
Renal Cell Carcinoma: A Study through NMR-Based Metabolomics Combined with Transcriptomics

 ,
,  ,
,  and
and
Abstract
:1. Introduction
2. Experimental Section
2.1. Sample Collection

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ccRCC-BN | ccRCC-AN | HC | |
|---|---|---|---|
| Number | 40 | 9 | 29 |
| Male/Female | 27/13 | 7/2 | 21/8 |
| Age (years) | 62.35 ± 11.65 | 64.11 ± 10.91 | 56 ± 5.81 |
| Histological classification | 18/40 G1 | 6/9 G1 | |
| 11/40 G2 | 3/9 G2 | ||
| 11/40 G3 | 0/9 G3 |
2.2. Sample Preparation
2.3. NMR Experiments
2.4. Data Processing and Analysis
2.5. Multivariate Statistical Analyses of 1H-NMR Data and Metabolite Identification
2.6. Metabolic Pathway Analysis
3. Results and Discussion
3.1. Results
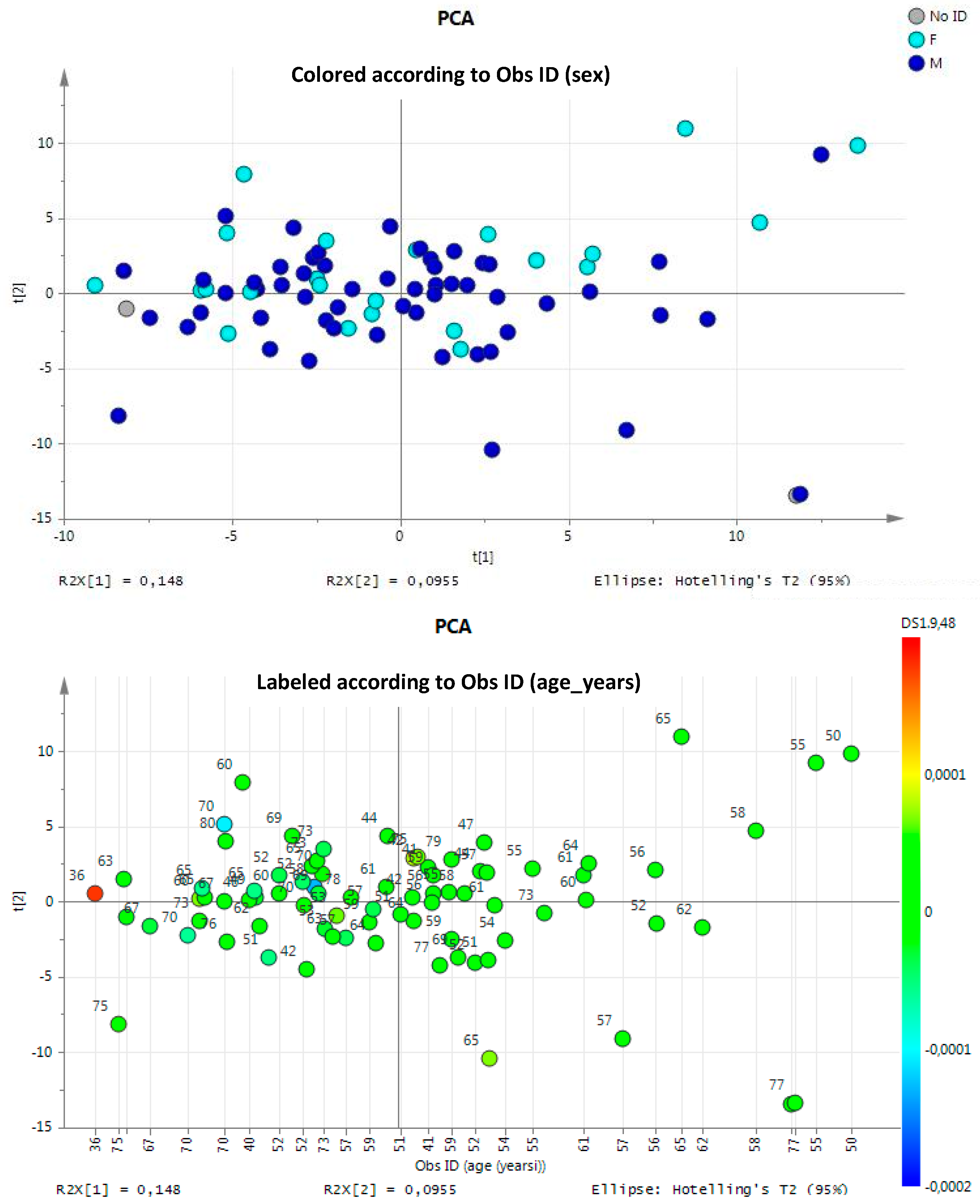
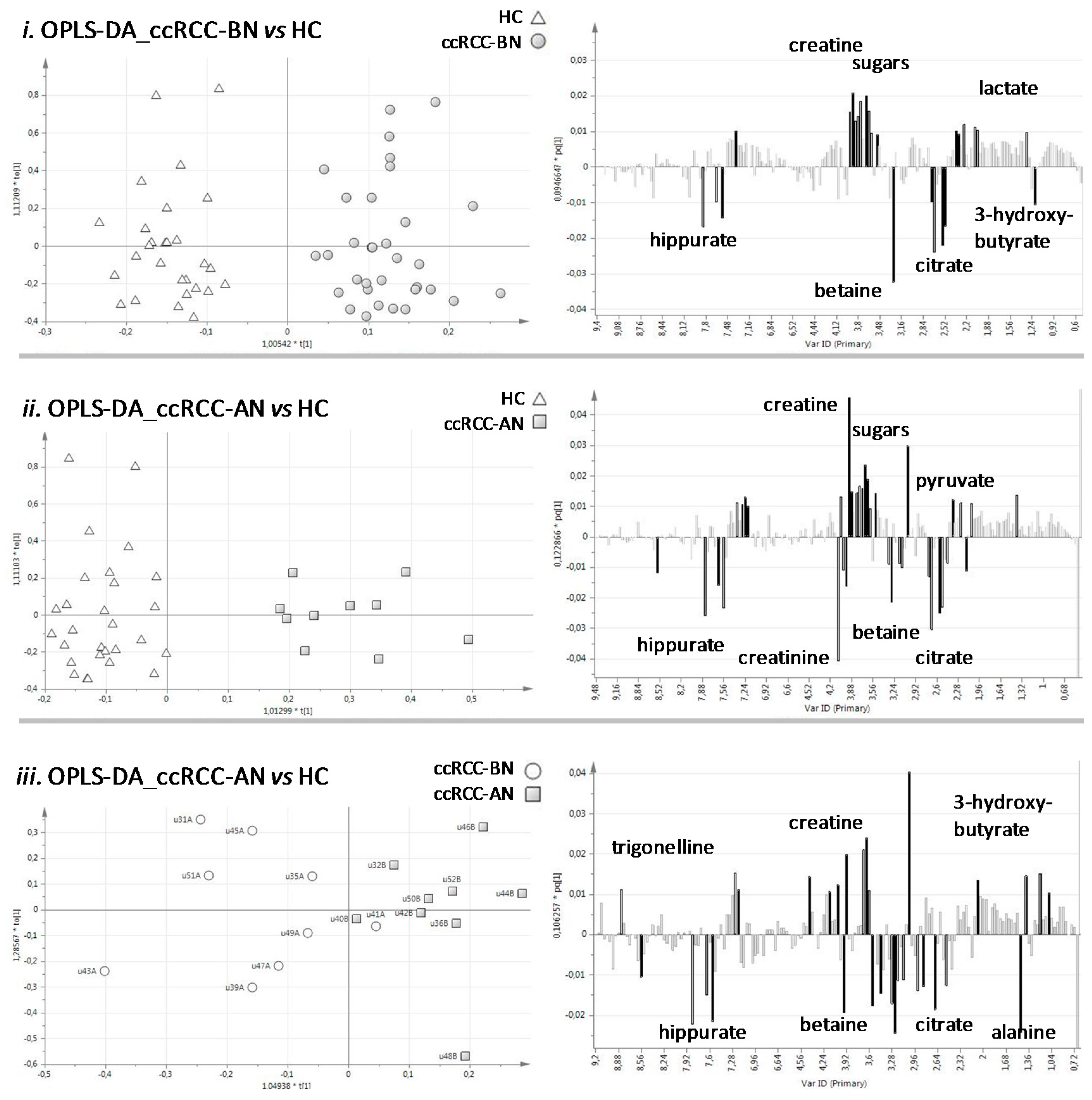
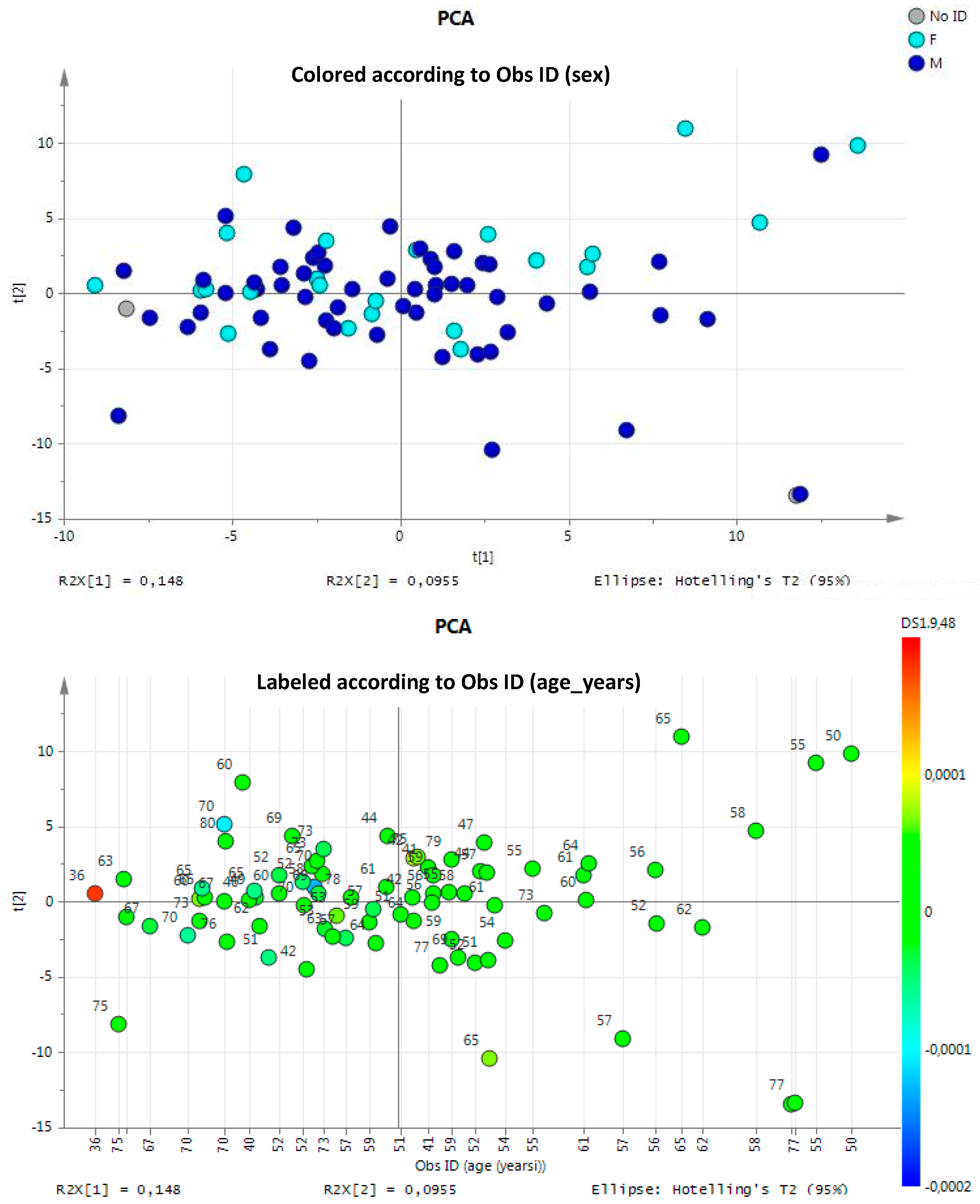
3.1.1. Multivariate Statistical Analysis of Urine NMR Profiles
| OPLS-DA | ccRCC-BN vs. HC | ccRCC-AN vs. HC | ccRCC-BN vs. ccRCC-AN | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Component | R2X(cum) | R2(cum) | Q2(cum) | R2X(cum) | R2(cum) | Q2(cum) | R2X(cum) | R2(cum) | Q2(cum) |
| Model | 0.474 | 0.765 | 0.615 | 0.429 | 0.874 | 0.685 | 0.256 | 0.699 | −0.267 |
| Predictive | 0.077 | 0.765 | 0.615 | 0.106 | 0.874 | 0.685 | 0.102 | 0.699 | −0.267 |
| P1 | 0.077 | 0.765 | 0.615 | 0.106 | 0.874 | 0.685 | 0.102 | 0.699 | −0.267 |
| Orthogonal in X | 0.397 | 0 | 0.323 | 0 | 0.155 | 0 | |||
| O1 | 0.305 | 0 | 0.234 | 0 | 0.155 | 0 | |||
| O2 | 0.397 | 0 | 0.323 | 0 | |||||
| Specificity | 0.893 | 0.964 | 0.778 | ||||||
| Sensitivity | 0.939 | 0.9 | 0.444 | ||||||
| Accuracy | 0.918 | 0.947 | 0.611 | ||||||
| Cohen’s K in cross-validation | 0.835 | 0.864 | 0.222 | ||||||
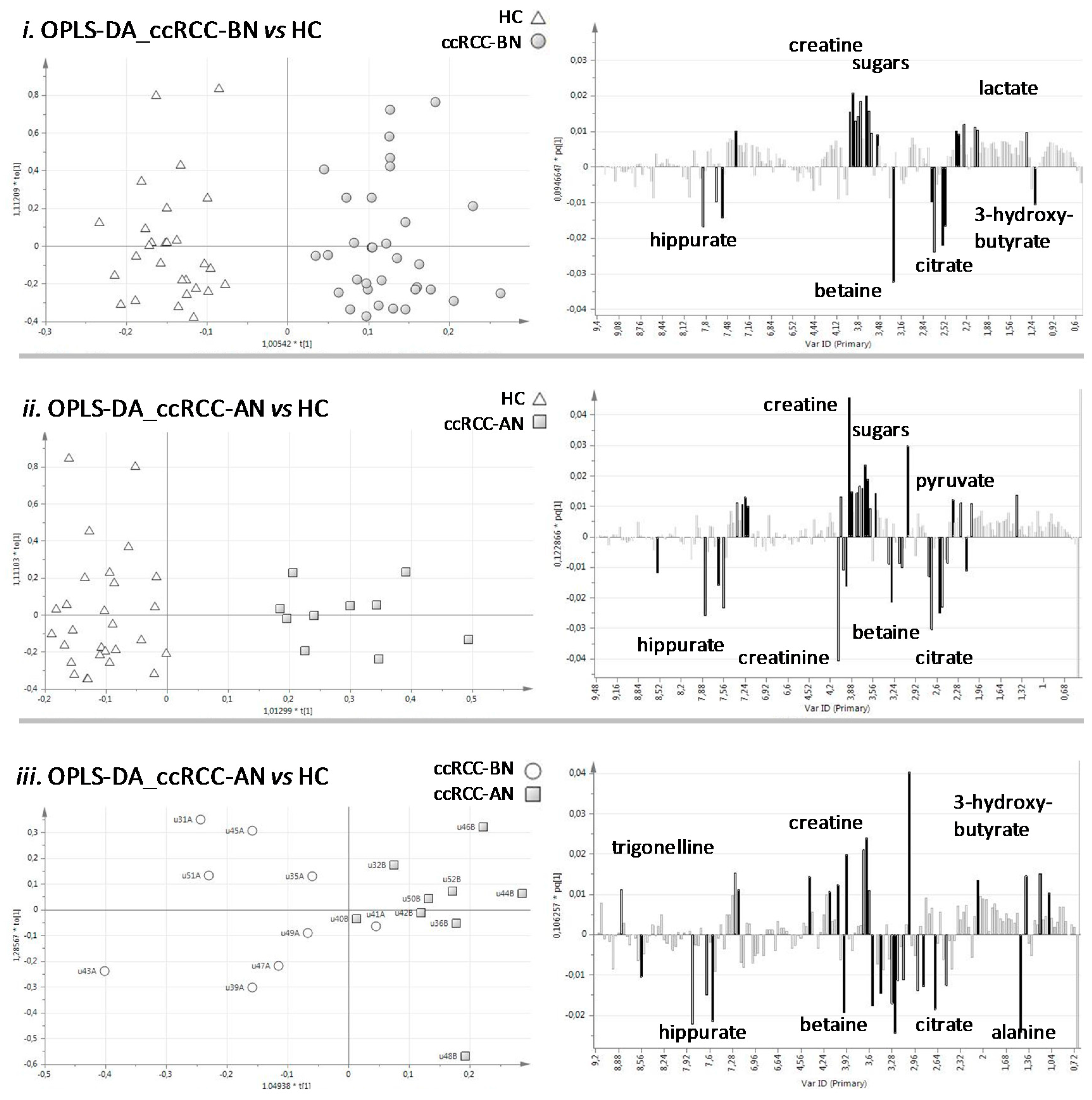
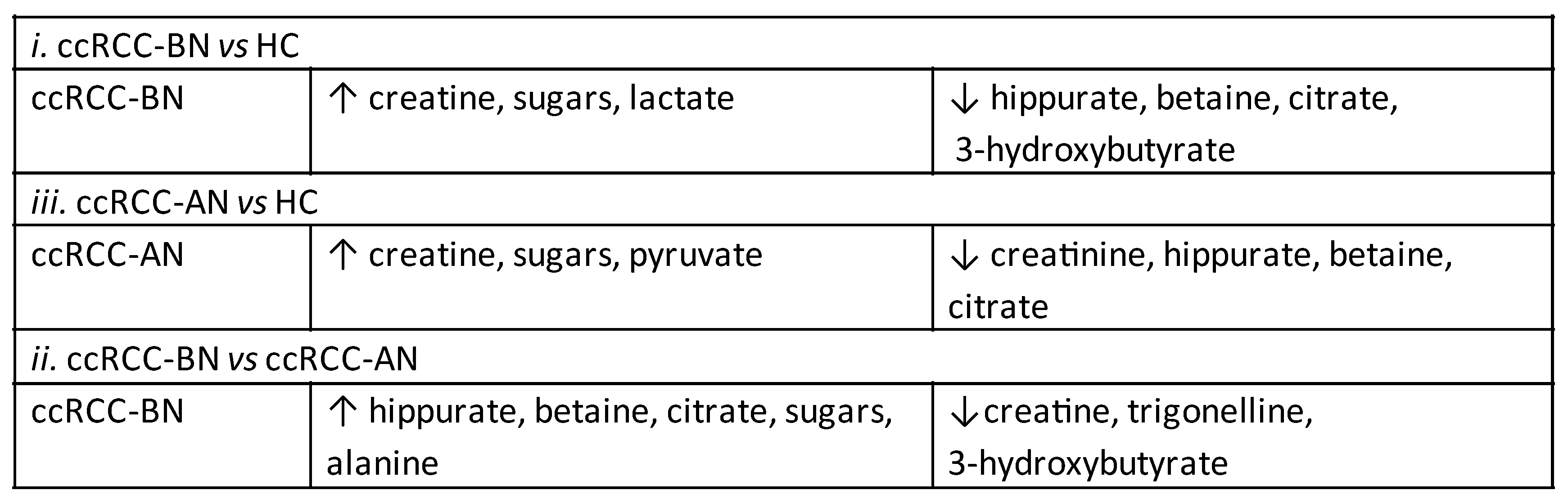
