
Tumor Necrosis Factor-Alpha’s Role in the Pathophysiology of Colon Cancer

 , , , ,
, , , , 
Abstract
1. Introduction
2. Role of Tumor Necrosis Factor-Alpha in Colon Cancer Pathophysiology
2.1. Structure and Function of TNF-α
2.2. Tumor Necrosis Factor Alpha (TNF-α) and Colorectal Carcinoma
2.3. Tumor Necrosis Factor Alpha (TNF-α) and Immune Response
2.3.1. Pro-Inflammatory Role and Immune Activation
2.3.2. Immune Evasion
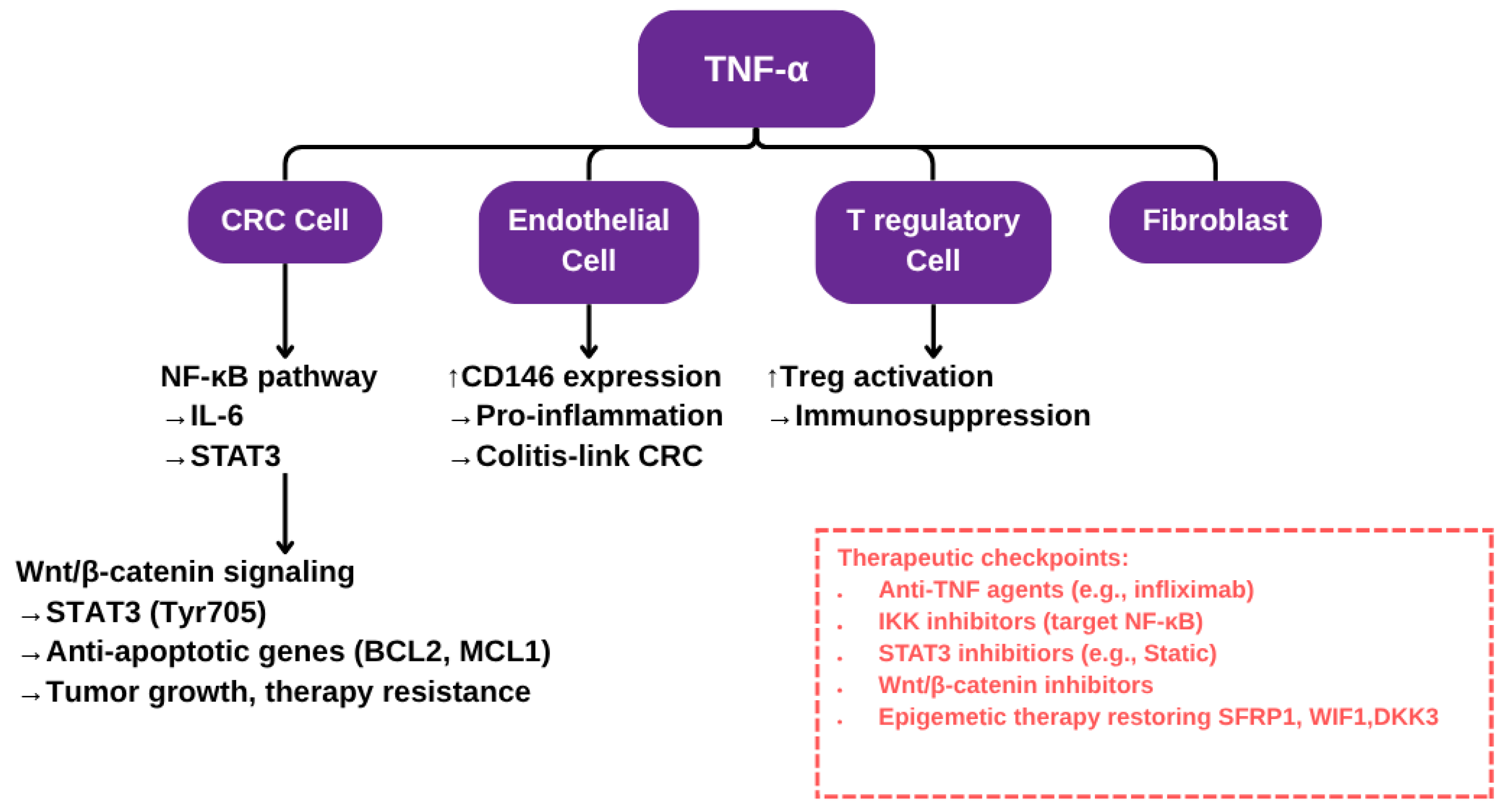
3. Mechanistic Insights of Pathways
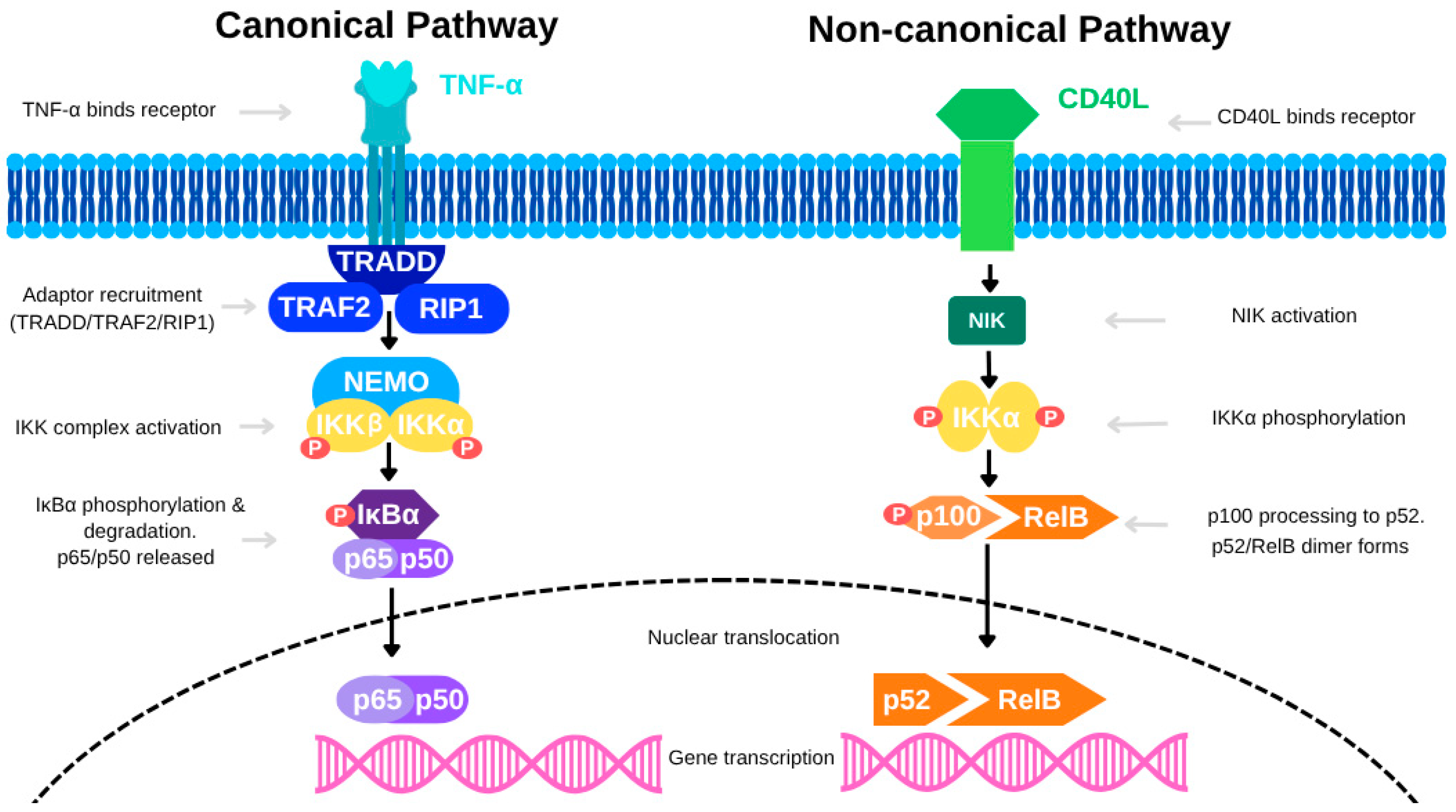
3.1. NF-κB Pathway Activation
3.2. TNF-α and Endothelial Cell Interactions
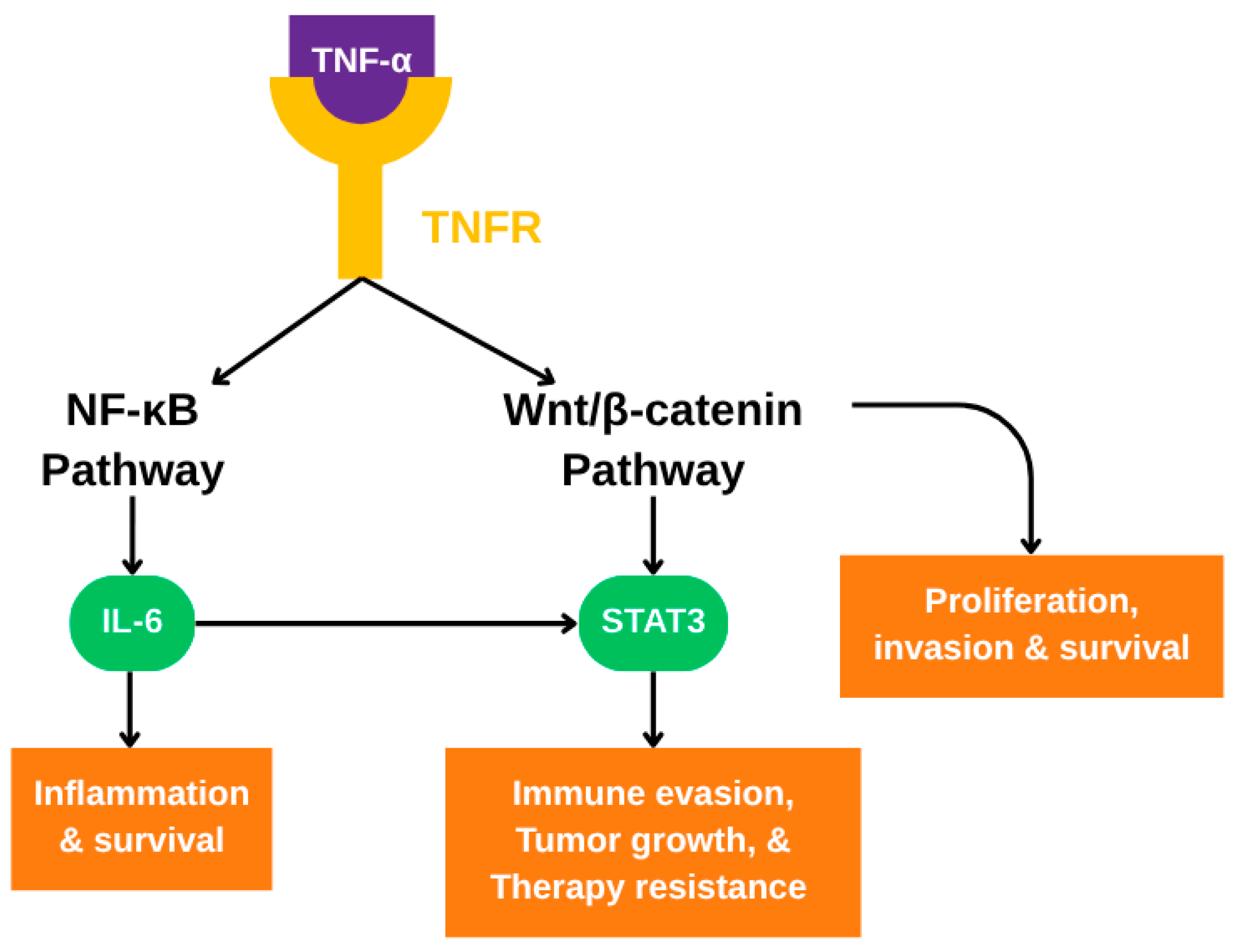
3.3. Crosstalk with Wnt/β-catenin and STAT3 Signaling Pathways
4. TNF-α as a Therapeutic Target in Colon Cancer
5. Limitations and Future Directions
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| TNF-α | Tumor necrosis factor alpha |
| NF-κB | Nuclear factor kappa-light-chain-enhancer of activated B cells |
| STAT3 | Signal transducer and activator of transcription 3 |
| CRC | Colorectal cancer |
| DNA | Deoxyribonucleic acid |
| ROS | Reactive oxygen species |
| Wnt | Wingless-type |
| EMT | Epithelial mesenchymal transition |
| TNFR1 | TNF-α type 1 receptor |
| TNFR2 | TNF-α type 2 receptor |
| HT-29 | Human colorectal adenocarcinoma cell line |
| IBD | Inflammatory bowel disease |
| IKK | IκB kinase |
| TRAF2 | Tumor necrosis factor receptor-associated factor 2 |
| RIP1 | Kinase receptor interacting protein 1 |
| NEMO | Nuclear factor-kappa B essential modulator |
| 8-OHdG | 8-hydroxydeoxyguanosine |
| GG-NER | Global Genome NER |
| DNMTs | DNA methyltransferases |
| TROP-2 | Tumor-associated calcium signal transducer 2 |
References
- Farinha, P.; Pinho, J.O.; Matias, M.; Gaspar, M.M. Nanomedicines in the treatment of colon cancer: A focus on metallodrugs. Drug Deliv. Transl. Res. 2021, 12, 49–66. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, A.; Pathak, S.; Subramanium, V.D.; Dharanivasan, G.; Murugesan, R.; Verma, R.S. Strategies for targeted drug delivery in treatment of colon cancer: Current trends and future perspectives. Drug Discov. Today 2017, 22, 1224–1232. [Google Scholar] [CrossRef]
- Cheng, E.; Shi, Q.; Shields, A.F.; Nixon, A.B.; Shergill, A.P.; Ma, C.; Guthrie, K.A.; Couture, F.; Kuebler, P.; Kumar, P.; et al. Association of Inflammatory Biomarkers With Survival Among Patients With Stage III Colon Cancer. JAMA Oncol. 2023, 9, 404–413. [Google Scholar] [CrossRef]
- Jang, D.-I.; Lee, A.-H.; Shin, H.-Y.; Song, H.-R.; Park, J.-H.; Kang, T.-B.; Lee, S.-R.; Yang, S.-H. The Role of Tumor Necrosis Factor Alpha (TNF-α) in Autoimmune Disease and Current TNF-α Inhibitors in Therapeutics. Int. J. Mol. Sci. 2021, 22, 2719. [Google Scholar] [CrossRef] [PubMed]
- You, K.; Gu, H.; Yuan, Z.; Xu, X. Tumor Necrosis Factor Alpha Signaling and Organogenesis. Front. Cell Dev. Biol. 2021, 9, 727075. [Google Scholar] [CrossRef] [PubMed]
