The study was conducted in two phases: the first involved preprocessing the dataset, and the second consisted of implementing path analysis to build the causal graphical model. All analyses were performed in R version 4.3.2 (Windows 11), using the following functions and packages:
The dataset and R scripts are available upon request.
3.1. Data Preprocessing
Data preprocessing is essential for any statistical analysis or machine learning model, as it includes steps such as imputation of missing values, handling of outliers, removal of irrelevant variables, among others. These procedures enhance dataset quality, reduce bias, and ensure more reliable and reproducible results [
27,
28]. It is important to note that the dataset does not exclusively contain variables related to bacterial vaginosis, as it was designed to support multiple types of studies.
As shown in
Figure 2, data preprocessing involved five steps: (a) understanding the source of the dataset, (b) analyzing its structure, (c) removing irrelevant variables, (d) addressing outliers, and (e) imputing missing values to ensure consistency and analytical readiness.
(a) Dataset: Data were collected by a researcher from the Multidisciplinary Academic Division of Comalcalco (DAMC), Universidad Juárez Autónoma de Tabasco (UJAT). This cross-sectional dataset included one sample per participant, collected at varying gestational weeks (4–24), without longitudinal follow-up, and was not generated by the authors of the present study. Sample collection followed the standardized clinical protocol previously described by [
4], conducted at the Laboratory of Research in Metabolic and Infectious Diseases (UJAT). Vaginal and cervical samples were obtained by a trained gynecologist using sterile swabs from the ectocervix and posterior vaginal fornix, and stored at 4 °C until genomic DNA extraction. Bacterial detection and quantification were performed using standardized molecular methods applied consistently across all samples. The dataset contains information from pregnant women participating in healthy pregnancy campaigns (August 2018–January 2020) across rural and urban communities in Tabasco, Mexico. It includes sociodemographic data, bacterial presence, Human Papillomavirus, and BV diagnosis.
(b) Data Structure: To understand the structure of the dataset, an exploratory analysis was performed.
Table 2 summarizes its main aspects, showing an imbalance between classes, with BV− as the majority class. However, this imbalance does not affect PA methodology, as PA relies on covariance or correlation matrices rather than class proportions [
13,
18].
The data set comprises three classes of BV diagnosis:
BV+: Positive BV, indicating presence of the condition.
Indeterminate: Cases without a clear diagnosis.
BV−: Normal microbiota.
In addition to class distribution, missing values and outliers were examined as part of the exploratory analysis. Box-and-whisker diagrams were used to identify outliers by visualizing distribution through minimum, first quartile, median, third quartile, and maximum values [
29], see
Figure 3.
(c) Variable Elimination: Next, consultations were held with the clinical-biological team responsible for data creation to identify variables unrelated to BV. Variables associated with Human Papillomavirus (HPV) were removed, as previous studies found no significant association with BV [
4]. This refinement reduced the dataset to 72 attributes and 132 instances, retaining only variables relevant to the study objectives.
(d) Resolving Outliers: To ensure result quality and reliability, missing values and outliers were addressed [
30]. A detailed analysis based on boxplot charts, identified outliers in Glucose (Glu), Cholesterol (Col), Triglycerides (Trig), and Homocysteine (Hcy). These attributes could be real values and may indicate additional patient conditions; thus, the clinical-biological expert recommends retaining them for a more comprehensive analysis.
Figure 3 presents box plots illustrating their distribution and preserved out-of-range values.
(e) Imputation of Values: Subsequently, missing values were addressed through imputation, replacing null entries to prevent gaps from affecting the analysis. For numerical variables (e.g., age, salary), missing values were substituted with the mean of the attribute, while for categorical variables (e.g., gender, color), the most frequent value (mode) was used [
31,
32].
In this study, imputation was performed based on the BV diagnosis. For categorical variables, complete instances of each attribute were grouped by diagnosis (BV+, Indeterminate, or BV−), and the mode of each group was assigned to the missing values corresponding to that specific diagnosis. For numerical variables, the mean was applied following a similar process.
