
Emergency Presentations of Pediatric Sickle Cell Disease in French Guiana



Abstract
1. Introduction
2. Patients and Methods
2.1. Study Setting
- The long dry season runs from mid-July until the end of November;
- December to February is a short rainy season;
- March to mid-April is a short summer;
- The main rainy season occurs from mid-April until the end of June.
2.2. Type of Study
2.3. Study Population
- -
- Patients living outside French Guiana;
- -
- Age;
- -
- Sex.
- -
- Medical history: type of SCD, history of >3 annual vaso-occlusive crises (VOCs), acute chest syndrome (ACS), stroke, splenectomy, cholecystectomy;
- -
- Associated chronic pathology: asthma, autoimmune disease, obstructive sleep apnea syndrome (OSAS), Gougerot–Sjögren syndrome, osteonecrosis of the femoral head, hypertension;
- -
- Date of consultation;
- -
- Length of hospital stay: treatment data;
2.4. Data Collection
2.5. Statistical Analysis
2.6. Ethical and Regulatory Status
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| SCD | Sickle cell disease |
| HbS | Sickle cell hemoglobin |
| DROM | Département et Région d’outre mer/French Guiana is an overseas French department and region |
| INSEE | Institut National de la Statistique et des Etudes Economiques/French National Institute |
| CHC | Centre Hospitalier de Cayenne |
| ED | Emergency department |
| VOCs | Vaso-occlusive crises |
| ACS | Acute chest syndrome |
| OSAS | Obstructive sleep apnea syndrome |
| TE | Transfusion exchange |
| IQR | Interquartile range |
| CNIL | Commission Nationale de l’ Informatique et des libertés |
| VAS | Visual Analogue Scale |
References
- Kato, G.J.; Piel, F.B.; Reid, C.D.; Gaston, M.H.; Ohene-Frempong, K.; Krishnamurti, L.; Smith, W.R.; Panepinto, J.A.; Weatherall, D.J.; Costa, F.F.; et al. Sickle cell disease. Nat. Rev. Dis. Primers. 2018, 4, 18010. [Google Scholar] [CrossRef] [PubMed]
- Leleu, H.; Arlet, J.B.; Habibi, A.; Etienne-Julan, M.; Khellaf, M.; Adjibi, Y.; Pirenne, F.; Pitel, M.; Granghaud, A.; Sinniah, C.; et al. Epidemiology and disease burden of sickle cell disease in France: A descriptive study based on a French nationwide claims database. PLoS ONE 2021, 16, e0253986. [Google Scholar] [CrossRef]
- Elenga, N.; Ro, V.; Missindu, J.M.; Boizan, N.T.; Vaz, T.; Lucarelli, A. La drépanocytose en Guyane: Bilan de 30 années de dépistage néonatal (1992–2021) [Sickle cell disease in French Guiana: Assessing 30 years of neonatal screening (1992–2021)]. Med. Trop. Sante. Int. 2024, 4, 488. [Google Scholar]
- Étienne-Julan, M.; Elana, G.; Loko, G.; Elenga, N.; Vaz, T.; Muszlak, M. La drépanocytose dans les départements français d’outre-mer (Antilles, Guyane, la Réunion, Mayotte). Données descriptives et organisation de la prise en charge. Bull. Epidemiol. Hebd. 2012, 27–28, 322–325. [Google Scholar]
- Brandow, A.M.; Liem, R.I. Advances in the diagnosis and treatment of sickle cell disease. J. Hematol. Oncol. 2022, 15, 20. [Google Scholar] [CrossRef] [PubMed]
- Sundd, P.; Gladwin, M.T.; Novelli, E.M. Pathophysiology of Sickle Cell Disease. Annu. Rev. Pathol. 2019, 14, 263–292. [Google Scholar] [CrossRef]
- Insee Population Estimates6all-French Guiana. Available online: https://www.insee.fr/en/statistiques/serie/001760178 (accessed on 2 January 2025).
