
Study of Adult and Pediatric Spanish Patients with Cryptogenic Splenomegaly and Splenectomy


 , and
, and
Abstract
1. Introduction
- -
- To carry out educational training in centers of the Spanish public health network where there are doctors of different specialties with patients who present unaffiliated splenomegaly;
- -
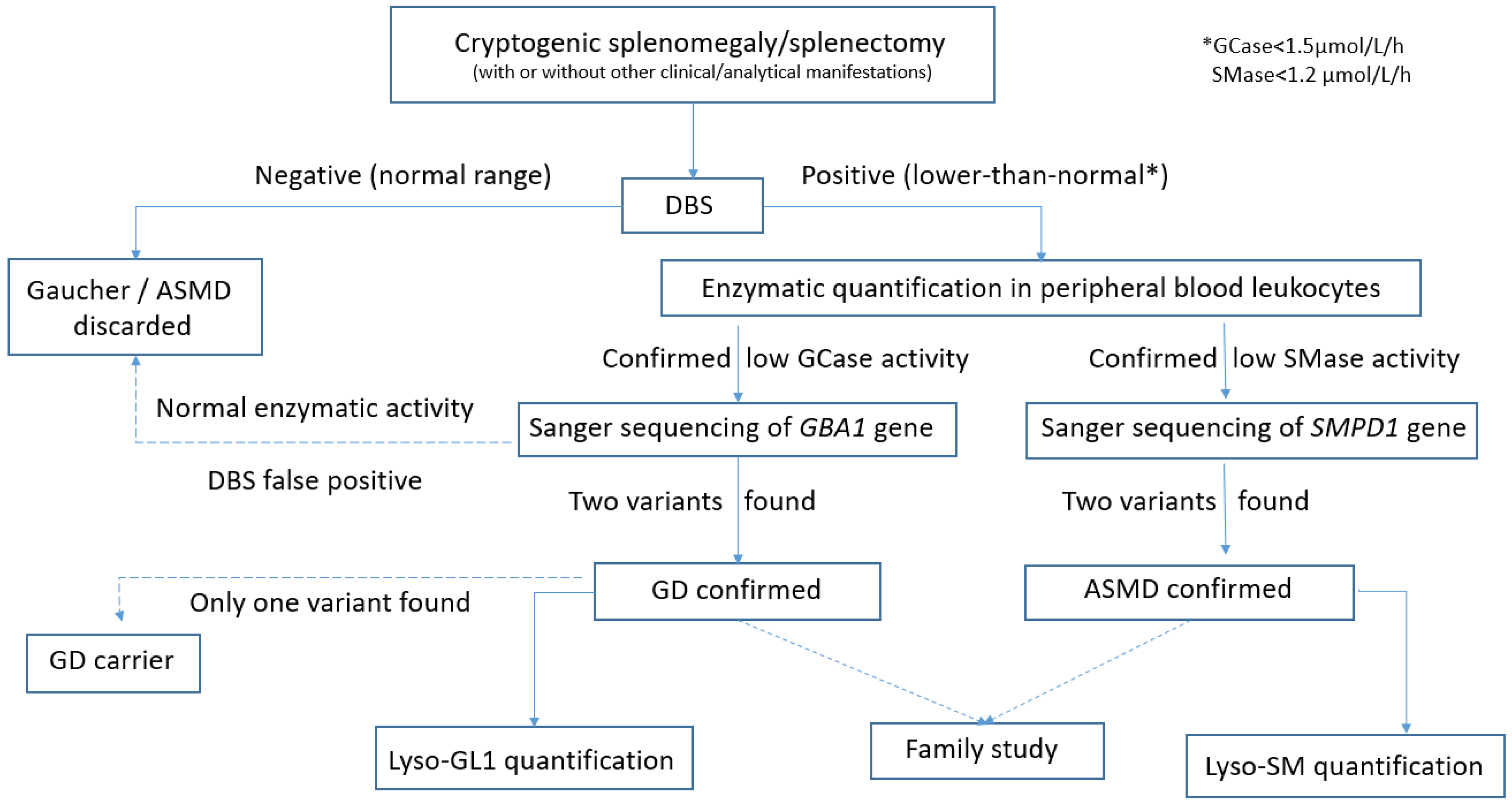
- To investigate if any of these patients could be affected by the GD and/or ASMD, by using enzymatic quantification with DBS testing as a diagnostic tool. Furthermore, we want to prove the usefulness of DBS testing as a simple, fast, and effective method for diagnosing and/or ruling out these two diseases. Analysis of the allelic variants found in confirmed GD or ASMD patients was performed.