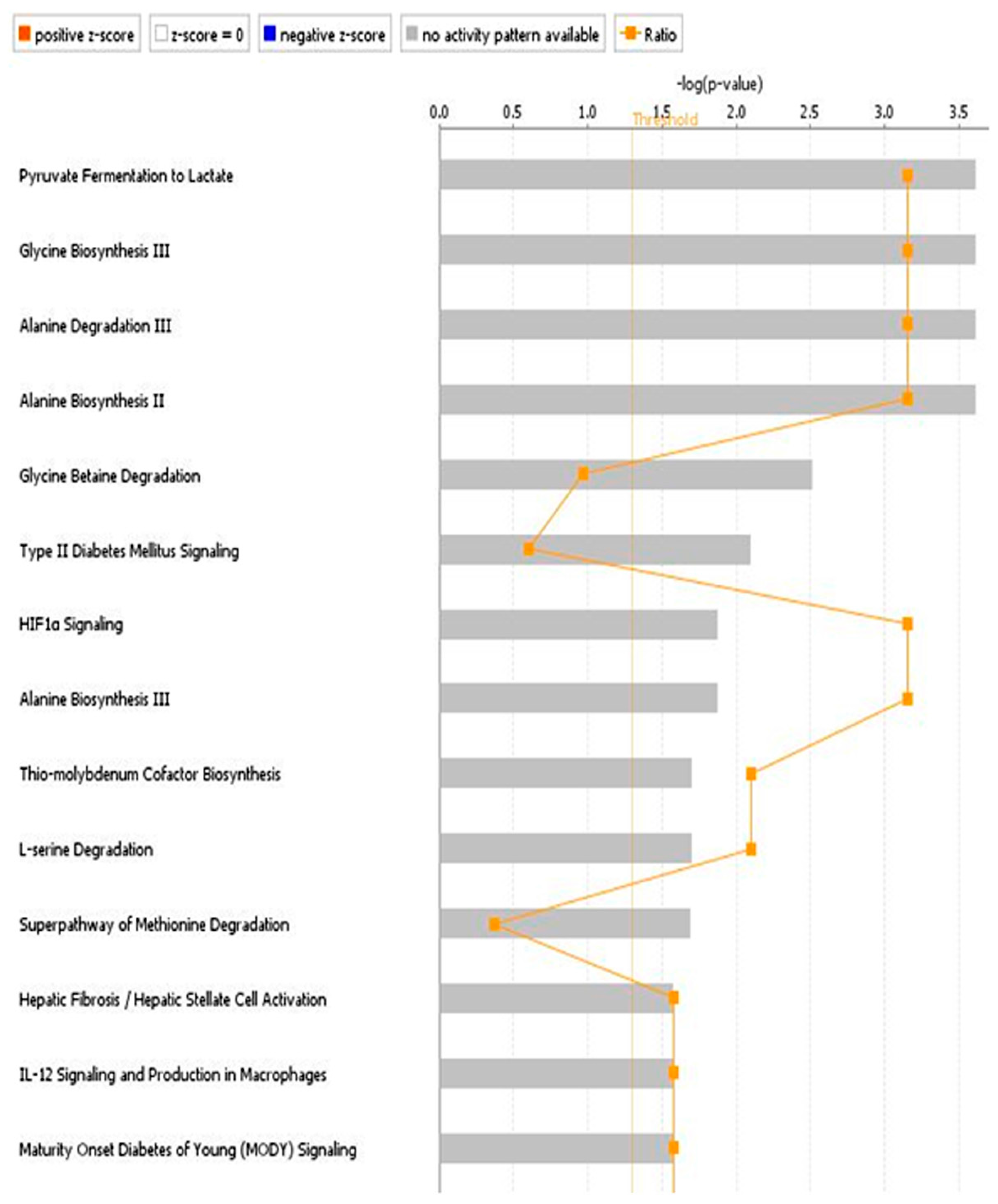
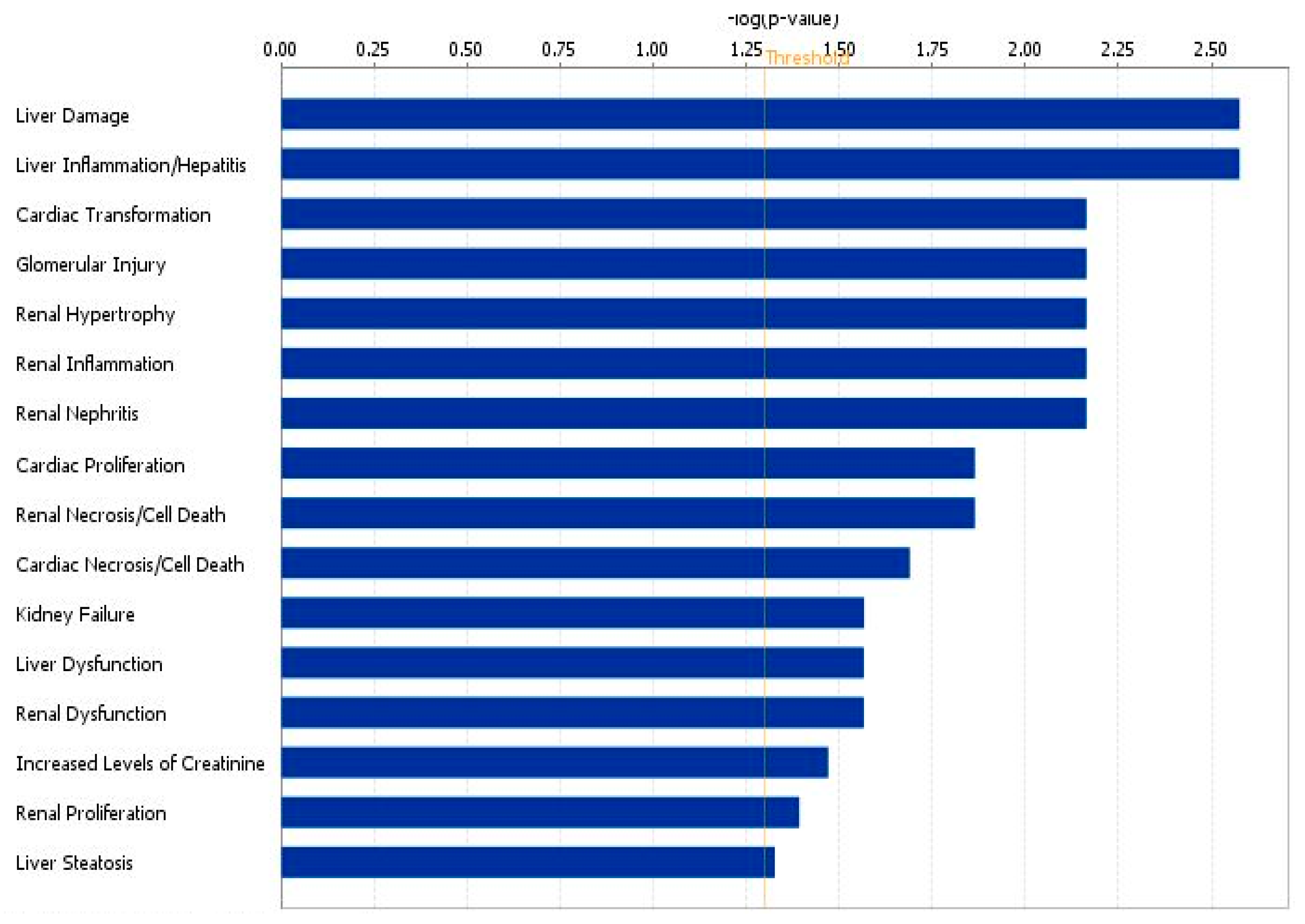
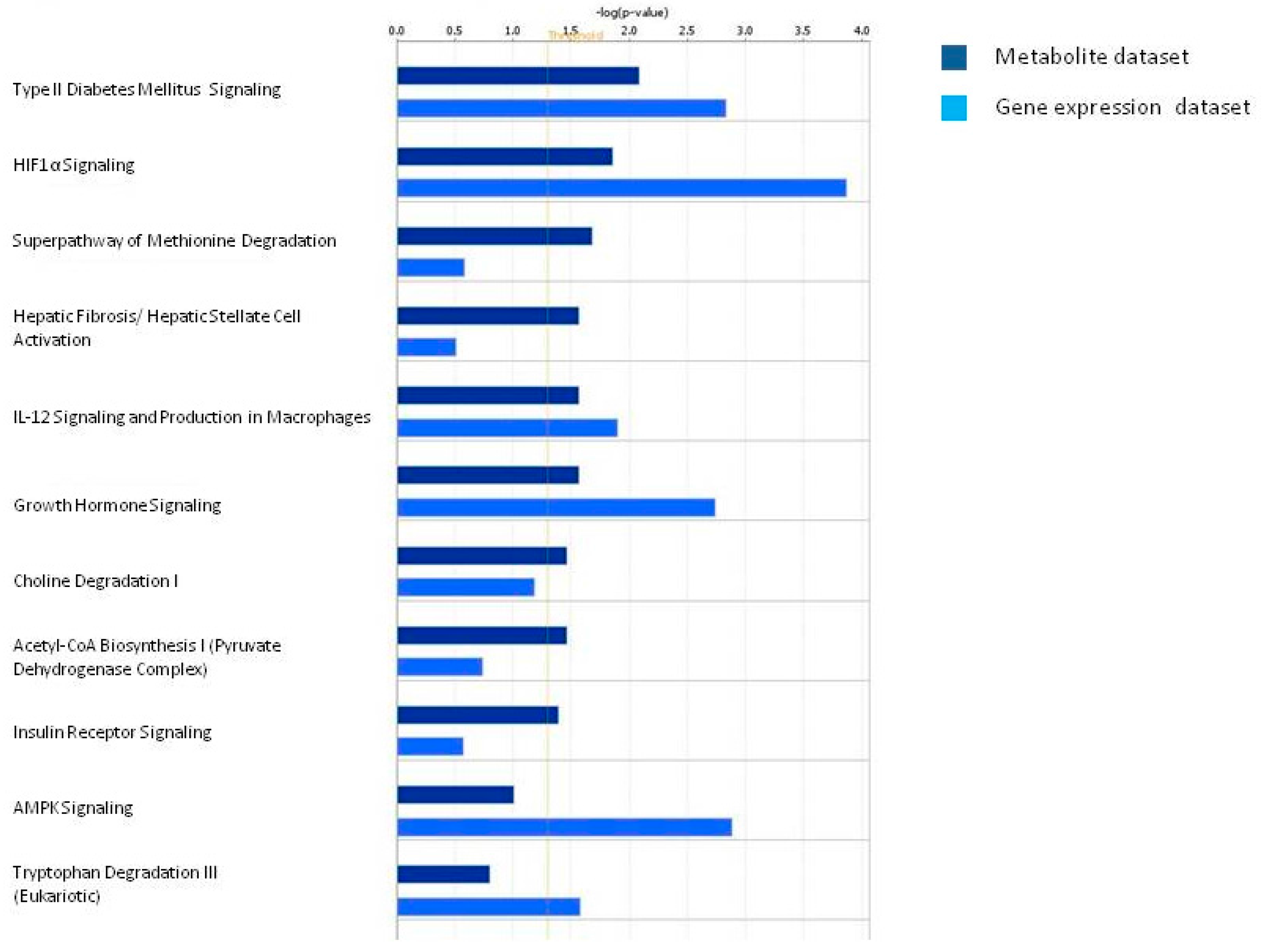
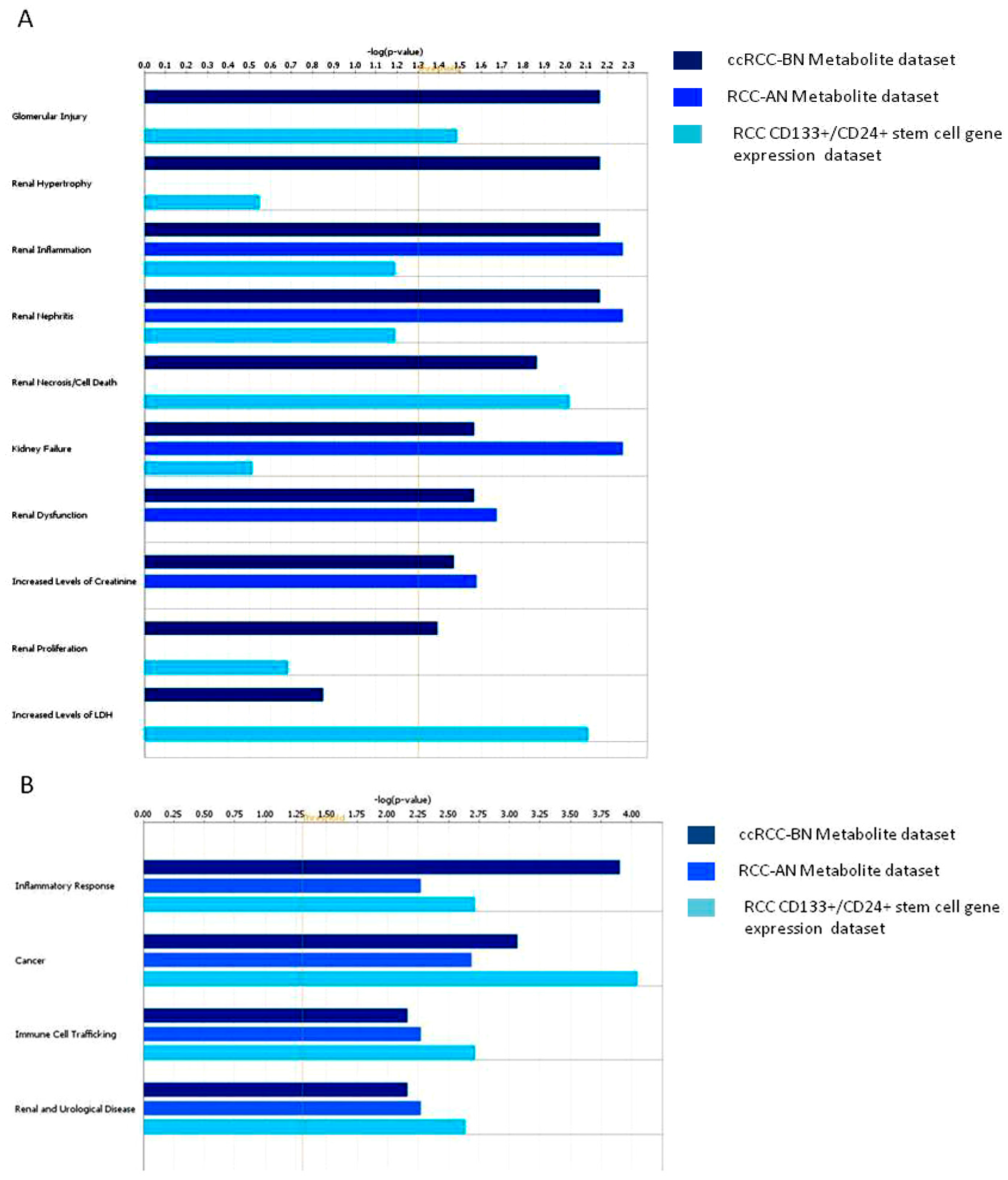
3.1.2. Network Analysis
| Metabolites | ppm |
|---|---|
| Trigonelline | 9.13 |
| Hippurate | 7.84 |
| N-phenylacetylglycine | 7.40 |
| Sucrose | 5.25 |
| Glucose | 4.65 |
| Creatinine | 4.06 |
| Creatine | 3.93 |
| Glycine | 3.58 |
| Carnitine | 3.23 |
| Betaine | 3.28 |
| Citrate | 2.55 |
| Pyruvate | 2.35 |
| Alanine | 1.49 |
| Lactate | 1.34 |
| 3-hydroxybutyrate | 1.20 |
| 3-hydroxyisobutyrate | 1.37 |
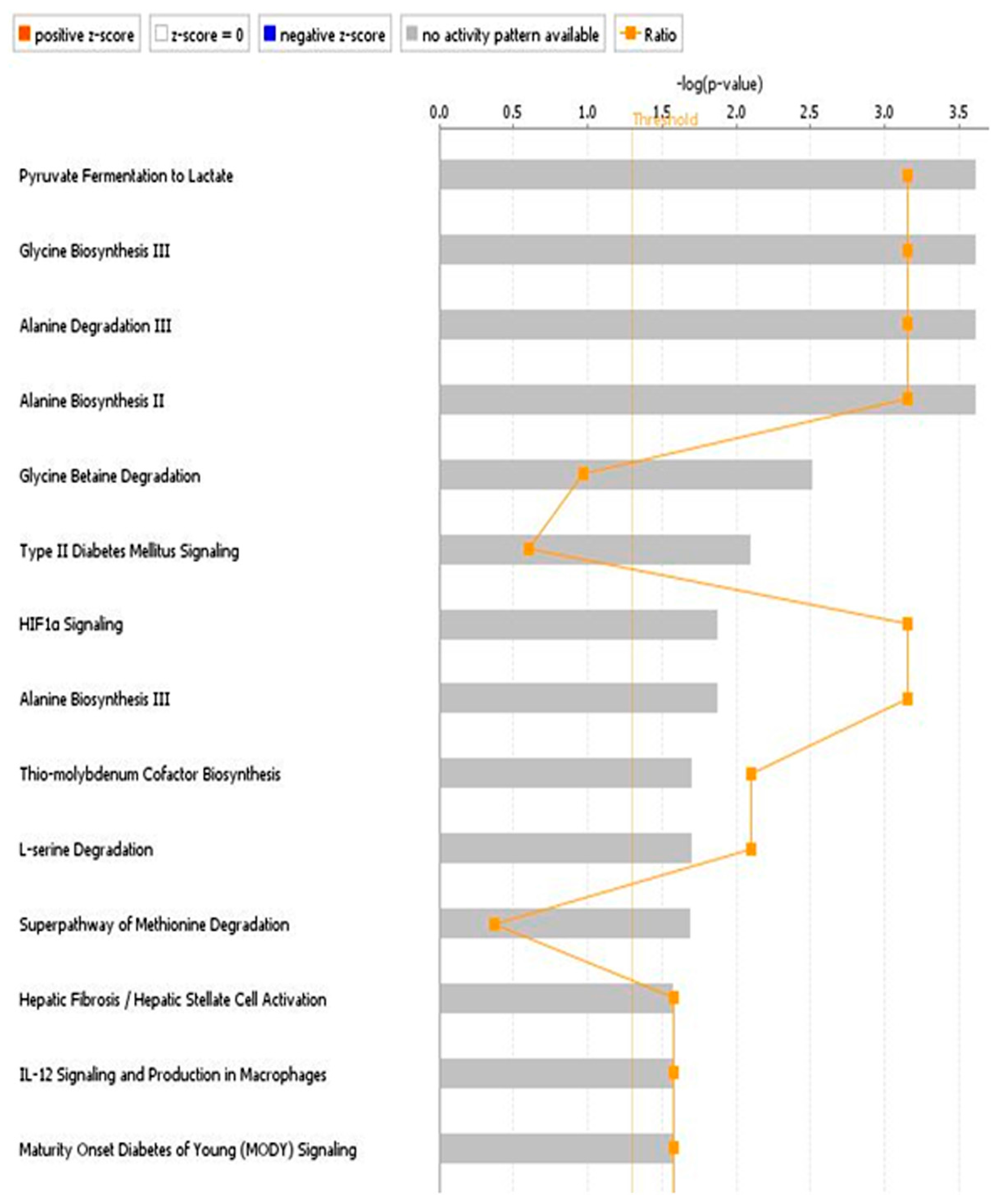
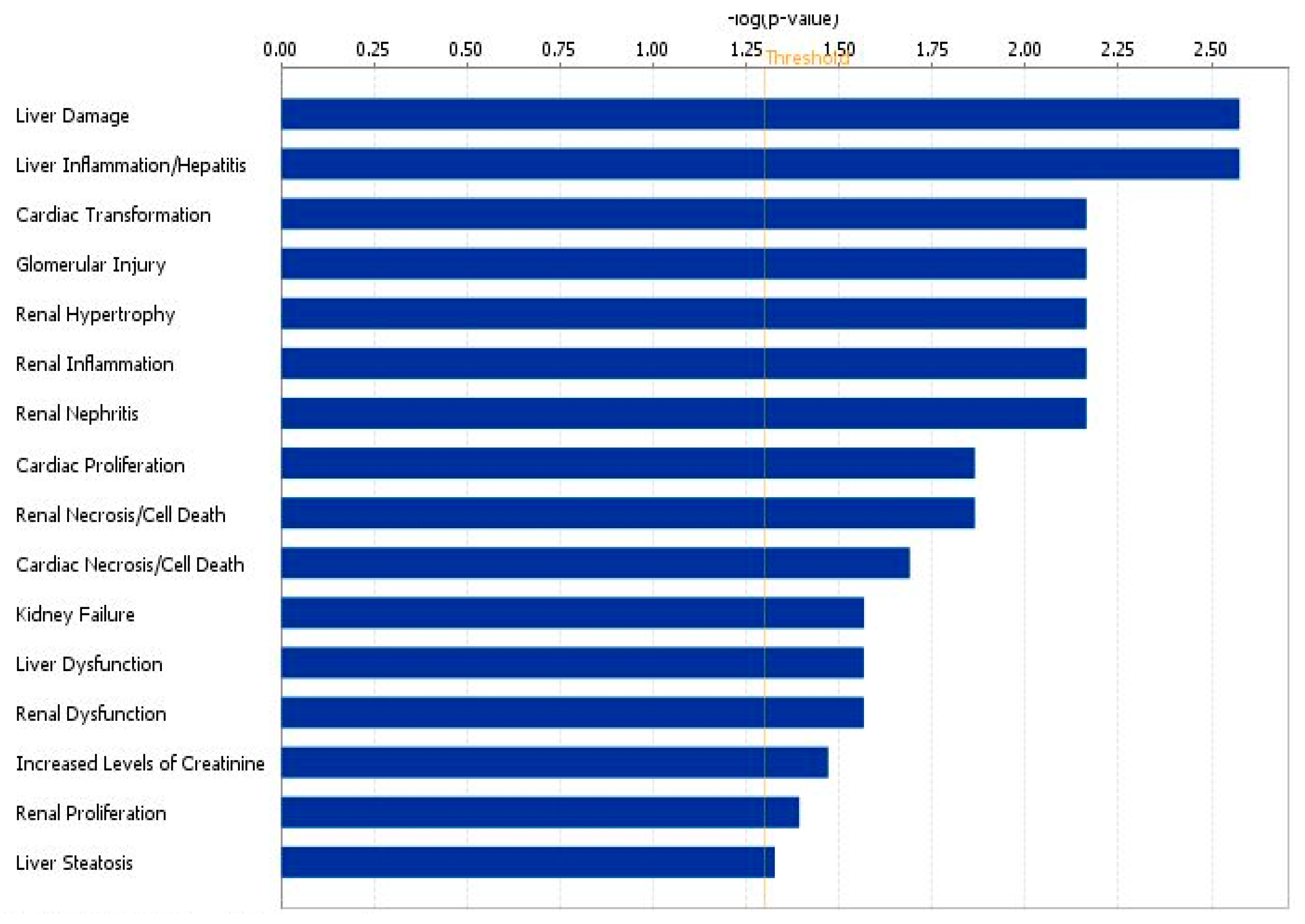
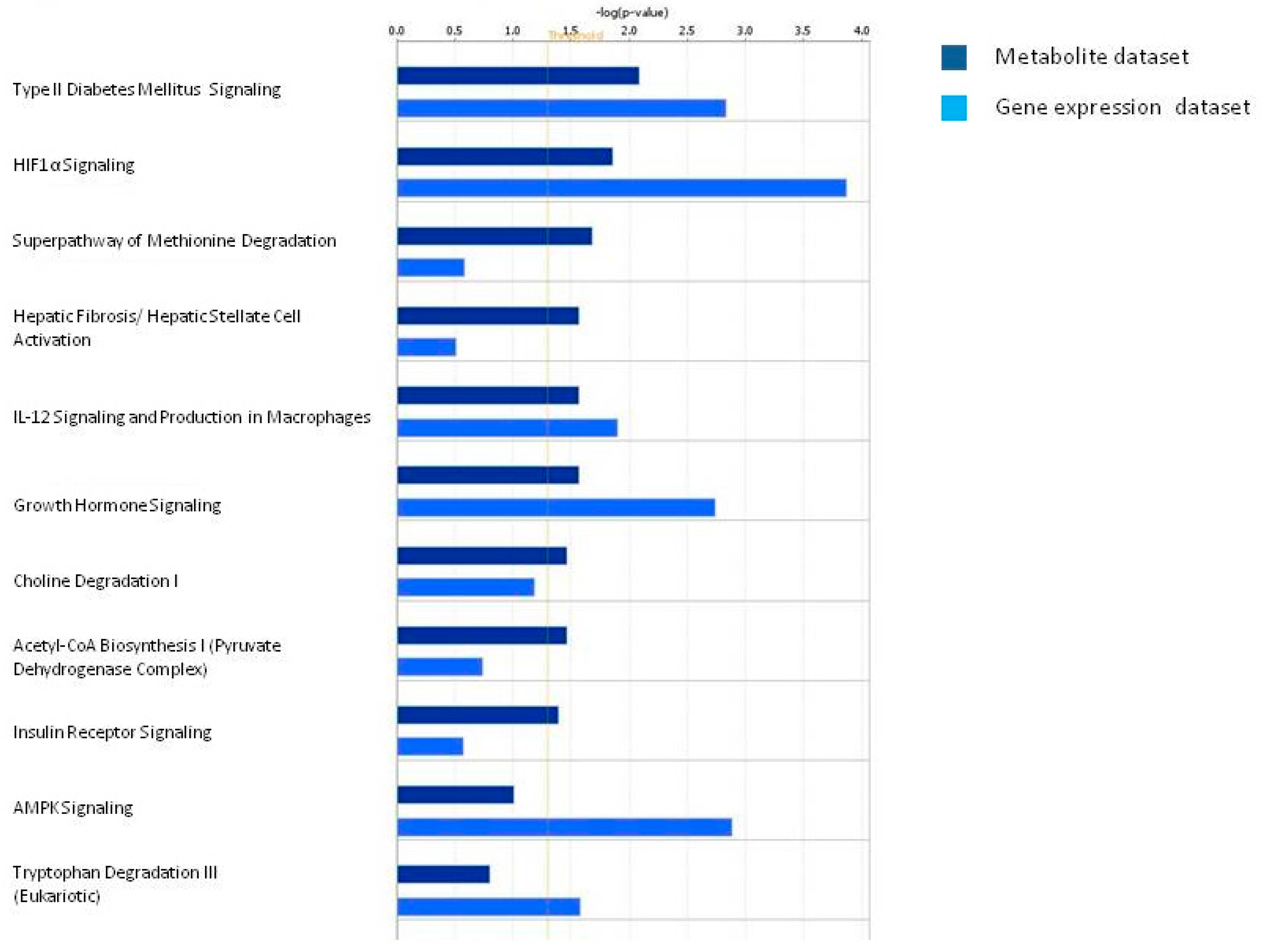
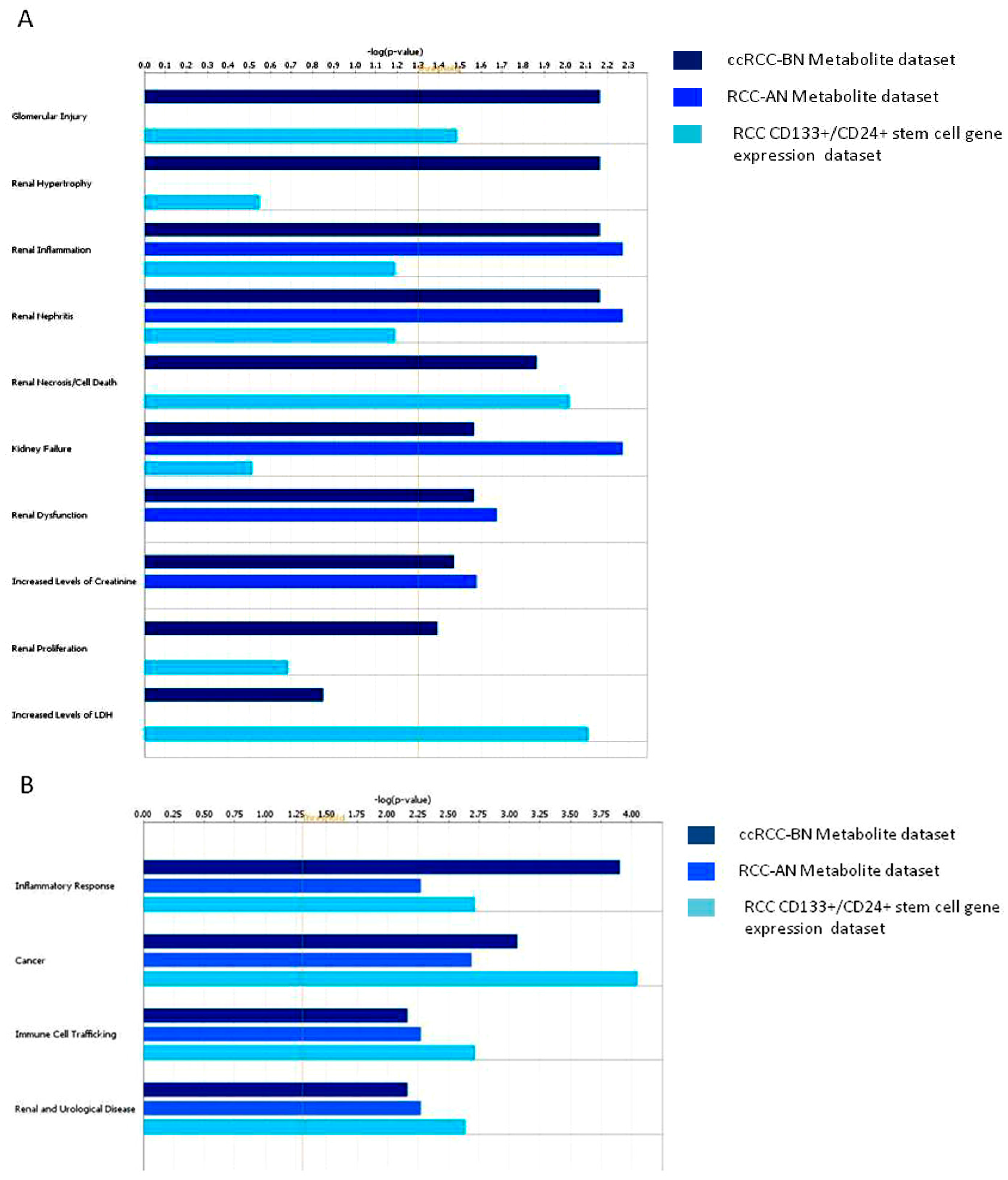
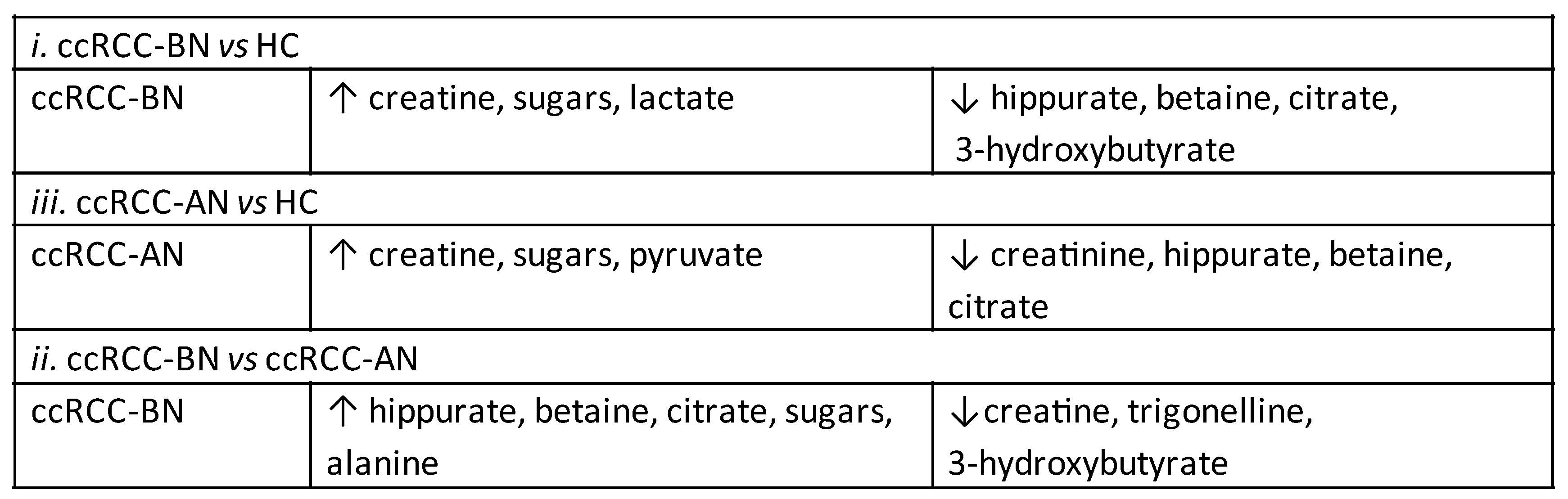
3.2. Discussion
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Ljungberg, B.; Campbell, S.C.; Choi, H.Y.; Jacqmin, D.; Lee, J.E.; Weikert, S.; Kiemeney, L.A. The epidemiology of renal cell carcinoma. Eur. Urol. 2011, 60, 615–621. [Google Scholar] [CrossRef] [PubMed]
- Figlin, R.A. Renal cell carcinoma: Management of advanced disease. J. Urol. 1999, 161, 381–387. [Google Scholar] [CrossRef]
- Furniss, D.; Harnden, P.; Ali, N.; Royston, P.; Eisen, T.; Oliver, R.T.; Hancock, B.W. Prognostic factors for renal cell carcinoma. Cancer Treat. Rev. 2008, 34, 407–426. [Google Scholar] [CrossRef] [PubMed]
- Mancini, V.; Battaglia, M.; Ditonno, P.; Palazzo, S.; Lastilla, G.; Montironi, R.; Bettocchi, C.; Cavalcanti, E.; Ranieri, E.; Selvaggi, F.P. Current insights in renal cell cancer pathology. Urol. Oncol. 2008, 26, 225–238. [Google Scholar] [CrossRef] [PubMed]
- Zira, A.N.; Theocharis, S.E.; Mitropoulos, D.; Migdalis, V.; Mikros, E. (1)H-NMR metabonomic analysis in renal cell carcinoma: A possible diagnostic tool. J. Prot. Res. 2010, 9, 4038–4044. [Google Scholar] [CrossRef] [PubMed]