- Yusuf, H.R.; Atrash, H.K.; Grosse, S.D.; Parker, C.S.; Grant, A.M. Emergency department visits by patients with sickle cell disease: A descriptive study, 1999–2007. Am. J. Prev. Med. 2010, 38 (Suppl. 4), S536–S541. [Google Scholar] [CrossRef]
- Attell, B.K.; Barrett, P.M.; Pace, B.S.; McLemore, M.L.; McGee, B.T.; Oshe, R.; DiGirolamo, A.M.; Cohen, L.L.; Snyder, A.B. Characteristics of emergency department visits made by individuals with Sickle Cell Disease in the U.S., 1999–2020. AJPM Focus 2023, 3, 100158. [Google Scholar] [CrossRef]
- Lovett, P.B.; Sule, H.P.; Lopez, B.L. Sickle cell disease in the emergency department. Emerg. Med. Clin. N. Am. 2014, 32, 629–647. [Google Scholar] [CrossRef]
- Ballas, S.K.; Dampier, C. Risk factors associated with increased emergency department utilization in patients with sickle cell disease: A systematic literature review. Ann. Hematol. 2020, 99, 2483–2495. [Google Scholar] [CrossRef]
- Parriault, M.-C.; Cropet, C.; Fahrasmane, A.; Rogier, S.; Parisot, M.; Nacher, M.; Elenga, N. Air Drep—A Retrospective Study Evaluating the Influence of Weather Conditions and Viral Epidemics on Vaso-Occlusive Crises in Sickle Cell Disease Patients Living in French Guiana. Int. J. Environ. Health 2019, 16, 2724. [Google Scholar] [CrossRef] [PubMed]
- Rogovik, A.L.; Persaud, J.; Friedman, J.N.; Kirby, M.A.; Goldman, R.D. Pediatric vasoocclusive crisis and weather conditions. J. Emerg. Med. 2011, 41, 559–565. [Google Scholar] [CrossRef] [PubMed]
- Smith, W.R.; Coyne, P.; Smith, V.S.; Mercier, B. Temperature changes, temperature extremes, and their relationship to emergency department visits and hospitalizations for sickle cell crisis. Pain Manag. Nurs. 2003, 4, 106–111. [Google Scholar] [CrossRef]
- Crego, N.P.; Masese, R.M.; Bonnabeau, E.B.; Douglas, C.D.; Rains, G.B.; Shah, N.; Tanabe, P.P. Patient Perspectives of Sickle Cell Management in the Emergency Department. Crit. Care Nurs. Q. 2021, 44, 160–174. [Google Scholar] [CrossRef] [PubMed]
- Masese, R.V.; Bulgin, D.; Douglas, C.; Shah, N.; Tanabe, P. Barriers and facilitators to care for individuals with sickle cell disease in central North Carolina: The emergency department providers’ perspective. PLoS ONE 2019, 14, e0216414. [Google Scholar] [CrossRef]