- Laha, D.; Grant, R.; Mishra, P.; Nilubol, N. The Role of Tumor Necrosis Factor in Manipulating the Immunological Response of Tumor Microenvironment. Front. Immunol. 2021, 12, 656908. [Google Scholar] [CrossRef]
- Pakdemirli, A.; Kocal, G.C. TNF-alpha Induces Pro-Inflammatory Factors in Colorectal Cancer Microenvironment. Med. Sci. Discov. 2020, 7, 466–469. [Google Scholar] [CrossRef]
- Alotaibi, A.G.; Li, J.V.; Gooderham, N.J. Tumour Necrosis Factor-Alpha (TNF-α)-Induced Metastatic Phenotype in Colorectal Cancer Epithelial Cells: Mechanistic Support for the Role of MicroRNA-21. Cancers 2023, 15, 627. [Google Scholar] [CrossRef]
- Wang, H.; Wang, H.-S.; Zhou, B.-H.; Li, C.-L.; Zhang, F.; Wang, X.-F.; Zhang, G.; Bu, X.-Z.; Cai, S.-H.; Du, J. Epithelial–Mesenchymal Transition (EMT) Induced by TNF-α Requires AKT/GSK-3β-Mediated Stabilization of Snail in Colorectal Cancer. PLoS ONE 2013, 8, e56664. [Google Scholar] [CrossRef]
- Stanilov, N.; Miteva, L.; Dobreva, Z.; Stanilova, S. Colorectal cancer severity and survival in correlation with tumour necrosis factor-alpha. Biotechnol. Biotechnol. Equip. 2014, 28, 911–917. [Google Scholar] [CrossRef]
- Wei, W.; Wang, J.; Huang, P.; Gou, S.; Yu, D.; Zong, L. Tumor necrosis factor-α induces proliferation and reduces apoptosis of colorectal cancer cells through STAT3 activation. Immunogenetics 2023, 75, 161–169. [Google Scholar] [CrossRef] [PubMed]
- Niture, S.; Dong, X.; Arthur, E.; Chimeh, U.; Niture, S.S.; Zheng, W.; Kumar, D. Oncogenic Role of Tumor Necrosis Factor α-Induced Protein 8 (TNFAIP8). Cells 2018, 8, 9. [Google Scholar] [CrossRef]
- Li, X.-M.; Su, J.-R.; Yan, S.-P.; Cheng, Z.-L.; Yang, T.-T.; Zhu, Q. A novel inflammatory regulator TIPE2 inhibits TLR4-mediated development of colon cancer via caspase-8. Cancer Biomark. 2014, 14, 233–240. [Google Scholar] [CrossRef] [PubMed]
- Al Obeed, O.A.; Alkhayal, K.A.; Al Sheikh, A.; Zubaidi, A.M.; Vaali-Mohammed, M.-A.; Boushey, R.; McKerrow, J.H.; Abdulla, M.-H. Increased expression of tumor necrosis factor-α is associated with advanced colorectal cancer stages. World J. Gastroenterol. 2014, 20, 18390–18396. [Google Scholar] [CrossRef]
- Kaminska, J.; Nowacki, M.; Kowalska, M.; Rysinska, A.; Chwalinski, M.; Fuksiewicz, M.; Michalski, W.; Chechlinska, M. Clinical Significance of Serum Cytokine Measurements in Untreated Colorectal Cancer Patients: Soluble Tumor Necrosis Factor Receptor Type I—An Independent Prognostic Factor. Tumor Biol. 2005, 26, 186–194. [Google Scholar] [CrossRef]
- Kemik, O.; Sumer, A.; Kemik, A.S.; Hasirci, I.; Purisa, S.; Dulger, A.C.; Demiriz, B.; Tuzun, S. The relationship among acute-phase response proteins, cytokines and hormones in cachectic patients with colon cancer. World J. Surg. Oncol. 2010, 8, 85. [Google Scholar] [CrossRef]
