Shiah, S.G.; Chen, P.S.; Jeng, Y.M.; Cheng, T.Y.; et al. H3K9 histone methyltransferase G9a promotes lung cancer invasion and metastasis by silencing the cell adhesion molecule Ep-CAM. Cancer Res. 2010, 70, 7830–7840. [Google Scholar] [CrossRef] [PubMed]
- Kondo, Y.; Shen, L.; Ahmed, S.; Boumber, Y.; Sekido, Y.; Haddad, B.R.; Issa, J.P. Downregulation of histone H3 lysine 9 methyltransferase G9a induces centrosome disruption and chromosome instability in cancer cells. PLoS ONE 2008, 3, e2037. [Google Scholar] [CrossRef] [PubMed]
- Kubicek, S.; O’Sullivan, R.J.; August, E.M.; Hickey, E.R.; Zhang, Q.; Teodoro, M.L.; Rea, S.; Mechtler, K.; Kowalski, J.A.; Homon, C.A.; et al. Reversal of H3K9me2 by a small-molecule inhibitor for the G9a histone methyltransferase. Mol. Cell 2007, 25, 473–481. [Google Scholar] [CrossRef] [PubMed]
- Lu, Z.; Tian, Y.; Salwen, H.R.; Chlenski, A.; Godley, L.A.; Raj, J.U.; Yang, Q. Histone-lysine methyltransferase EHMT2 is involved in proliferation, apoptosis, cell invasion, and DNA methylation of human neuroblastoma cells. Anticancer Drugs 2013, 24, 484–493. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Wang, Q.; Paulk, J.; Kubicek, S.; Kemp, M.M.; Adams, D.J.; Shamji, A.F.; Wagner, B.K.; Schreiber, S.L. A small-molecule probe of the histone methyltransferase G9a induces cellular senescence in pancreatic adenocarcinoma. ACS Chem. Biol. 2012, 7, 1152–1157. [Google Scholar] [CrossRef] [PubMed]
- De Souza Rocha Simonini, P.; Breiling, A.; Gupta, N.; Malekpour, M.; Youns, M.; Omranipour, R.; Malekpour, F.; Volinia, S.; Croce, C.M.; Najmabadi, H.; et al. Epigenetically deregulated microRNA-375 is involved in a positive feedback loop with estrogen receptor alpha in breast cancer cells. Cancer Res. 2010, 70, 9175–9184. [Google Scholar] [CrossRef] [PubMed]
- Du, T.; Zamore, P.D. microPrimer: The biogenesis and function of microRNA. Development 2005, 132, 4645–4652. [Google Scholar] [CrossRef] [PubMed]
- Davis-Dusenbery, B.N.; Hata, A. MicroRNA in Cancer: The Involvement of Aberrant MicroRNA Biogenesis Regulatory Pathways. Genes Cancer 2011, 1, 1100–1114. [Google Scholar] [CrossRef] [PubMed]
- Mulrane, L.; McGee, S.F.; Gallagher, W.M.; O’Connor, D.P. miRNA Dysregulation in Breast Cancer. Cancer Res. 2013, 73, 6554–6562. [Google Scholar] [CrossRef] [PubMed]
- Davoren, P.A.; McNeill, R.E.; Lowery, A.J.; Kerin, M.J.; Miller, N. Identification of suitable endogenous control genes for microRNA gene expression analysis in human breast cancer. BMC Mol. Biol. 2008, 9, 76. [Google Scholar] [CrossRef] [PubMed]
- Frankel, L.B.; Christoffersen, N.R.; Jacobsen, A.; Lindow, M.; Krogh, A.; Lund, A.H. Programmed cell death 4 (PDCD4) is an important functional target of the microRNA miR-21 in breast cancer cells. J. Biol. Chem. 2008, 283, 1026–1033. [Google Scholar] [CrossRef] [PubMed]
- Iorio, M.V.; Ferracin, M.; Liu, C.G.; Veronese, A.; Spizzo, R.; Sabbioni, S.; Magri, E.; Pedriali, M.; Fabbri, M.; Campiglio, M.; et al. MicroRNA gene expression deregulation in human breast cancer. Cancer Res. 2005, 65, 7065–7070. [Google Scholar] [CrossRef] [PubMed]