2. Materials and Methods
2.1. Study Design
2.2. Medical Educational Part
2.3. Investigative Part
2.3.1. Inclusion and Exclusion Criteria
2.3.2. Procedures
2.3.3. Statistical Analysis
3. Results
3.1. Medical Educational Part
3.2. Investigative Part
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| GD | Gaucher disease |
| ASMD | Acid sphingomyelinase deficiency |
| LD | Lysosomal diseases |
| DBS | Dried blood spot |
| GCase | Glucocerebrosidase |
| SMase | Sphingomyelinase |
| SD | Standard deviation |
| IQR | Interquartile range |
References
- Pozo, A.L.; Godfrey, E.M.; Bowles, K.M. Splenomegaly: Investigation, diagnosis and management. Blood Rev. 2009, 23, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Pelizzo, G.; Guazzotti, M.; Klersy, C.; Nakib, G.; Costanzo, F.; Andreatta, E.; Bassotti, G.; Calcaterra, V. Spleen size evaluation in children: Time to define splenomegaly for pediatric surgeons and pediatricians. PLoS ONE 2018, 13, e0202741. [Google Scholar] [CrossRef]
- Arkles, L.B.; Gill, G.D.; Molan, M.P. A palpable spleen is not necessarily enlarged or pathological. Med. J. Aust. 1986, 145, 15–17. [Google Scholar] [CrossRef] [PubMed]
- Suttorp, M.; Classen, C.F. Splenomegaly in Children and Adolescents. Front. Pediatr. 2021, 9, 704635. [Google Scholar] [CrossRef] [PubMed]
- Curovic Rotbain, E.; Lund Hansen, D.; Schaffalitzky de Muckadell, O.; Wibrand, F.; Meldgaard Lund, A.; Frederiksen, H. Splenomegaly—Diagnostic validity, work-up, and underlying causes. PLoS ONE 2017, 12, e0186674. [Google Scholar] [CrossRef] [PubMed]
- Motta, I.; Filocamo, M.; Poggiali, E.; Stroppiano, M.; Dragani, A.; Consonni, D.; Warcellini, W.; Gaidano, G.; Facchini, L.; Specchia, G.; et al. Splenomegaly Gaucher Disease study group. A multicentre observational study for early diagnosis of Gaucher disease in patients with Splenomegaly and/or Thrombocytopenia. Eur. J. Haematol. 2016, 96, 352–359. [Google Scholar] [CrossRef] [PubMed]
- O’Reilly, R.A. Splenomegaly in 2505 patients at a large university medical center from 1913 to 1995. 1913 to 1962: 2056 patients. West. J. Med. 1998, 169, 78–87. [Google Scholar] [PubMed]
- Sjoberg, B.P.; Menias, C.O.; Lubner, M.G.; Mellnick, V.M.; Pickhardt, P.J. Splenomegal: A combined clinical and Radiologic approach to differential diagnosis. Gastroenterol. Clin. N. Am. 2018, 47, 643–666. [Google Scholar] [CrossRef] [PubMed]
- Eskes, E.C.B.; van Dussen, L.; Aerts, J.M.F.G.; van der Lienden, M.J.C.; Maas, M.; Akkerman, E.M.; van Kuilenburg, A.B.P.; Sjouke, B.; Hollak, C.E.M. Acid sphingomyelinase deficiency and Gaucher disease in adults: Similarities and differences in two macrophage storage disorders. JIMD Rep. 2024, 65, 330–340. [Google Scholar] [CrossRef] [PubMed]
- Capellini, M.D.; Motta, I.; Barbato, A.; Giuffrida, G.; Manna, R.; Carubbi, F.; Giona, F. Similarities and differences between Gaucher disease and acid sphingomyelinase deficiency: An algorithm to support the diagnosis. Eur. J. Intern. Med. 2023, 108, 81–84. [Google Scholar] [CrossRef]
- Oliva, P.; Schwarz, M.; Mechtler, T.P.; Sansen, S.; Keutzer, J.; Prusa, A.R.; Streubel, B.; Kasper, D.C. Importance to include differential diagnostics for acid sphingomyelinase deficiency (ASMD) in patients suspected to have to Gaucher disease. Mol. Genet. Metab. 2023, 139, 107563. [Google Scholar] [CrossRef] [PubMed]