- Walsh, M.C.; Nugent, A.; Brennan, L.; Gibney, M.J. Understanding the metabolome—Challenges for metabolomics. Nutr. Bull. 2008, 33, 316–323. [Google Scholar] [CrossRef]
- Takahashi, M.; Rhodes, D.R.; Furge, K.A.; Kanayama, H.; Kagawa, S.; Haab, B.B.; Teh, B.T. Gene expression profiling of clear cell renal cell carcinoma: Gene identification and prognostic classification. Proc. Natl. Acad. Sci. USA 2001, 98, 9754–9759. [Google Scholar] [CrossRef] [PubMed]
- Jones, J.; Otu, H.; Spentzos, D.; Kolia, S.; Inan, M.; Beecken, W.D.; Fellbaum, C.; Gu, X.; Joseph, M.; Pantuck, A.J.; et al. Gene signatures of progression and metastasis in renal cell cancer. Clin. Cancer Res. 2005, 11, 5730–5739. [Google Scholar] [CrossRef] [PubMed]
- Ibanez de Caceres, I.; Dulaimi, E.; Hoffman, A.M.; Al-Saleem, T.; Uzzo, R.G.; Cairns, P. Identification of novel target genes by an epigenetic reactivation screen of renal cancer. Cancer Res. 2006, 66, 5021–5028. [Google Scholar] [CrossRef] [PubMed]
- Jung, M.; Mollenkopf, H.J.; Grimm, C.; Wagner, I.; Albrecht, M.; Waller, T.; Pilarsky, C.; Johannsen, M.; Stephan, C.; Lehrach, H.; et al. Microrna profiling of clear cell renal cell cancer identifies a robust signature to define renal malignancy. J. Cell. Mol. Med. 2009, 13, 3918–3928. [Google Scholar] [CrossRef] [PubMed]
- Nakada, C.; Matsuura, K.; Tsukamoto, Y.; Tanigawa, M.; Yoshimoto, T.; Narimatsu, T.; Nguyen, L.T.; Hijiya, N.; Uchida, T.; Sato, F.; et al. Genome-wide microrna expression profiling in renal cell carcinoma: Significant down-regulation of mir-141 and mir-200c. J. Pathol. 2008, 216, 418–427. [Google Scholar] [CrossRef] [PubMed]
- Sato, Y.; Yoshizato, T.; Shiraishi, Y.; Maekawa, S.; Okuno, Y.; Kamura, T.; Shimamura, T.; Sato-Otsubo, A.; Nagae, G.; Suzuki, H.; et al. Integrated molecular analysis of clear-cell renal cell carcinoma. Nat. Genet. 2013, 45, 860–867. [Google Scholar] [CrossRef] [PubMed]