- Babic, A.; Shah, S.M.; Song, M.; Wu, K.; A Meyerhardt, J.; Ogino, S.; Yuan, C.; Giovannucci, E.L.; Chan, A.T.; Stampfer, M.J.; et al. Soluble tumour necrosis factor receptor type II and survival in colorectal cancer. Br. J. Cancer 2016, 114, 995–1002. [Google Scholar] [CrossRef] [PubMed]
- Chan, A.T.; Ogino, S.; Giovannucci, E.L.; Fuchs, C.S. Inflammatory Markers Are Associated With Risk of Colorectal Cancer and Chemopreventive Response to Anti-Inflammatory Drugs. Gastroenterology 2011, 140, 799–808.e2. [Google Scholar] [CrossRef]
- Kapitanović, S.; Čačev, T.; Ivković, T.C.; Lončar, B.; Aralica, G. TNFα gene/protein in tumorigenesis of sporadic colon adenocarcinoma. Exp. Mol. Pathol. 2014, 97, 285–291. [Google Scholar] [CrossRef]
- Zhao, P.; Zhang, Z. TNF-α promotes colon cancer cell migration and invasion by upregulating TROP-2. Oncol. Lett. 2018, 15, 3820–3827. [Google Scholar] [CrossRef]
- Li, W.; Xu, J.; Zhao, J.; Zhang, R. Oxaliplatin and Infliximab Combination Synergizes in Inducing Colon Cancer Regression. Med. Sci. Monit. 2017, 23, 780–789. [Google Scholar] [CrossRef]
- Grimm, M.; Lazariotou, M.; Kircher, S.; Höfelmayr, A.; Germer, C.T.; von Rahden, B.H.A.; Waaga-Gasser, A.M.; Gasser, M. Tumor necrosis factor-α is associated with positive lymph node status in patients with recurrence of colorectal cancer—Indications for anti-TNF-α agents in cancer treatment. Cell. Oncol. 2011, 34, 315–326. [Google Scholar] [CrossRef]
- Guo, Y.; Xie, F.; Liu, X.; Ke, S.; Chen, J.; Zhao, Y.; Li, N.; Wang, Z.; Yi, G.; Shen, Y.; et al. Blockade of TNF-α/TNFR2 signalling suppresses colorectal cancer and enhances the efficacy of anti-PD1 immunotherapy by decreasing CCR8+T regulatory cells. J. Mol. Cell Biol. 2023, 16, mjad067. [Google Scholar] [CrossRef]
- Hamilton, K.E.; Simmons, J.G.; Ding, S.; Van Landeghem, L.; Lund, P.K. Cytokine Induction of Tumor Necrosis Factor Receptor 2 Is Mediated by STAT3 in Colon Cancer Cells. Mol. Cancer Res. 2011, 9, 1718–1731. [Google Scholar] [CrossRef] [PubMed]
- Alotaibi, A.G.; Li, J.V.; Gooderham, N.J. Tumour necrosis factor-α (TNF-α) enhances dietary carcinogen-induced DNA damage in colorectal cancer epithelial cells through activation of JNK signaling pathway. Toxicology 2021, 457, 152806. [Google Scholar] [CrossRef]
- Zhu, M.; Zhu, Y.; Lance, P. TNFα-activated stromal COX-2 signalling promotes proliferative and invasive potential of colon cancer epithelial cells. Cell Prolif. 2013, 46, 374–381. [Google Scholar] [CrossRef] [PubMed]
- Romagny, S.; Bouaouiche, S.; Lucchi, G.; Ducoroy, P.; Bertoldo, J.B.; Terenzi, H.; Bettaieb, A.; Plenchette, S. S-Nitrosylation of cIAP1 Switches Cancer Cell Fate from TNFα/TNFR1-Mediated Cell Survival to Cell Death. Cancer Res. 2018, 78, 1948–1957. [Google Scholar] [CrossRef] [PubMed]