- Si, M.L.; Zhu, S.; Wu, H.; Lu, Z.; Wu, F.; Mo, Y.Y. miR-21-mediated tumor growth. Oncogene 2007, 26, 2799–2803. [Google Scholar] [CrossRef] [PubMed]
- Lowery, A.J.; Miller, N.; Devaney, A.; McNeill, R.E.; Davoren, P.A.; Lemetre, C.; Benes, V.; Schmidt, S.; Blake, J.; Ball, G.; et al. MicroRNA signatures predict oestrogen receptor, progesterone receptor and HER2/neu receptor status in breast cancer. Breast Cancer Res. 2009, 11, R27. [Google Scholar] [CrossRef] [PubMed]
- Mattie, M.D.; Benz, C.C.; Bowers, J.; Sensinger, K.; Wong, L.; Scott, G.K.; Fedele, V.; Ginzinger, D.; Getts, R.; Haqq, C. Optimized high-throughput microRNA expression profiling provides novel biomarker assessment of clinical prostate and breast cancer biopsies. Mol. Cancer 2006, 5, 24. [Google Scholar] [CrossRef] [PubMed]
- Gregory, P.A.; Bracken, C.P.; Bert, A.G.; Goodall, G.J. MicroRNAs as regulators of epithelial-mesenchymal transition. Cell Cycle 2008, 7, 3112–3118. [Google Scholar] [CrossRef] [PubMed]
- Sempere, L.F.; Christensen, M.; Silahtaroglu, A.; Bak, M.; Heath, C.V.; Schwartz, G.; Wells, W.; Kauppinen, S.; Cole, C.N. Altered MicroRNA expression confined to specific epithelial cell subpopulations in breast cancer. Cancer Res. 2007, 67, 11612–11620. [Google Scholar] [CrossRef] [PubMed]
- Meng, F.; Henson, R.; Wehbe-Janek, H.; Ghoshal, K.; Jacob, S.T.; Patel, T. MicroRNA-21 regulates expression of the PTEN tumor suppressor gene in human hepatocellular cancer. Gastroenterology 2007, 133, 647–658. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.; Yao, H.; Zhu, P.; Zhang, X.; Pan, Q.; Gong, C.; Huang, Y.; Hu, X.; Su, F.; Lieberman, J.; et al. let-7 regulates self renewal and tumorigenicity of breast cancer cells. Cell 2007, 131, 1109–1123. [Google Scholar] [CrossRef] [PubMed]
- Iliopoulos, D.; Polytarchou, C.; Hatziapostolou, M.; Kottakis, F.; Maroulakou, I.G.; Struhl, K.; Tsichlis, P.N. MicroRNAs differentially regulated by Akt isoforms control EMT and stem cell renewal in cancer cells. Sci. Signal 2009, 2, ra62. [Google Scholar] [CrossRef] [PubMed]
- Gandellini, P.; Doldi, V.; Zaffaroni, N. microRNAs as players and signals in the metastatic cascade: Implications for the development of novel anti-metastatic therapies. Semin. Cancer Biol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Cava, C.; Bertoli, G.; Castiglioni, I. Integrating genetics and epigenetics in breast cancer: Biological insights, experimental, computational methods and therapeutic potential. BMC Syst. Biol. 2015, 9, 62. [Google Scholar] [CrossRef] [PubMed]
- Mann, M.; Cortez, V.; Vadlamudi, R.K. Epigenetics of estrogen receptor signaling: Role in hormonal cancer progression and therapy. Cancers (Basel) 2011, 3, 1691–1707. [Google Scholar] [CrossRef] [PubMed]
- Hervouet, E.; Cartron, P.F.; Jouvenot, M.; Delage-Mourroux, R. Epigenetic regulation of estrogen signaling in breast cancer. Epigenetics 2013, 8, 237–245. [Google Scholar] [CrossRef] [PubMed]