- Crispen, P.L.; Boorjian, S.A.; Lohse, C.M.; Leibovich, B.C.; Kwon, E.D. Predicting disease progression after nephrectomy for localized renal cell carcinoma: The utility of prognostic models and molecular biomarkers. Cancer 2008, 113, 450–460. [Google Scholar] [CrossRef] [PubMed]
- Nogueira, M.; Kim, H.L. Molecular markers for predicting prognosis of renal cell carcinoma. Urol. Oncol. 2008, 26, 113–124. [Google Scholar] [CrossRef] [PubMed]
- Bollard, M.E.; Holmes, E.; Lindon, J.C.; Mitchell, S.C.; Branstetter, D.; Zhang, W.; Nicholson, J.K. Investigations into biochemical changes due to diurnal variation and estrus cycle in female rats using high-resolution (1)H-NMR spectroscopy of urine and pattern recognition. Anal. Biochem. 2001, 295, 194–202. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.; Dong, B.; Liu, X.; Xuan, H.; Huang, Y.; Lin, D. Metabonomic profiling of renal cell carcinoma: High-resolution proton nuclear magnetic resonance spectroscopy of human serum with multivariate data analysis. Anal. Chim. Acta 2008, 624, 269–277. [Google Scholar] [CrossRef] [PubMed]
- Makinen, V.P.; Soininen, P.; Forsblom, C.; Parkkonen, M.; Ingman, P.; Kaski, K.; Groop, P.H.; Ala-Korpela, M. Diagnosing diabetic nephropathy by 1H-NMR metabonomics of serum. MAGMA 2006, 19, 281–296. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Y.; Cai, G.; Su, M.; Chen, T.; Liu, Y.; Xu, Y.; Ni, Y.; Zhao, A.; Cai, S.; Xu, L.X.; et al. Urinary metabonomic study on colorectal cancer. J. Prot. Res. 2010, 9, 1627–1634. [Google Scholar] [CrossRef] [PubMed]
- Catchpole, G.; Platzer, A.; Weikert, C.; Kempkensteffen, C.; Johannsen, M.; Krause, H.; Jung, K.; Miller, K.; Willmitzer, L.; Selbig, J.; et al. Metabolic profiling reveals key metabolic features of renal cell carcinoma. J. Cell. Mol. Med. 2011, 15, 109–118. [Google Scholar] [CrossRef] [PubMed]