- Nair, H.G.; Jurkiewicz, A.; Graczyk, D. Inhibition of RNA Polymerase III Augments the Anti-Cancer Properties of TNFα. Cancers 2023, 15, 1495. [Google Scholar] [CrossRef]
- Burgos-Molina, A.M.; Santana, T.T.; Redondo, M.; Romero, M.J.B. The Crucial Role of Inflammation and the Immune System in Colorectal Cancer Carcinogenesis: A Comprehensive Perspective. Int. J. Mol. Sci. 2024, 25, 6188. [Google Scholar] [CrossRef]
- Meckawy, G.R.; Mohamed, A.M.; Zaki, W.K.; Khattab, M.A.; Amin, M.M.; ElDeeb, M.A.; El-Najjar, M.R.; Safwat, N.A. Natural killer NKG2A and NKG2D in patients with colorectal cancer. J. Gastrointest. Oncol. 2019, 10, 218–225. [Google Scholar] [CrossRef]
- Lin, Y.; Bai, L.; Chen, W.; Xu, S. The NF-κB activation pathways, emerging molecular targets for cancer prevention and therapy. Expert Opin. Ther. Targets 2009, 14, 45–55. [Google Scholar] [CrossRef]
- Mitchell, S.; Vargas, J.; Hoffmann, A. Signaling via the NFκB system. Wiley Interdiscip. Rev. Syst. Biol. Med. 2016, 8, 227–241. [Google Scholar] [CrossRef] [PubMed]
- A Baeuerle, P.; Henkel, T. Function and Activation of NF-kappaB in the Immune System. Annu. Rev. Immunol. 1994, 12, 141–179. [Google Scholar] [CrossRef]
- Bhat, A.A.; Nisar, S.; Singh, M.; Ashraf, B.; Masoodi, T.; Prasad, C.P.; Sharma, A.; Maacha, S.; Karedath, T.; Hashem, S.; et al. Cytokine- and chemokine-induced inflammatory colorectal tumor microenvironment: Emerging avenue for targeted therapy. Cancer Commun. 2022, 42, 689–715. [Google Scholar] [CrossRef]
- Zinatizadeh, M.R.; Schock, B.; Chalbatani, G.M.; Zarandi, P.K.; Jalali, S.A.; Miri, S.R. The Nuclear Factor Kappa B (NF-kB) signaling in cancer development and immune diseases. Genes Dis. 2021, 8, 287–297. [Google Scholar] [CrossRef]
- Hong, Y.; Boiti, A.; Vallone, D.; Foulkes, N.S. Reactive Oxygen Species Signaling and Oxidative Stress: Transcriptional Regulation and Evolution. Antioxidants 2024, 13, 312. [Google Scholar] [CrossRef] [PubMed]
- Klaunig, J.E.; Kamendulis, L.M.; Hocevar, B.A. Oxidative Stress and Oxidative Damage in Carcinogenesis. Toxicol. Pathol. 2009, 38, 96–109. [Google Scholar] [CrossRef]
- Jridi, I.; Canté-Barrett, K.; Pike-Overzet, K.; Staal, F.J.T. Inflammation and Wnt Signaling: Target for Immunomodulatory Therapy? Front. Cell Dev. Biol. 2021, 8, 615131. [Google Scholar] [CrossRef] [PubMed]
- Fragoso, M.A.; Patel, A.K.; Nakamura, R.E.I.; Yi, H.; Surapaneni, K.; Hackam, A.S. The Wnt/β-Catenin Pathway Cross-Talks with STAT3 Signaling to Regulate Survival of Retinal Pigment Epithelium Cells. PLoS ONE 2012, 7, e46892. [Google Scholar] [CrossRef]
- Yoshimatsu, Y.; Wakabayashi, I.; Kimuro, S.; Takahashi, N.; Takahashi, K.; Kobayashi, M.; Maishi, N.; Podyma-Inoue, K.A.; Hida, K.; Miyazono, K.; et al. TNF-α enhances TGF-β-induced endothelial-to-mesenchymal transition via TGF-β signal augmentation. Cancer Sci. 2020, 111, 2385–2399. [Google Scholar] [CrossRef]