- Do, Y.R.; Choi, Y.; Heo, M.H.; Kim, J.S.; Yoon, J.H.; Lee, J.H.; Park, J.S.; Sohn, S.K.; Kim, S.H.; Lim, S.; et al. Early diagnosis of Gaucher disease in Korean patients with unexplained splenomegaly: A multicenter observational study. Blood Res. 2022, 57, 207–215. [Google Scholar] [CrossRef] [PubMed]
- Di Rocco, M.; Vici, C.D.; Burlina, A.; Venturelli, F.; Fiumara, A.; Fecarotta, S.; Donatl, M.A.; Spada, M.; Concolino Pession, A. Screening for lysosomal diseases in a selected pediatric population: The case of Gaucher disease and acid sphingomyelinase deficiency. Orphanet J. Rare Dis. 2023, 18, 197. [Google Scholar] [CrossRef] [PubMed]
- Pession, A.; Di Rocco, M.; Venturelli, F.; Tappino, B.; Morello, W.; Santoro, N.; Giodano, P.; Filippini, B.; Rinieri, S.; Russo, G.; et al. GAU-PED study for early diagnosis of Gaucher disease in children with splenomegaly and cytopenia. Orphanet J. Rare Dis. 2023, 18, 151. [Google Scholar] [CrossRef] [PubMed]
- Giacomarra, M.; Colomba, P.; Francofonte, D.; Zora, M.; Caocci, G.; Diomede, D.; Guiffrida, G.; Fiori, L.; Montanari, C.; Sapuppo, A.; et al. Gaucher Disease or Acid Sphingomyelinase Deficiency? The Importance of Differential Diagnosis. J. Clin. Med. 2024, 13, 1487. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Scott, C.R.; Chamoles, N.A.; Ghavami, A.; Pinto, B.M.; Turecek, F.; Gelb, M.H. Direct multiplex assay of lysosomal enzymes in dried blood spots for newborn screening. Clin. Chem. 2004, 50, 1785–1796. [Google Scholar] [CrossRef] [PubMed]
- Motta, I.; Consoni, D.; Stroppiano, M.; Benedetto, C.; Cassinerio, E.; Tappino, B.; Ranalli, P.; Borin, L.; Facchini, L.; Patriarca, A.; et al. Predicting the probability of Gaucher disease in subjects with splenomegaly and thrombocytopenia. Sci. Rep. 2021, 11, 2594. [Google Scholar] [CrossRef] [PubMed]
- Torralba, M.A.; Alfonso, P.; Pérez-Calvo, J.I.; Cenarro, A.; Pastores, G.M.; Giraldo, P.; Civeira, F.; Pocoví, M. High prevalence of the 55-bp deletion (c.1263del55) in exon 9 of the glucocerebrosidase gene causing misdiagnosis (for homozygous N370S (c.1226A > G) mutation) in Spanish Gaucher disease patients. Blood Cells Mol. Dis. 2002, 29, 35–40. [Google Scholar] [CrossRef] [PubMed]
- Weinreb, N.J.; Goker-Alpan, O.; Kishnani, P.S.; Longo, N.; Burrow, T.A.; Bernat, J.A.; Gupta, P.; Henderson, N.; Pedro, H.; Prada, C.E.; et al. The diagnosis and management of Gaucher disease in pediatric patients: Where do we go from here? Mol. Genet. Metab. 2022, 136, 4–21. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| Total Cohort of Patients N = 220 | Patients Without Diagnosis N = 212 | Patients Diagnosed GD/ASMD N = 8 | |
|---|---|---|---|
| Gender | |||
| Male n (%) | 158 (72) | 154 (73) | 4 (50) |
| Female n (%) | 62 (28) | 58 (27) | 4 (50) |
| Age at study inclusion | |||
| Adults n (%) | 208 (95) | 202 (95) | 6 (75) |
| Children n(%) | 12 (5) | 10 | 2 (25) |
| Reason for screening: | |||
| -Crytogenic splenomegaly n (%) | 208 (95) | 200 (94) | 8 (100) |
| Adults n (%) | 196 (94) | 190 (95) | 6 (75) |
| Children n (%) | 12 (6) | 10 (5) | 2 (25) |
| -Splenectomized n (%) | 12 (5) | 12 (6) | 0 |