- Kind, T.; Tolstikov, V.; Fiehn, O.; Weiss, R.H. A comprehensive urinary metabolomic approach for identifying kidney cancerr. Anal. Biochem. 2007, 363, 185–195. [Google Scholar] [CrossRef] [PubMed]
- Perroud, B.; Lee, J.; Valkova, N.; Dhirapong, A.; Lin, P.Y.; Fiehn, O.; Kultz, D.; Weiss, R.H. Pathway analysis of kidney cancer using proteomics and metabolic profiling. Mol. Cancer 2006, 5, 136–141. [Google Scholar] [CrossRef] [PubMed]
- Galleggiante, V.; Rutigliano, M.; Sallustio, F.; Ribatti, D.; Ditonno, P.; Bettocchi, C.; Selvaggi, F.P.; Lucarelli, G.; Battaglia, M. Ctr2 identifies a population of cancer cells with stem cell-like features in patients with clear cell renal cell carcinoma. J. Urol. 2014, 192, 1831–1841. [Google Scholar] [CrossRef] [PubMed]
- Pelantova, H.; Buganova, M.; Anyz, J.; Zelezna, B.; Maletinska, L.; Novak, D.; Haluzik, M.; Kuzma, M. Strategy for nmr metabolomic analysis of urine in mouse models of obesity--from sample collection to interpretation of acquired data. J. Pharm. Biomed. Anal. 2015, 115, 225–235. [Google Scholar] [CrossRef] [PubMed]
- Schicho, R.; Shaykhutdinov, R.; Ngo, J.; Nazyrova, A.; Schneider, C.; Panaccione, R.; Kaplan, G.G.; Vogel, H.J.; Storr, M. Quantitative metabolomic profiling of serum, plasma, and urine by (1)H-NMR spectroscopy discriminates between patients with inflammatory bowel disease and healthy individuals. J. Prot. Res. 2012, 11, 3344–3357. [Google Scholar] [CrossRef] [PubMed]
- Eriksson, L.B.T.; Johansson, E.; Trygg, J.; Vikstrom, C. Multi- and megavariate data analysis. Technometrics 2003, 45. [Google Scholar] [CrossRef]
- Clark, W.R.; Mueller, B.A.; Kraus, M.A.; Macias, W.L. Quantification of creatinine kinetic parameters in patients with acute renal failure. Kidney Int. 1998, 54, 554–560. [Google Scholar] [CrossRef] [PubMed]