- Linton, E.A.; Goodin, D.A.; Hankins, J.S.; Kanter, J.; Preiss, L.; Simon, J.; Souffront, K.; Tanabe, P.; Gibson, R.; Hsu, L.L.; et al. A survey-based needs assessment of barriers to optimal sickle cell disease care in the Emergency Department. Ann. Emerg. Med. 2020, 76, S64–S72. [Google Scholar] [CrossRef]
- Lapite, A.; Lavina, I.; Goel, S.; Umana, J.; Ellison, A.M. A Qualitative Systematic Review of Pediatric Patient and Caregiver Perspectives on Pain Management for Vaso-Occlusive Episodes in the Emergency Department. Pediatr. Emerg. Care 2023, 39, 162–166. [Google Scholar] [CrossRef]
- Eisenbrown, K.; Ellison, A.M.; Nimmer, M.; Badaki-Makun, O.; Brousseau, D.C. Practice variation in emergency department management of children with Sickle Cell Disease who present with fever. Pediatr. Emerg. Care 2018, 34, 574–577. [Google Scholar] [CrossRef]
- Shenoi, R.; Ma, L.; Syblik, D.; Yusuf, S. Emergency department crowding and analgesic delay in pediatric sickle cell pain crises. Pediatr. Emerg. Care 2011, 27, 911–917. [Google Scholar] [CrossRef]
- Muslu, C.S.; Kopetsky, M.; Nimmer, M.; Visotcky, A.; Fraser, R.; Brousseau, D.C. Association between timely opioid administration and hospitalization in children with sickle cell disease presenting to the emergency department with acute pain. Pediatr. Blood Cancer 2020, 67, e28268. [Google Scholar] [CrossRef]
- Awe, M.; Robbins, A.B.; Chandi, M.B.; Cortright, L.M.; Tumin, D.; Whitfield, A.D. Provider communication and fever protocol for children with Sickle Cell Disease in the emergency department. Pediatr. Emerg. Care 2022, 38, 376–379. [Google Scholar] [CrossRef] [PubMed]
- Thein, S.L.; Pirenne, F.; Fasano, R.M.; Habibi, A.; Bartolucci, P.; Chonat, S.; Hendrickson, J.E.; Stowell, S.R. Hemolytic transfusion reactions in sickle cell disease: Underappreciated and potentially fatal. Haematologica 2020, 105, 539–544. [Google Scholar] [CrossRef]
- Umana, J.; Lapite, A.; Ellison, A.M. New Horizons in Emergency Department Management of Pediatric Sickle Cell Disease. Pediatr. Emerg. Care 2024, 40, 406–411. [Google Scholar] [CrossRef]
- Lin, S.M.; Strouse, J.J.; Whiteman, L.N.; Anders, J.; Stewart, R.W. Improving the quality of care for sick patients in the pediatric emergency department. Pediatr. Emerg. Care 2016, 32, 14–16. [Google Scholar] [CrossRef]
- Reparaz, P.; Serrano, I.; Adan-Pedroso, R.; Astigarraga, I.; de Pedro Olabarri, J.; Echebarria-Barona, A. Clinical management of acute complications of sickle cell anemia: Eleven years of experience in a tertiary hospital. Pediatrics 2022, 97, 4–11. [Google Scholar] [CrossRef] [PubMed]