- Musgrove, E.A.; Sutherland, R.L. Biological determinants of endocrine resistance in breast cancer. Nat. Rev. Cancer 2009, 9, 631–643. [Google Scholar] [CrossRef] [PubMed]
- Green, K.A.; Carroll, J.S. Oestrogen-receptor-mediated transcription and the influence of co-factors and chromatin state. Nat. Rev. Cancer 2007, 7, 713–722. [Google Scholar] [CrossRef] [PubMed]
- Girault, I.; Bieche, I.; Lidereau, R. Role of estrogen receptor alpha transcriptional coregulators in tamoxifen resistance in breast cancer. Maturitas 2006, 54, 342–351. [Google Scholar] [CrossRef] [PubMed]
- Fleming, F.J.; Myers, E.; Kelly, G.; Crotty, T.B.; McDermott, E.W.; O’Higgins, N.J.; Hill, A.D.; Young, L.S. Expression of SRC-1, AIB1, and PEA3 in HER2 mediated endocrine resistant breast cancer; a predictive role for SRC-1. J. Clin. Pathol. 2004, 57, 1069–1074. [Google Scholar] [CrossRef] [PubMed]
- Iwase, H.; Omoto, Y.; Toyama, T.; Yamashita, H.; Hara, Y.; Sugiura, H.; Zhang, Z. Clinical significance of AIB1 expression in human breast cancer. Breast Cancer Res. Treat. 2003, 80, 339–345. [Google Scholar] [CrossRef] [PubMed]
- Wong, M.M.; Guo, C.; Zhang, J. Nuclear receptor corepressor complexes in cancer: Mechanism, function and regulation. Am. J. Clin. Exp. Urol. 2014, 2, 169–187. [Google Scholar] [PubMed]
- Legare, S; Basik, M. Minireview: The Link Between ERalpha Corepressors and Histone Deacetylases in Tamoxifen Resistance in Breast Cancer. Mol. Endocrinol. 2016, 30, 965–976. [Google Scholar]
- Zou, Z.; Luo, X.; Nie, P.; Wu, B.; Zhang, T.; Wei, Y.; Wang, W.; Geng, G.; Jiang, J.; Mi, Y. Inhibition of SRC-3 enhances sensitivity of human cancer cells to histone deacetylase inhibitors. Biochem. Biophys. Res. Commun. 2016, 478, 227–233. [Google Scholar] [CrossRef] [PubMed]
- Phan, N.L.; Trinh, N.V.; Pham, P.V. Low concentrations of 5-aza-2′-deoxycytidine induce breast cancer stem cell differentiation by triggering tumor suppressor gene expression. Onco Targets Ther. 2016, 9, 49–59. [Google Scholar] [PubMed]
- Lorico, A.; Rappa, G. Phenotypic heterogeneity of breast cancer stem cells. J. Oncol. 2011, 2011, 135039. [Google Scholar]
- Dalerba, P.; Cho, R.W.; Clarke, M.F. Cancer stem cells: Models and concepts. Annu. Rev. Med. 2007, 58, 267–284. [Google Scholar] [CrossRef] [PubMed]
- van Vlerken, L.E.; Hurt, E.M.; Hollingsworth, R.E. The role of epigenetic regulation in stem cell and cancer biology. J. Mol. Med. (Berl.) 2012, 90, 791–801. [Google Scholar] [CrossRef] [PubMed]
- Clarke, M.F.; Fuller, M. Stem cells and cancer: Two faces of eve. Cell 2006, 124, 1111–1115. [Google Scholar] [CrossRef] [PubMed]
- Pajonk, F.; Vlashi, E.; McBride, W.H. Radiation resistance of cancer stem cells: The 4 R’s of radiobiology revisited. Stem Cells 2010, 28, 639–648. [Google Scholar] [CrossRef] [PubMed]