- Buhrmann, C.; Yazdi, M.; Popper, B.; Kunnumakkara, A.B.; Aggarwal, B.B.; Shakibaei, M. Induction of the Epithelial-to-Mesenchymal Transition of Human Colorectal Cancer by Human TNF-β (Lymphotoxin) and its Reversal by Resveratrol. Nutrients 2019, 11, 704. [Google Scholar] [CrossRef] [PubMed]
- Shen, Z.; Zhou, R.; Liu, C.; Wang, Y.; Zhan, W.; Shao, Z.; Liu, J.; Zhang, F.; Xu, L.; Zhou, X.; et al. MicroRNA-105 is involved in TNF-α-related tumor microenvironment enhanced colorectal cancer progression. Cell Death Dis. 2017, 8, 3213. [Google Scholar] [CrossRef] [PubMed]
- Kang, A.-R.; Kim, J.-L.; Kim, Y.; Kang, S.; Oh, S.-C.; Park, J.K. A novel RIP1-mediated canonical WNT signaling pathway that promotes colorectal cancer metastasis via β -catenin stabilization-induced EMT. Cancer Gene Ther. 2023, 30, 1403–1413. [Google Scholar] [CrossRef]
- Kobelt, D.; Zhang, C.; Clayton-Lucey, I.A.; Glauben, R.; Voss, C.; Siegmund, B.; Stein, U. Pro-inflammatory TNF-α and IFN-γ Promote Tumor Growth and Metastasis via Induction of MACC1. Front. Immunol. 2020, 11, 980. [Google Scholar] [CrossRef] [PubMed]
- Sammons, R.D.; Gaines, T.A. Glyphosate resistance: State of knowledge. Pest Manag. Sci. 2005, 70, 1367–1377. [Google Scholar] [CrossRef]
- Kim, S.-J.; Kang, H.-G.; Kim, K.; Kim, H.; Zetterberg, F.; Park, Y.S.; Cho, H.-S.; Hewitt, S.M.; Chung, J.-Y.; Nilsson, U.J.; et al. Crosstalk between WNT and STAT3 is mediated by galectin-3 in tumor progression. Gastric Cancer 2021, 24, 1050–1062. [Google Scholar] [CrossRef]
- Kawada, M.; Seno, H.; Uenoyama, Y.; Sawabu, T.; Kanda, N.; Fukui, H.; Shimahara, Y.; Chiba, T. Signal Transducers and Activators of Transcription 3 Activation Is Involved in Nuclear Accumulation of β-Catenin in Colorectal Cancer. Cancer Res. 2006, 66, 2913–2917. [Google Scholar] [CrossRef]
- Li, Q.; Geng, S.; Luo, H.; Wang, W.; Mo, Y.-Q.; Luo, Q.; Wang, L.; Song, G.-B.; Sheng, J.-P.; Xu, B. Signaling pathways involved in colorectal cancer: Pathogenesis and targeted therapy. Signal Transduct. Target. Ther. 2024, 9, 266. [Google Scholar] [CrossRef]
- Zidi, I.; Mestiri, S.; Bartegi, A.; Ben Amor, N. TNF-α and its inhibitors in cancer. Med Oncol. 2009, 27, 185–198. [Google Scholar] [CrossRef]
- Totzke, J.; Gurbani, D.; Raphemot, R.; Hughes, P.F.; Bodoor, K.; Carlson, D.A.; Loiselle, D.R.; Bera, A.K.; Eibschutz, L.S.; Perkins, M.M.; et al. Takinib, a Selective TAK1 Inhibitor, Broadens the Therapeutic Efficacy of TNF-α Inhibition for Cancer and Autoimmune Disease. Cell Chem. Biol. 2017, 24, 1029–1039.e7. [Google Scholar] [CrossRef]
- He, M.M.; Smith, A.S.; Oslob, J.D.; Flanagan, W.M.; Braisted, A.C.; Whitty, A.; Cancilla, M.T.; Wang, J.; Lugovskoy, A.A.; Yoburn, J.C.; et al. Small-Molecule Inhibition of TNF-α. Science 2005, 310, 1022–1025. [Google Scholar] [CrossRef] [PubMed]