- Strunk, C.; Tartaglione, I.; Piccone, C.M.; Colombatti, R.; Andemariam, B.; Manwani, D.; Smith, A.; Haile, H.; Kim, E.; Wilson, S.; et al. Global geographic differences in healthcare utilization for sickle cell disease pain crises in the CASiRe cohort. Blood Cells Mol. Dis. 2021, 92, 102612. [Google Scholar] [CrossRef] [PubMed]
- Williams, S.-A.; Henson, S.; Trimmingham, S.; Newman, J.; Kanter, J. Emergency department utilization for sickle cell disease in St. Vincent and the Grenadines. Pan. Afr. Med. J. 2021, 38, 100. [Google Scholar] [CrossRef]
- Newman, T.V.; Yang, J.; Suh, K.; Jonassaint, C.R.; Kane-Gill, S.L.; Novelli, E.M. Use of disease-modifying treatments in patients with Sickle Cell Disease. JAMA Netw. Open 2023, 6, e2344546. [Google Scholar] [CrossRef]
- Dick, M.H.; Abdelgadir, A.; Kulkarni, V.V.; Akram, H.; Chatterjee, A.; Pokhrel, S.; Khan, S. Comparing the safety and efficacy of L-glutamine, voxelotor, and crizanlizumab for reducing the frequency of vasoocclusive crisis in Sickle Cell Disease: A systematic review. Cureus 2022, 14, e24920. [Google Scholar] [CrossRef]
- Elenga, N.; Kayemba-Kay, S.; Nacher, M.; Archer, N. A call to start hydroxyurea by 6 months of age and before the advent of sickle cell disease complications. Pediatr. Blood Cancer 2022, 69, e29423. [Google Scholar] [CrossRef]
- Piel, F.B.; Rees, D.C.; DeBaun, M.R.; Nnodu, O.; Ranque, B.; Thompson, A.A.; Ware, R.E.; Abboud, M.R.; Abraham, A.; Ambrose, E.E.; et al. Defining global strategies to improve outcomes in sickle cell disease: A lancet hematology commission. Lancet Haematol. 2023, 10, e633–e686. [Google Scholar] [CrossRef] [PubMed]
- Dexter, D.; McGann, P.T. Hydroxyurea for children with sickle cell disease in sub-Saharan Africa: A summary of the evidence, opportunities, and challenges. Pharmacotherapy 2023, 43, 430–441. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variables | Number (%) |
|---|---|
| Age distribution | |
| <5 years | 109 (13%) |
| 5–10 years | 217 (25%) |
| >10 years | 531 (62%) |
| Gender | |
| Famale | 441 (51) |
| Male | 416 (49) |
| Sickle cell genotype | |
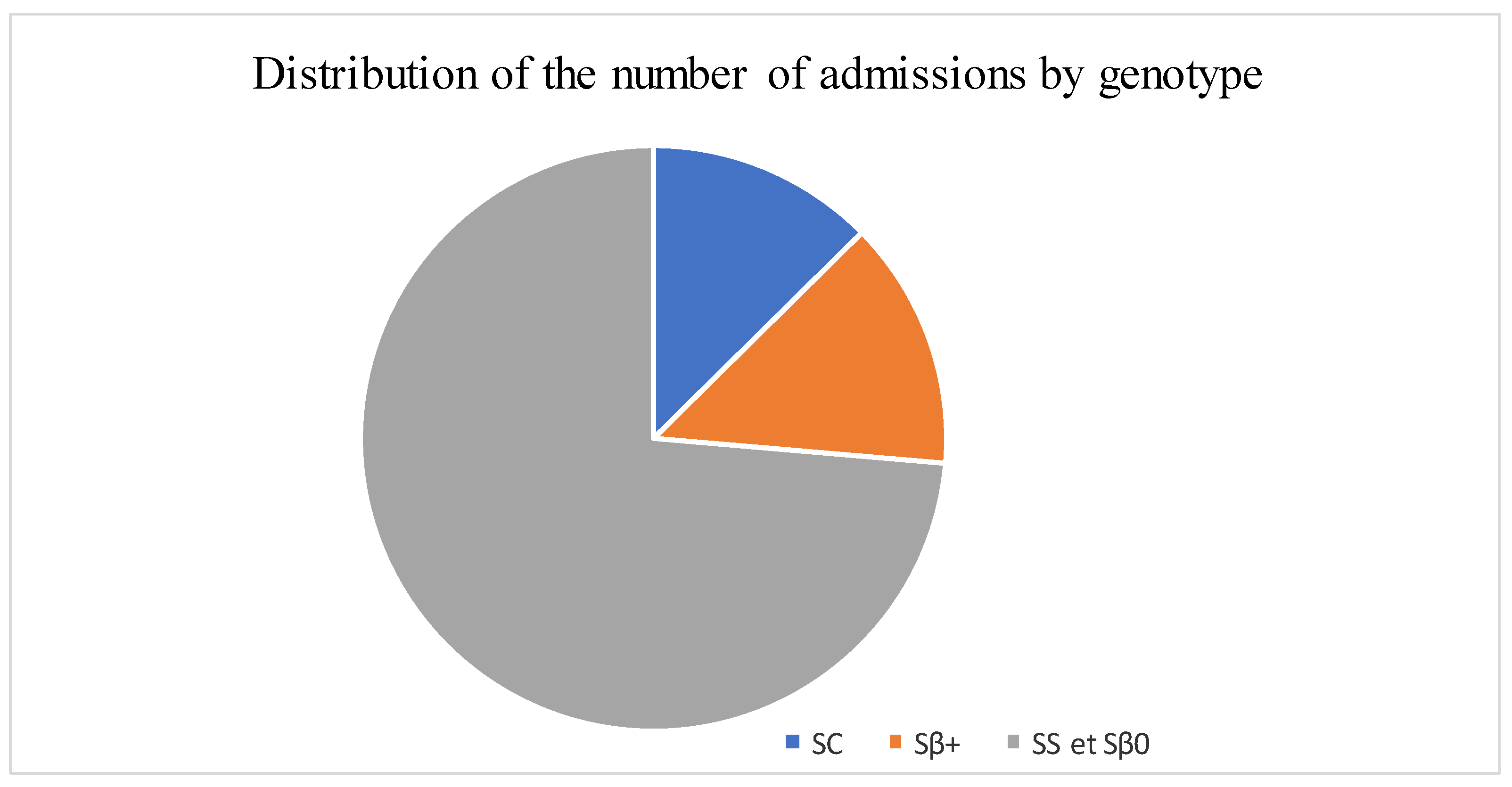
| SC | 107 (12) |
| Sß+thalassemia | 117 (14) |
| SS/Sß°thalassemia | 626 (74) |
| History | |
| >3 VOCs per year | 585 (71) |
| Sepsis | 234 (28) |
| ACS | 239 (29) |
| Transfusion/TE | 318 (39) |