- Moncharmont, C.; Levy, A.; Gilormini, M.; Bertrand, G.; Chargari, C.; Alphonse, G.; Ardail, D.; Rodriguez-Lafrasse, C.; Magne, N. Targeting a cornerstone of radiation resistance: Cancer stem cell. Cancer Lett. 2012, 322, 139–147. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.M.; Kim, Y.S.; Kang, M.J.; Farrar, W.L.; Hurt, E.M. Long-term recovery of irradiated prostate cancer increases cancer stem cells. Prostate 2012, 72, 1746–1756. [Google Scholar] [CrossRef] [PubMed]
- Koch, U.; Krause, M.; Baumann, M. Cancer stem cells at the crossroads of current cancer therapy failures--radiation oncology perspective. Semin. Cancer Biol. 2010, 20, 116–124. [Google Scholar] [CrossRef] [PubMed]
- Phillips, T.M.; McBride, W.H.; Pajonk, F. The response of CD24(-/low)/CD44+ breast cancer-initiating cells to radiation. J. Natl. Cancer Inst. 2006, 98, 1777–1785. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.; Glass, J. The phenotypic radiation resistance of CD44+/CD24(-or low) breast cancer cells is mediated through the enhanced activation of ATM signaling. PLoS ONE 2011, 6, e24080. [Google Scholar] [CrossRef] [PubMed]
- Maugeri-Sacca, M.; Vigneri, P.; De Maria, R. Cancer stem cells and chemosensitivity. Clin. Cancer Res. 2011, 17, 4942–4947. [Google Scholar] [CrossRef] [PubMed]
- Eramo, A.; Ricci-Vitiani, L.; Zeuner, A.; Pallini, R.; Lotti, F.; Sette, G.; Pilozzi, E.; Larocca, L.M.; Peschle, C.; De Maria, R. Chemotherapy resistance of glioblastoma stem cells. Cell Death Differ. 2006, 13, 1238–1241. [Google Scholar] [CrossRef] [PubMed]
- Eramo, A.; Lotti, F.; Sette, G.; Pilozzi, E.; Biffoni, M.; Di Virgilio, A.; Conticello, C.; Ruco, L.; Peschle, C.; De Maria, R. Identification and expansion of the tumorigenic lung cancer stem cell population. Cell Death Differ. 2008, 15, 504–514. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.; Lee, T.K.; Zheng, B.J.; Chan, K.W.; Guan, X.Y. CD133+ HCC cancer stem cells confer chemoresistance by preferential expression of the Akt/PKB survival pathway. Oncogene 2008, 27, 1749–1758. [Google Scholar] [CrossRef] [PubMed]
- Zeppernick, F.; Ahmadi, R.; Campos, B.; Dictus, C.; Helmke, B.M.; Becker, N.; Lichter, P.; Unterberg, A.; Radlwimmer, B.; Herold-Mende, C.C. Stem cell marker CD133 affects clinical outcome in glioma patients. Clin. Cancer Res. 2008, 14, 123–129. [Google Scholar] [CrossRef] [PubMed]
- Balic, M.; Lin, H.; Young, L.; Hawes, D.; Giuliano, A.; McNamara, G.; Datar, R.H.; Cote, R.J. Most early disseminated cancer cells detected in bone marrow of breast cancer patients have a putative breast cancer stem cell phenotype. Clin. Cancer Res. 2006, 12, 5615–5621. [Google Scholar] [CrossRef] [PubMed]
- Witt, A.E.; Lee, C.W.; Lee, T.I.; Azzam, D.J.; Wang, B.; Caslini, C.; Petrocca, F.; Grosso, J.; Jones, M.; Cohick, E.B.; et al. Identification of a cancer stem cell-specific function for the histone deacetylases, HDAC1 and HDAC7, in breast and ovarian cancer. Oncogene 2016, 36, 1707–1720. [Google Scholar] [CrossRef] [PubMed]