- Bates, R.C.; Mercurio, A.M. Tumor Necrosis Factor-α Stimulates the Epithelial-to-Mesenchymal Transition of Human Colonic Organoids. Mol. Biol. Cell 2003, 14, 1790–1800. [Google Scholar] [CrossRef]
- Vaculová, A.; Hofmanova, J.; Soucek, K.; Kovariková, M.; Kozubík, A. Tumor necrosis factor-alpha induces apoptosis associated with poly(ADP-ribose) polymerase cleavage in HT-29 colon cancer cells. Anticancer Res. 2002, 22, 1635–1639. [Google Scholar]
- Mercogliano, M.F.; Bruni, S.; Mauro, F.; Elizalde, P.V.; Schillaci, R. Harnessing Tumor Necrosis Factor Alpha to Achieve Effective Cancer Immunotherapy. Cancers 2021, 13, 564. [Google Scholar] [CrossRef] [PubMed]
- Mozooni, Z.; Ghadyani, R.; Soleimani, S.; Ahangar, E.R.; Sheikhpour, M.; Haghighi, M.; Motallebi, M.; Movafagh, A.; Aghaei-Zarch, S.M. TNF-α, and TNFRs in gastrointestinal cancers. Pathol. Res. Pract. 2024, 263, 155665. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Ai, F.; Tian, L.; Liu, S.; Zhao, L.; Wang, X. Infliximab enhances the therapeutic effects of 5-fluorouracil resulting in tumor regression in colon cancer. OncoTargets Ther. 2016, 9, 5999–6008. [Google Scholar] [CrossRef]
- Zins, K.; Abraham, D.; Sioud, M.; Aharinejad, S. Colon Cancer Cell–Derived Tumor Necrosis Factor-α Mediates the Tumor Growth–Promoting Response in Macrophages by Up-regulating the Colony-Stimulating Factor-1 Pathway. Cancer Res. 2007, 67, 1038–1045. [Google Scholar] [CrossRef]
- Liu, Z.-G.; Yang, Y.-Q.; Zhang, Z.-G.; Wang, X.-L.; Li, Y.-L.; Sun, R.-F. Role of tumor necrosis factor alpha-induced protein 6 (TNFAIP6) in tumors: A pan-cancer analysis. Oncol. Transl. Med. 2023, 10, 22–29. [Google Scholar] [CrossRef]
- Cornista, A.M.; Giolito, M.V.; Baker, K.; Hazime, H.; Dufait, I.; Datta, J.; Khumukcham, S.S.; De Ridder, M.; Roper, J.; Abreu, M.T.; et al. Colorectal Cancer Immunotherapy: State of the Art and Future Directions. Gastro Hep Adv. 2023, 2, 1103–1119. [Google Scholar] [CrossRef]
- Johnson, D.; Chee, C.E.; Wong, W.; Lam, R.C.; Tan, I.B.H.; Ma, B.B. Current advances in targeted therapy for metastatic colorectal cancer—Clinical translation and future directions. Cancer Treat. Rev. 2024, 125, 102700. [Google Scholar] [CrossRef]
- Tsai, K.-Y.; Huang, P.-S.; Chu, P.-Y.; Nguyen, T.N.A.; Hung, H.-Y.; Hsieh, C.-H.; Wu, M.-H. Current Applications and Future Directions of Circulating Tumor Cells in Colorectal Cancer Recurrence. Cancers 2024, 16, 2316. [Google Scholar] [CrossRef] [PubMed]
- Ben-Baruch, A. Tumor Necrosis Factor α: Taking a Personalized Road in Cancer Therapy. Front. Immunol. 2022, 13, 903679. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
| Study | Study Design | Sample Size | TNFα Status | Findings and Associated Outcomes |
|---|---|---|---|---|
| Al Obeed et al., 2014 [14] | Retrospective cohort study | 30 colorectal cancer patients | ↑ TNF-α (mRNA and protein) | Elevated expression associated with advanced stages (Stage III/IV) and contribute to tumor progression and immune evasion |