| Adults n (%) | 12 (100) | 12 (100) | 0 |
| Children n (%) | 0 (0) | 0 (0) | 0 |
| Diagnosis | Clinical Features | GCase μmol/L/h (N ≥ 1.5) | SMase umol/L/h (N ≥ 1.2) | LGL-1 ng/mL (N < 14) | L-SM ng/mL (N < 70) | GBA1 Gene Variants | SMPD1 Gene Variants |
|---|---|---|---|---|---|---|---|
| ASMD | 60 y-o. female Splenomegaly diagnosis at age 22 y-o ASMD diagnosis after 39 years of cryptogenic splenomegaly | 0.3 | 327 | c.1829_1831delGCC; p.Arg610del c.1547A>G; p.His516Arg | |||
| ASMD | 69 y-o male Splenomegaly diagnosis at 68 y-o ASMD diagnosis after 3months of cryptogenic splenomegaly | 0.6 | 41 | c.1744C>A; p.Pro582Thr c.1744C>A; p.Pro582Thr | |||
| GD III | 4 month old male Splenomegaly diagnosis at 2 m-o Initially suspected pancytopenia secondary to CMV infection GD type III diagnosis after 2 months | 0.9 | 2.4 | c.1265_1319del55; p.Leu422Pro*fs c.(1448T>C; 1483G>C; 1497G>C); p.(Leu483Pro; Ala495Pro; Val499=) | |||
| GD I | 53 y-o female Splenomegaly diagnosis at age 53 y Associated with pancytopenia GD type I diagnosis after 2 months | 0.0 | 851.8 | c.1226A>G; p.Asn409Ser c.706C>T; p.Leu236Phe | |||
| GD I | 14 y-o female Splenomegaly diagnosis at 13 y-o Associated with thrombocytopenia Bone marrow morphology: Gaucher cells GD type I diagnosis after 14 months | 0.0 | 839.7 | c.1226A>G; p.Asn409Ser c.(475C>T+667T>C+681T>G+689T>G+703T>C+721G>A+754T>A); p.(Arg159Trp+Trp223Arg+Asn227Lys+Val230Gly+Ser235Pro+Gly241 Arg+Phe252lle) | |||
| GD I | 71 y-o. male Splenomegaly diagnosis at 66 y-o GD type I diagnosis after 5 years of cryptogenic splenomegaly | 0.0 | 229.9 | c.1226A>G; p.Asn409Ser c.1226A>G; p.Asn409Ser | |||
| GD I | 65 y-o male Splenomegaly diagnosis at 64 y-o Associated with thrombocytopenia GD type I diagnosis after 2 years | 0.4 | 17.60 | c.1226A>G; p.Asn409Ser c.1448T>C; p.Leu483Pro | |||
| GD I | 20 y-o female Splenomegaly diagnosis at age 20 y Associated with pancytopenia GD type I diagnosis after 1 month | 0.8 | 348.0 | c.1265_1319del55; p.Leu422Pro*fs c.1604G>A p.Arg535His |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Morado Arias, M.; Villarrubia Espinosa, J.; Vitoria Miñana, I.; Calderón Sandubete, E.; Quintero, V.; Torralba-Cabeza, M.Á. Study of Adult and Pediatric Spanish Patients with Cryptogenic Splenomegaly and Splenectomy. Diseases 2025, 13, 102. https://doi.org/10.3390/diseases13040102
Morado Arias M, Villarrubia Espinosa J, Vitoria Miñana I, Calderón Sandubete E, Quintero V, Torralba-Cabeza MÁ. Study of Adult and Pediatric Spanish Patients with Cryptogenic Splenomegaly and Splenectomy. Diseases. 2025; 13(4):102. https://doi.org/10.3390/diseases13040102
Chicago/Turabian StyleMorado Arias, Marta, Jesús Villarrubia Espinosa, Isidro Vitoria Miñana, Enrique Calderón Sandubete, Víctor Quintero, and Miguel Ángel Torralba-Cabeza. 2025. "Study of Adult and Pediatric Spanish Patients with Cryptogenic Splenomegaly and Splenectomy" Diseases 13, no. 4: 102. https://doi.org/10.3390/diseases13040102
APA StyleMorado Arias, M., Villarrubia Espinosa, J., Vitoria Miñana, I., Calderón Sandubete, E., Quintero, V., & Torralba-Cabeza, M. Á. (2025). Study of Adult and Pediatric Spanish Patients with Cryptogenic Splenomegaly and Splenectomy. Diseases, 13(4), 102. https://doi.org/10.3390/diseases13040102