- Gualano, B.; Roschel, H.; Lancha, A.H., Jr.; Brightbill, C.E.; Rawson, E.S. In sickness and in health: The widespread application of creatine supplementation. Amino Acids 2012, 43, 519–529. [Google Scholar] [CrossRef] [PubMed]
- Nicholls, A.W.; Mortishire-Smith, R.J.; Nicholson, J.K. NMR spectroscopic-based metabonomic studies of urinary metabolite variation in acclimatizing germ-free rats. Chem. Res. Toxicol. 2003, 16, 1395–1404. [Google Scholar] [CrossRef] [PubMed]
- Williams, R.E.; Eyton-Jones, H.W.; Farnworth, M.J.; Gallagher, R.; Provan, W.M. Effect of intestinal microflora on the urinary metabolic profile of rats: A (1)h-nuclear magnetic resonance spectroscopy study. Xenobiotica 2002, 32, 783–794. [Google Scholar] [CrossRef] [PubMed]
- Lundina, T.A.; Knubovets, T.L.; Sedov, K.R.; Markova, S.A.; Sibeldin, L.A. Variability of kidney tubular interstitial distortions in glomerulonephritis as measured by 1h-nmr urinalysis. Clin. Chim. Acta 1993, 214, 165–173. [Google Scholar] [CrossRef]
- Christians, U.; Schmitz, V.; Schoning, W.; Bendrick-Peart, J.; Klawitter, J.; Haschke, M. Toxicodynamic therapeutic drug monitoring of immunosuppressants: Promises, reality, and challenges. Ther. Drug Monit. 2008, 30, 151–158. [Google Scholar] [CrossRef] [PubMed]
- Romick-Rosendale, L.E.; Brunner, H.I.; Bennett, M.R.; Mina, R.; Nelson, S.; Petri, M.; Kiani, A.; Devarajan, P.; Kennedy, M.A. Identification of urinary metabolites that distinguish membranous lupus nephritis from proliferative lupus nephritis and focal segmental glomerulosclerosis. Arthritis Res. Ther. 2011, 13, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Del Coco, L.; Assfalg, M.; D’Onofrio, M.; Sallustio, F.; Pesce, F.; Fanizzi, F.P.; Schena, F.P. A proton nuclear magnetic resonance-based metabolomic approach in iga nephropathy urinary profiles. Metabolomics 2013, 9, 740–751. [Google Scholar] [CrossRef]