- Challen, G.A.; Sun, D.; Jeong, M.; Luo, M.; Jelinek, J.; Berg, J.S.; Bock, C.; Vasanthakumar, A.; Gu, H.; Xi, Y.; et al. Dnmt3a is essential for hematopoietic stem cell differentiation. Nat. Genet. 2011, 44, 23–31. [Google Scholar] [CrossRef] [PubMed]
- Leis, O.; Eguiara, A.; Lopez-Arribillaga, E.; Alberdi, M.J.; Hernandez-Garcia, S.; Elorriaga, K.; Pandiella, A.; Rezola, R.; Martin, A.G. Sox2 expression in breast tumours and activation in breast cancer stem cells. Oncogene 2011, 31, 1354–1365. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Liu, S.; Wang, P.; Zhao, S.; Wang, F.; Bing, L.; Zhang, Y.; Ling, E.A.; Gao, J.; Hao, A. Expression profile of embryonic stem cell-associated genes Oct4, Sox2 and Nanog in human gliomas. Histopathology 2011, 59, 763–775. [Google Scholar] [CrossRef] [PubMed]
- Jeter, C.R.; Liu, B.; Liu, X.; Chen, X.; Liu, C.; Calhoun-Davis, T.; Repass, J.; Zaehres, H.; Shen, J.J.; Tang, D.G. NANOG promotes cancer stem cell characteristics and prostate cancer resistance to androgen deprivation. Oncogene 2011, 30, 3833–3845. [Google Scholar] [CrossRef] [PubMed]
- Leung, E.L.; Fiscus, R.R.; Tung, J.W.; Tin, V.P.; Cheng, L.C.; Sihoe, A.D.; Fink, L.M.; Ma, Y.; Wong, M.P. Non-small cell lung cancer cells expressing CD44 are enriched for stem cell-like properties. PLoS ONE 2010, 5, e14062. [Google Scholar] [CrossRef] [PubMed]
- Baba, T.; Convery, P.A.; Matsumura, N.; Whitaker, R.S.; Kondoh, E.; Perry, T.; Huang, Z.; Bentley, R.C.; Mori, S.; Fujii, S.; et al. Epigenetic regulation of CD133 and tumorigenicity of CD133 + ovarian cancer cells. Oncogene 2009, 28, 209–218. [Google Scholar] [CrossRef] [PubMed]
- Tabu, K.; Sasai, K.; Kimura, T.; Wang, L.; Aoyanagi, E.; Kohsaka, S.; Tanino, M.; Nishihara, H.; Tanaka, S. Promoter hypomethylation regulates CD133 expression in human gliomas. Cell Res. 2008, 18, 1037–1046. [Google Scholar] [CrossRef] [PubMed]
- Yi, J.M.; Tsai, H.C.; Glockner, S.C.; Lin, S.; Ohm, J.E.; Easwaran, H.; James, C.D.; Costello, J.F.; Riggins, G.; Eberhart, C.G.; et al. Abnormal DNA methylation of CD133 in colorectal and glioblastoma tumors. Cancer Res. 2008, 68, 8094–8103. [Google Scholar] [CrossRef] [PubMed]
- You, H.; Ding, W.; Rountree, C.B. Epigenetic regulation of cancer stem cell marker CD133 by transforming growth factor-beta. Hepatology 2010, 51, 1635–1644. [Google Scholar] [CrossRef] [PubMed]
- Fouse, S.D.; Shen, Y.; Pellegrini, M.; Cole, S.; Meissner, A.; Van Neste, L.; Jaenisch, R.; Fan, G. Promoter CpG methylation contributes to ES cell gene regulation in parallel with Oct4/Nanog, PcG complex, and histone H3 K4/K27 trimethylation. Cell Stem Cell 2008, 2, 160–169. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Bi, C.; Cheong, L.L.; Mahara, S.; Liu, S.C.; Tay, K.G.; Koh, T.L.; Yu, Q.; Chng, W.J. The histone methyltransferase inhibitor, DZNep, up-regulates TXNIP, increases ROS production, and targets leukemia cells in AML. Blood 2011, 118, 2830–2839. [Google Scholar] [CrossRef] [PubMed]