| Stroke | 64 (8) |
| Splenectomy | 83 (10) |
| Cholecystectomy/cholelithiasis | 222 (27) |
| Associated pathology | 310 (37) |
| First quarter | 233 (27) |
| Second quarter | 218 (26) |
| Third quarter | 189 (22) |
| Fourth quarter | 217 (25) |
| Emergency department visit reason | |
| Pain (VOC) | 573 (67) |
| Fever (infection) | 99 (12) |
| Pallor (anemia) | 70 (8) |
| Difficulty breathing (ACS) | 19 (2) |
| Other * | 93 (11) |
| Hospitalizations | 285 (37) |
| Length of hospital stay | |
| <5 days | 239 (84) |
| ≥5 days | 46 (16) |
| Definitive diagnosis | |
| Vaso-occlusive crisis | 513 (68) |
| Anemia or splenic sequestration s | 98 (13) |
| Sepsis/infection | 104 (14) |
| Acute chest syndrome | 21 (3) |
| Other | 24 (3) |
| Treatments | |
| IV Hydration | 858 (100) |
| Morphine | 85 (11) |
| Antibiotics | 113 (15) |
| Oxygen | 40 (5) |
| Variables. | Children Hospitalized | Children not Hospitalized | AOR | p |
|---|---|---|---|---|
| Number (%) | Number (%) | |||
| Age distribution | 0.7 [0.6–0.9] | 0.01 | ||
| <5 years | 44 (42) | 60 (58) | ||
| 5–10 years | 93 (46) | 111 (54) | ||
| >10 years | 148 (33) | 306 (67) | ||
| Gender | 0.8 [0.6–1.1] | 0.2 | ||
| Famale | 157 (40) | 231 (60) | ||
| Male | 128 (34) | 246 (66) | ||
| History | ||||
| >3 Annual VOCs | 216 (41) | 313 (59) | 2 [1.3–3.1] | 0.001 |
| ACS | 95 (44) | 116 (56) | 1.6 [1.1–2.5] | 0.02 |
| Transfusion/TE | 95 (32) | 203 (68) | 0.7 [0.4–1] | 0.07 |
| VCA | 9 (14) | 55 (86) | 0.5 [0.2–0.9] | 0.04 |
| Cholecystectomy/cholelithiasis | 59 (26) | 154 (74) | 0.5 [0.4–0.8] | 0.03 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Djomo, C.F.; Sile, S.N.; Elenga, N. Emergency Presentations of Pediatric Sickle Cell Disease in French Guiana. Diseases 2025, 13, 142. https://doi.org/10.3390/diseases13050142
Djomo CF, Sile SN, Elenga N. Emergency Presentations of Pediatric Sickle Cell Disease in French Guiana. Diseases. 2025; 13(5):142. https://doi.org/10.3390/diseases13050142
Chicago/Turabian StyleDjomo, Carine Fankep, Souam Nguele Sile, and Narcisse Elenga. 2025. "Emergency Presentations of Pediatric Sickle Cell Disease in French Guiana" Diseases 13, no. 5: 142. https://doi.org/10.3390/diseases13050142
APA StyleDjomo, C. F., Sile, S. N., & Elenga, N. (2025). Emergency Presentations of Pediatric Sickle Cell Disease in French Guiana. Diseases, 13(5), 142. https://doi.org/10.3390/diseases13050142