- Psihogios, N.G.; Kalaitzidis, R.G.; Dimou, S.; Seferiadis, K.I.; Siamopoulos, K.C.; Bairaktari, E.T. Evaluation of tubulointerstitial lesions' severity in patients with glomerulonephritides: An nmr-based metabonomic study. J. Prot. Res. 2007, 6, 3760–3770. [Google Scholar] [CrossRef] [PubMed]
- Walenta, S.; Schroeder, T.; Mueller-Klieser, W. Lactate in solid malignant tumors: Potential basis of a metabolic classification in clinical oncology. Curr. Med. Chem. 2004, 11, 2195–2204. [Google Scholar] [CrossRef] [PubMed]
- Beck Gooz, M.; Maldonado, E.N.; Dang, Y.; Amria, M.Y.; Higashiyama, S.; Abboud, H.E.; Lemasters, J.J.; Bell, P.D. Adam17 promotes proliferation of collecting duct kidney epithelial cells through erk activation and increased glycolysis in polycystic kidney disease. Am. J. Physiol. Renal Physiol. 2014, 307, F551–F559. [Google Scholar] [CrossRef] [PubMed]
- Munoz-Clares, R.A.; Diaz-Sanchez, A.G.; Gonzalez-Segura, L.; Montiel, C. Kinetic and structural features of betaine aldehyde dehydrogenases: Mechanistic and regulatory implications. Arch. Biochem. Biophys. 2010, 493, 71–81. [Google Scholar] [CrossRef] [PubMed]
- Lever, M.; Slow, S. The clinical significance of betaine, an osmolyte with a key role in methyl group metabolism. Clin. Biochem. 2010, 43, 732–744. [Google Scholar] [CrossRef] [PubMed]
- Ema, M.; Hirota, K.; Mimura, J.; Abe, H.; Yodoi, J.; Sogawa, K.; Poellinger, L.; Fujii-Kuriyama, Y. Molecular mechanisms of transcription activation by hlf and hif1alpha in response to hypoxia: Their stabilization and redox signal-induced interaction with cbp/p300. EMBO J. 1999, 18, 1905–1914. [Google Scholar] [PubMed]