| Li et al., 2011 [13] | Case–control study | 180 colon cancer patients and 180 control subjects | ↑ TNF-α in 308AA genotype | Patients with 308AA genotype were associated with ↑ TNF-alpha levels and a ↑ risk of colon cancer |
| Kaminska et al., 2005 [15] | Observational study | 157 untreated colorectal cancer patients and 50 healthy volunteers | ↑ sTNF RI in 69% of CRC patients | sTNF RI is strongly associated with tumor grade, invasion, and worse prognosis and is an independent prognostic marker for overall survival |
| Kemik et al., 2010 [16] | Observational study | 126 colon cancer patients and 36 controls | ↑ TNF-α | Elevated TNF-α levels were observed in cachectic patients and this elevation contributes to the development and severity of cancer cachexia |
| Babic et al., 2016 [17] | Prospective cohort study | 544 CRC patients (225 males and 319 females) | ↑ sTNFR2 (TNF-alpha activity) | Poor overall survival and ↓ colorectal cancer-specific survival as sTNFR2 is linked to TNF-α pathway activation |
| Chan et al., 2011 [18] | Case–control study | 280 cases of colorectal cancer and 555 matched controls | ↑ inflammatory markers | Inflammatory markers increase the risk of CRC and anti-inflammatory drugs reduce inflammation leading to decreased colorectal cancer risk, particularly in those with elevated inflammatory markers |
| Kapitanovic, 2014 [19] | Case–control study | 91 patients with sporadic colon adenocarcinoma and 100 healthy controls | ↑ TNFα gene/protein | TNFα gene and protein expression levels were significantly resulting in escalated tumor progression and tumor growth in colon adenocarcinoma |
| Zhao and Zhang, 2018 [20] | Experimental study | HCT-116 colon cancer cells treated with different TNF-α concentrations | ↑ TNF-α at low concentration | ↑ TROP-2 promotes cancer cell motility and invasion |
| Li et al., 2017 [21] | Tissue sample analysis study | 108 human colon cancer tissue samples | ↑ TNF-α in colon cancer tissues and cell lines | Blocking TNF-α with infliximab negates the effect of TNF-α-driven tumor-promoting inflammation, it enhances chemotherapy effectiveness |
| M. Grimm et al., 2011 [22] | In vitro study | 104 patients | ↑ TNF-α in 94% of CRC patients | Increased TNF-α levels correlated with more aggressive disease and lymph node metastases |
| Guo et al., 2023 [23] | In vitro study | 54 CRC and 60 gastric cancer samples. | ↑ TNF-α/TNFR2 signaling | TNF-α contributes to Tregs activation and blocking TNF-α improves the immunotherapy response |
| Study | TNF-α Effect | Mechanism/Pathway | Pro- or Anti-Tumorigenic | Human Models Used |
|---|---|---|---|---|
| Zhao & Zhang, 2018 [20]; Hamilton et al., 2011 [24] | Proliferation/Survival | NF-κB, STAT3 activation; TNFR2 upregulation | Pro-tumorigenic | Human colon cancer cell lines (e.g., HCT-116) |
