
Mechanical Properties of Glioblastoma: Perspectives for YAP/TAZ Signaling Pathway and Beyond


{kind=link}
{kind=link}
Abstract
1. Introduction
2. Mechanics of GBM
3. The Hippo Signaling Pathway
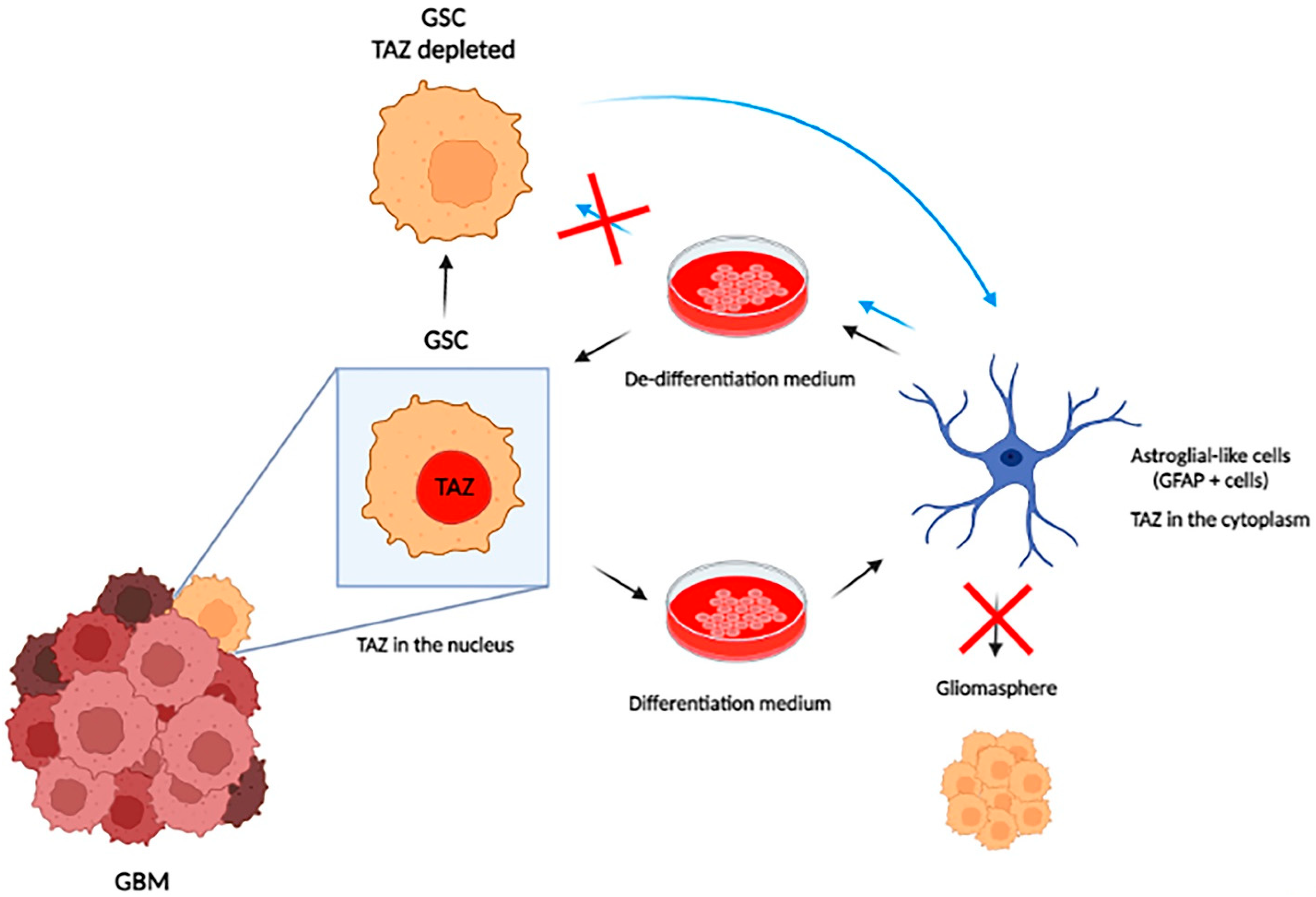
4. Hippo Signaling in GBM
5. Conclusions and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wirtz, D.; Konstantopoulos, K.; Searson, P.C. The physics of cancer: The role of physical interactions and mechanical forces in metastasis. Nat. Rev. Cancer 2011, 11, 512–522. [Google Scholar] [CrossRef] [PubMed]
- Roma-Rodrigues, C.; Mendes, R.; Baptista, P.V.; Fernandes, A.R. Targeting Tumor Microenvironment for Cancer Therapy. Int. J. Mol. Sci. 2019, 20, 840. [Google Scholar] [CrossRef] [PubMed]
- Nia, H.T.; Munn, L.L.; Jain, R.K. Physical traits of cancer. Science 2020, 370, eaaz0868. [Google Scholar] [CrossRef] [PubMed]
- Seano, G.; Nia, H.T.; Emblem, K.E.; Datta, M.; Ren, J.; Krishnan, S.; Kloepper, J.; Pinho, M.C.; Ho, W.W.; Ghosh, M.; et al. Solid stress in brain tumours causes neuronal loss and neurological dysfunction and can be reversed by lithium. Nat. Biomed. Eng. 2019, 3, 230–245. [Google Scholar] [CrossRef] [PubMed]
- Stylianopoulos, T.; Martin, J.D.; Snuderl, M.; Mpekris, F.; Jain, S.R.; Jain, R.K. Coevolution of solid stress and interstitial fluid pressure in tumors during progression: Implications for vascular collapse. Cancer Res. 2013, 73, 3833–3841. [Google Scholar] [CrossRef]
- Hoffman, B.D.; Crocker, J.C. Cell mechanics: Dissecting the physical responses of cells to force. Annu. Rev. Biomed. Eng. 2009, 11, 259–288. [Google Scholar] [CrossRef]
- Ayad, N.M.E.; Kaushik, S.; Weaver, V.M. Tissue mechanics, an important regulator of development and disease. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2019, 374, 20180215. [Google Scholar] [CrossRef]