- Chiba, T.; Suzuki, E.; Negishi, M.; Saraya, A.; Miyagi, S.; Konuma, T.; Tanaka, S.; Tada, M.; Kanai, F.; Imazeki, F.; et al. 3-Deazaneplanocin A is a promising therapeutic agent for the eradication of tumor-initiating hepatocellular carcinoma cells. Int. J. Cancer 2011, 130, 2557–2567. [Google Scholar] [CrossRef] [PubMed]
- Suva, M.L.; Riggi, N.; Janiszewska, M.; Radovanovic, I.; Provero, P.; Stehle, J.C.; Baumer, K.; Le Bitoux, M.A.; Marino, D.; Cironi, L.; et al. EZH2 is essential for glioblastoma cancer stem cell maintenance. Cancer Res. 2009, 69, 9211–9218. [Google Scholar] [CrossRef] [PubMed]
- Crea, F.; Hurt, E.M.; Mathews, L.A.; Cabarcas, S.M.; Sun, L.; Marquez, V.E.; Danesi, R.; Farrar, W.L. Pharmacologic disruption of Polycomb Repressive Complex 2 inhibits tumorigenicity and tumor progression in prostate cancer. Mol. Cancer 2011, 10, 40. [Google Scholar] [CrossRef] [PubMed]
- Pietersen, A.M.; van Lohuizen, M. Stem cell regulation by polycomb repressors: Postponing commitment. Curr. Opin. Cell Biol. 2008, 20, 201–207. [Google Scholar] [CrossRef] [PubMed]
- van Vlerken, L.E.; Kiefer, C.M.; Morehouse, C.; Li, Y.; Groves, C.; Wilson, S.D.; Yao, Y.; Hollingsworth, R.E.; Hurt, E.M. EZH2 is required for breast and pancreatic cancer stem cell maintenance and can be used as a functional cancer stem cell reporter. Stem Cells Transl. Med. 2013, 2, 43–52. [Google Scholar] [CrossRef] [PubMed]
- Rizzo, S.; Hersey, J.M.; Mellor, P.; Dai, W.; Santos-Silva, A.; Liber, D.; Luk, L.; Titley, I.; Carden, C.P.; Box, G.; et al. Ovarian cancer stem cell-like side populations are enriched following chemotherapy and overexpress EZH2. Mol. Cancer Ther. 2011, 10, 325–335. [Google Scholar] [CrossRef] [PubMed]
- Crea, F.; Hurt, E.M.; Farrar, W.L. Clinical significance of Polycomb gene expression in brain tumors. Mol. Cancer 2010, 9, 265. [Google Scholar] [CrossRef] [PubMed]
- Paranjape, A.N.; Balaji, S.A.; Mandal, T.; Krushik, E.V.; Nagaraj, P.; Mukherjee, G.; Rangarajan, A. Bmi1 regulates self-renewal and epithelial to mesenchymal transition in breast cancer cells through Nanog. BMC Cancer 2014, 14, 785. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Lu, F.; Ren, Q.; Sun, H.; Xu, Z.; Lan, R.; Liu, Y.; Ward, D.; Quan, J.; Ye, T.; et al. Novel histone demethylase LSD1 inhibitors selectively target cancer cells with pluripotent stem cell properties. Cancer Res. 2011, 71, 7238–7249. [Google Scholar] [CrossRef] [PubMed]
- Duru, N.; Gernapudi, R.; Eades, G.; Eckert, R.; Zhou, Q. Epigenetic Regulation of miRNAs and Breast Cancer Stem Cells. Curr. Pharmacol. Rep. 2015, 1, 161–169. [Google Scholar] [CrossRef] [PubMed]
- Nalls, D.; Tang, S.N.; Rodova, M.; Srivastava, R.K.; Shankar, S. Targeting epigenetic regulation of miR-34a for treatment of pancreatic cancer by inhibition of pancreatic cancer stem cells. PLoS ONE 2011, 6, e24099. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.Y.; Fu, J.; Xiao, X.; Wu, J.; Wu, R.C. MiR-34a regulates therapy resistance by targeting HDAC1 and HDAC7 in breast cancer. Cancer Lett. 2014, 354, 311–319. [Google Scholar] [CrossRef] [PubMed]