Table 3 and
Table 4 display the imputed data for the first ten observations of the variables number of pregnancies (N.EMBARAZO) and vaginal discharge (FlujoV). The column DxVBNoMh (Diagnosis of Bacterial Vaginosis without
Mycoplasma hominis) indicates the BV diagnosis: 1 for BV+ (positive), 2 for Indeterminate, and 3 for BV− (normal microbiota).
The original dataset included 87 variables and 132 instances. After removing irrelevant attributes, 72 variables remained, with the number of instances unchanged. Outliers were identified and retained based on recommendations from the clinical-biological team, who deemed them relevant for interpretation and analysis. Missing values were imputed using the mode for categorical variables and the mean for numerical ones, ensuring completeness and coherence for subsequent analysis.
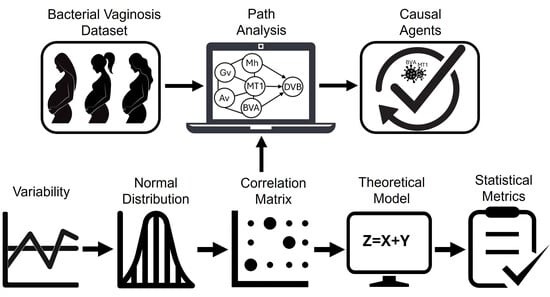
3.2. Application of Path Analysis
Figure 4 illustrates the steps of the second phase of the study, which focused on path analysis. This stage included (a) variability assessment, (b) distributional analysis, (c) exploration of the correlation structure, (d) theoretical modeling, (e) validation using statistical metrics, (f) construction of the causal graphical model and (g) review by the expert team.
(a) Variability: Ensuring data variability is crucial for PA validity, as parameter estimation in the path diagram depends on variance and covariances [
18,
22]. Thus, variance was calculated to confirm that the variables exhibited sufficient variability.
Table 5 shows the variance values for some variables in the dataset.
(b) Normal Distribution: After verifying variability, the distribution of the variables was analyzed to determine whether Pearson’s or Spearman’s correlation matrix would be appropriate for assessing correlations. The Q-Q plot and Kolmogorov-Smirnov normality test evaluated adherence to a normal distribution and were applied to each attribute in the dataset, revealing that none exhibited normality.
For example,
Figure 5 and
Figure 6 display the Q-Q plots for weeks of gestation (SemanaGesta) and
Atopobium vaginae (Av), both of which indicate a non-normal distribution. The test results showed
with
for SemanaGesta and
with
for Av, both with
p-values below 0.05, confirming a deviation from normality.
Different transformation techniques, including logarithmic, square root, reciprocal, and Box-Cox transformations, were applied to improve distribution characteristics. However, due to the nature of the data, no one successfully achieved normality.
Table 6 presents the
p-values of the Kolmogorov-Smirnov test applied to each illustrative variable: weeks of gestation (SemanasGesta),
Mycoplasma hominis (Mh),
Atopobium vaginae (Av),
Bacteria Associated with Bacterial Vaginosis Type 2 (BVAB2) and
Lactobacillus crispatus (Lcrispatus), before and after transformation. N/A values indicate that the transformation was not applicable due to the presence of zeros.
(c) Correlation Matrix: Since the dataset does not follow a normal distribution, the Spearman correlation matrix assessed relationships between variables and their association with BV diagnosis. Five key variables linked to the class attribute (DxVBNoMh) were identified:
Mycoplasma hominis (Mh),
Atopobium vaginae (Av),
Gardnerella vaginalis (Gv),
Megasphaera Type 1 (MT1), and
Bacteria Associated with Bacterial Vaginosis Type 2 (BVAB2) [
33].
The correlation matrix presents the coefficients between these variables and the BV diagnosis.
Figure 7 includes three diagnostic categories: BV+ (presence of condition), Indeterminate (unclear diagnosis), and BV− (normal microbiota), while
Figure 8 considers only BV+ and BV−. Comparing the matrices reveals that the correlation values are highly similar, suggesting that the Indeterminate class has minimal impact and can be omitted without affecting the results.