| Zhao & Zhang, 2018 [20] | Migration/Invasion | TROP-2, MMP-9, ERK1/2, EMT, CXCL10/CXCR3 axis | Pro-tumorigenic | Human colon cancer cell lines, patient samples |
| Alotaibi et al., 2021 [25] | DNA damage | JNK pathway, especially with dietary carcinogens | Pro-tumorigenic | Human colorectal epithelial cell lines |
| Zhu et al., 2013 [26] | Stromal Interaction | TNFα-activated stromal COX-2 increases invasiveness | Pro-tumorigenic | Human stromal/epithelial co-culture models |
| Romagny et al., 2018 [27] | Switch to cell death | S-nitrosylation of cIAP1 alters TNFR1 signaling to apoptosis | Anti-tumorigenic | Human colon cancer cells |
| Nair et al., 2023 [28] | Contextual sensitization | Inhibition of RNA Pol III enhances TNFα-mediated cell death | Anti-tumorigenic | Human colon cancer cells (e.g., HCT-116) |
| Aspect | Details |
|---|---|
| Biological Role of TNF-α | Plays a critical role in colon cancer progression. High expression of TNF-α correlates with poor prognosis [21,49]. |
| Mechanisms of Action | Stimulates macrophages to produce pro-tumorigenic factors such as CSF-1, VEGF-A, and MMP-2 [50]. Enhances epithelial-to-mesenchymal transition (EMT), increasing invasiveness [51]. |
| Therapeutic Targeting | TNF-α is a key target in colon cancer due to its involvement in tumor-stromal interactions and inflammatory pathways. |
| Monoclonal Antibody Therapy | Infliximab (anti-TNF-α monoclonal antibody) induces antibody-dependent cellular cytotoxicity (ADCC) and complement-dependent cytotoxicity (CDC), promoting tumor cell apoptosis [56]. |
| Combination Therapy | Synergistic effects when combined with chemotherapy agents like oxaliplatin or 5-fluorouracil, resulting in tumor regression in preclinical models [21,56]. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khan, S.; Aldawood, Y.; Shaikh, A.H.; Zobairi, A.; Nabilah, U.; Alqahtani, H.M.; Vaali-Mohammed, M.-A. Tumor Necrosis Factor-Alpha’s Role in the Pathophysiology of Colon Cancer. Diseases 2025, 13, 185. https://doi.org/10.3390/diseases13060185
Khan S, Aldawood Y, Shaikh AH, Zobairi A, Nabilah U, Alqahtani HM, Vaali-Mohammed M-A. Tumor Necrosis Factor-Alpha’s Role in the Pathophysiology of Colon Cancer. Diseases. 2025; 13(6):185. https://doi.org/10.3390/diseases13060185
Chicago/Turabian StyleKhan, Saleha, Yara Aldawood, Ayesha Hanin Shaikh, Aleena Zobairi, Urwa Nabilah, H. M. Alqahtani, and Mansoor-Ali Vaali-Mohammed. 2025. "Tumor Necrosis Factor-Alpha’s Role in the Pathophysiology of Colon Cancer" Diseases 13, no. 6: 185. https://doi.org/10.3390/diseases13060185
APA StyleKhan, S., Aldawood, Y., Shaikh, A. H., Zobairi, A., Nabilah, U., Alqahtani, H. M., & Vaali-Mohammed, M.-A. (2025). Tumor Necrosis Factor-Alpha’s Role in the Pathophysiology of Colon Cancer. Diseases, 13(6), 185. https://doi.org/10.3390/diseases13060185