- Schwarzenbacher, D.; Balic, M.; Pichler, M. The role of microRNAs in breast cancer stem cells. Int. J. Mol. Sci. 2013, 14, 14712–14723. [Google Scholar] [CrossRef] [PubMed]
- Gong, C.; Tan, W.; Chen, K.; You, N.; Zhu, S.; Liang, G.; Xie, X.; Li, Q.; Zeng, Y.; Ouyang, N.; et al. Prognostic Value of a BCSC-associated MicroRNA Signature in Hormone Receptor-Positive HER2-Negative Breast Cancer. EBioMedicine 2016, 11, 199–209. [Google Scholar] [CrossRef] [PubMed]
- Shimono, Y.; Zabala, M.; Cho, R.W.; Lobo, N.; Dalerba, P.; Qian, D.; Diehn, M.; Liu, H.; Panula, S.P.; Chiao, E.; et al. Downregulation of miRNA-200c links breast cancer stem cells with normal stem cells. Cell 2009, 138, 592–603. [Google Scholar] [CrossRef] [PubMed]
- Pal, A.; Valdez, K.E.; Carletti, M.Z.; Behbod, F. Targeting the perpetrator: Breast cancer stem cell therapeutics. Curr. Drug Targets 2010, 11, 1147–1156. [Google Scholar] [CrossRef] [PubMed]
- Park, S.M.; Gaur, A.B.; Lengyel, E.; Peter, M.E. The miR-200 family determines the epithelial phenotype of cancer cells by targeting the E-cadherin repressors ZEB1 and ZEB2. Genes Dev. 2008, 22, 894–907. [Google Scholar] [CrossRef] [PubMed]
- Heo, I.; Joo, C.; Kim, Y.K.; Ha, M.; Yoon, M.J.; Cho, J.; Yeom, K.H.; Han, J.; Kim, V.N. TUT4 in concert with Lin28 suppresses microRNA biogenesis through pre-microRNA uridylation. Cell 2009, 138, 696–708. [Google Scholar] [CrossRef] [PubMed]
- Martello, G.; Rosato, A.; Ferrari, F.; Manfrin, A.; Cordenonsi, M.; Dupont, S.; Enzo, E.; Guzzardo, V.; Rondina, M.; Spruce, T.; et al. A MicroRNA targeting dicer for metastasis control. Cell 2010, 141, 1195–1207. [Google Scholar] [CrossRef] [PubMed]
- Mani, S.A.; Guo, W.; Liao, M.J.; Eaton, E.N.; Ayyanan, A.; Zhou, A.Y.; Brooks, M.; Reinhard, F.; Zhang, C.C.; Shipitsin, M.; et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 2008, 133, 704–715. [Google Scholar] [CrossRef] [PubMed]
- Dagdemir, A.; Judes, G.; Lebert, A.; Echegut, M.; Karsli-Ceppioglu, S.; Rifai, K.; Daures, M.; Ngollo, M.; Dubois, L.; Penault-Llorca, F.; et al. Epigenetic Modifications with DZNep, NaBu and SAHA in Luminal and Mesenchymal-like Breast Cancer Subtype Cells. Cancer Genom. Proteom. 2016, 13, 291–303. [Google Scholar]

© 2017 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abdel-Hafiz, H.A. Epigenetic Mechanisms of Tamoxifen Resistance in Luminal Breast Cancer. Diseases 2017, 5, 16. https://doi.org/10.3390/diseases5030016
Abdel-Hafiz HA. Epigenetic Mechanisms of Tamoxifen Resistance in Luminal Breast Cancer. Diseases. 2017; 5(3):16. https://doi.org/10.3390/diseases5030016
Chicago/Turabian StyleAbdel-Hafiz, Hany A. 2017. "Epigenetic Mechanisms of Tamoxifen Resistance in Luminal Breast Cancer" Diseases 5, no. 3: 16. https://doi.org/10.3390/diseases5030016
APA StyleAbdel-Hafiz, H. A. (2017). Epigenetic Mechanisms of Tamoxifen Resistance in Luminal Breast Cancer. Diseases, 5(3), 16. https://doi.org/10.3390/diseases5030016