To investigate the indeterminate class further, future research will conduct an exploratory analysis to determine whether specific patterns justify its inclusion in refined diagnostic models. Thus,
Figure 8 is this study’s matrix of primary interest. The last column presents correlation coefficients between the variables and the BV diagnosis: DxVBNoMh with Av (−0.70), Gv (−0.41), MT1 (−0.89), BVAB2 (−0.83), and Mh (0.55). Although some values are low, their clinical relevance lies in their potential to provide essential information for diagnostic and therapeutic decisions [
34,
35].
(d) Theoretical Model: Based on the correlation matrix, a theoretical model was built as a prerequisite for obtaining the CGM from the dataset. This model is represented by the Equation (
7) which specifies the structure of the theoretical model used to construct the CGM. The symbol ∼ denotes a directional relationship, where the variable on the left is modeled as a function of those on the right. The first line indicates that the BV diagnosis without
Mycoplasma hominis (DxVBNoMh) is predicted using five bacterial variables: Mh, Av, Gv, MT1, and BVAB2. The subsequent lines model MT1 as a dependent variable influenced by each of the other bacteria individually, reflecting its mediating role in the causal pathway. This structure suggests that MT1 may act as a conduit through which other bacteria exert influence on BV diagnosis.
(e) Statistical Metrics: After obtaining the theoretical model, its fit must be evaluated.
Table 7 presents the values derived from applying the statistical metrics in
Table 1 to Equation (7).
(f) Causal Graphical Model: Based on the theoretical model in Equation (7) and statistical metrics in
Table 7, the CGM (
Figure 9) was constructed to visually represent causal and indirect relationships among the investigated variables and their impact on DVB. Due to R programming simplifications, some variables were abbreviated: DxVBNoMh as DVB and BVAB2 as BVA. The CGM illustrates causal and indirect relationships (solid lines) between all variables and DVB, as well as covariations (dashed lines). Path coefficients indicate association strength, with values near ±1 representing strong associations and values close to 0 indicating weak associations.
The strongest causal relationship observed was between MT1 and DVB, with a coefficient of 0.49, suggesting that an increase in MT1 level is associated with a higher probability of DVB diagnosis. The weakest causal relationship was between Gv and DVB, with a coefficient of −0.04, indicating a weak negative association. Additionally, indirect relationships were identified, such as the connection between BVAB2 and DVB through MT1, where MT1 acts as an intermediate variable, with coefficients of 0.50 and 0.49, respectively.
Total effects were calculated by summing direct and indirect effects using standardized path coefficients [
36,
37]. The direct effect corresponds to the path coefficient linking one variable to another, representing their immediate relationship. Indirect effects were determined by multiplying coefficients along causal pathways.
Table 8 summarizes the total effects of predictor variables on BV diagnosis.
An interpretation of
Table 8 is provided below:
Mh (−0.1937): Slightly reduces the probability of DVB, suggesting a modest protective effect.
Av (0.2627): Positively associated with DVB, indicating a higher likelihood of diagnosis.
Gv (0.0482): Weak positive correlation; a small increase in Gv slightly raises diagnosis probability.
MT1 (0.4900): Strong positive impact, significantly increasing DVB likelihood.
BVAB2 (0.5750): Highest positive correlation, indicating a strong association with DVB risk.
(g) Biologist Evaluation: Finally, the CGM was presented to the clinical-biological expert for interpretation, validating the identified causal relationships and assessing their relevance to BV diagnosis. The expert evaluated the causal links based on their biological plausibility, clinical coherence, and consistency with the specialized scientific literature.
Path analysis visualizes bacterial relationships through a diagram, illustrating their connection to BV. In this study, the selected bacteria (Av, Gv, MT1, BVAB2, and Mh) align with those most commonly associated with BV, as reported in specialized biologist literature. The diagram highlights direct and indirect interactions, allowing for calculating each bacterium’s total effect on the diagnosis and identifying the most influential one.


 ,
, 










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}