- Tendler, D.S.; Bao, C.; Wang, T.; Huang, E.L.; Ratovitski, E.A.; Pardoll, D.A.; Lowenstein, C.J. Intersection of interferon and hypoxia signal transduction pathways in nitric oxide-induced tumor apoptosis. Cancer Res. 2001, 61, 3682–3688. [Google Scholar] [PubMed]
- Zorzano, A.; Sevilla, L.; Camps, M.; Becker, C.; Meyer, J.; Kammermeier, H.; Munoz, P.; Guma, A.; Testar, X.; Palacin, M.; et al. Regulation of glucose transport, and glucose transporters expression and trafficking in the heart: Studies in cardiac myocytes. Am. J. Cardiol. 1997, 80, 65A–76A. [Google Scholar] [PubMed]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ragone, R.; Sallustio, F.; Piccinonna, S.; Rutigliano, M.; Vanessa, G.; Palazzo, S.; Lucarelli, G.; Ditonno, P.; Battaglia, M.; Fanizzi, F.P.; et al. Renal Cell Carcinoma: A Study through NMR-Based Metabolomics Combined with Transcriptomics. Diseases 2016, 4, 7. https://doi.org/10.3390/diseases4010007
Ragone R, Sallustio F, Piccinonna S, Rutigliano M, Vanessa G, Palazzo S, Lucarelli G, Ditonno P, Battaglia M, Fanizzi FP, et al. Renal Cell Carcinoma: A Study through NMR-Based Metabolomics Combined with Transcriptomics. Diseases. 2016; 4(1):7. https://doi.org/10.3390/diseases4010007
Chicago/Turabian StyleRagone, Rosa, Fabio Sallustio, Sara Piccinonna, Monica Rutigliano, Galleggiante Vanessa, Silvano Palazzo, Giuseppe Lucarelli, Pasquale Ditonno, Michele Battaglia, Francesco Paolo Fanizzi, and et al. 2016. "Renal Cell Carcinoma: A Study through NMR-Based Metabolomics Combined with Transcriptomics" Diseases 4, no. 1: 7. https://doi.org/10.3390/diseases4010007
APA StyleRagone, R., Sallustio, F., Piccinonna, S., Rutigliano, M., Vanessa, G., Palazzo, S., Lucarelli, G., Ditonno, P., Battaglia, M., Fanizzi, F. P., & Schena, F. P. (2016). Renal Cell Carcinoma: A Study through NMR-Based Metabolomics Combined with Transcriptomics. Diseases, 4(1), 7. https://doi.org/10.3390/diseases4010007