- DuFort, C.C.; Paszek, M.J.; Weaver, V.M. Balancing forces: Architectural control of mechanotransduction. Nat. Rev. Mol. Cell Biol. 2011, 12, 308–319. [Google Scholar] [CrossRef]
- Huang, H.; Kamm, R.D.; Lee, R.T. Cell mechanics and mechanotransduction: Pathways, probes, and physiology. Am. J. Physiol. Cell. Physiol. 2004, 287, C1–C11. [Google Scholar] [CrossRef]
- Eke, I.; Cordes, N. Focal adhesion signaling and therapy resistance in cancer. Semin. Cancer Biol. 2015, 31, 65–75. [Google Scholar] [CrossRef]
- Borghi, N.; Sorokina, M.; Shcherbakova, O.G.; Weis, W.I.; Pruitt, B.L.; Nelson, W.J.; Dunn, A.R. E-cadherin is under constitutive actomyosin-generated tension that is increased at cell-cell contacts upon externally applied stretch. Proc. Natl. Acad. Sci. USA 2012, 109, 12568–12573. [Google Scholar] [CrossRef]
- Price, A.J.; Cost, A.L.; Ungewiß, H.; Waschke, J.; Dunn, A.R.; Grashoff, C. Mechanical loading of desmosomes depends on the magnitude and orientation of external stress. Nat. Commun. 2018, 9, 5284. [Google Scholar] [CrossRef]
- Kirby, T.J.; Lammerding, J. Emerging views of the nucleus as a cellular mechanosensor. Nat. Cell Biol. 2018, 20, 373–381. [Google Scholar] [CrossRef]
- De Vecchis, D.; Beech, D.J.; Kalli, A.C. Molecular dynamics simulations of Piezo1 channel opening by increases in membrane tension. Biophys. J. 2021, 120, 1510–1521. [Google Scholar] [CrossRef]
- Liang, X.; Howard, J. Structural Biology: Piezo Senses Tension through Curvature. Curr. Biol. 2018, 28, R357–R359. [Google Scholar] [CrossRef]
- Lin, Y.C.; Guo, Y.R.; Miyagi, A.; Levring, J.; MacKinnon, R.; Scheuring, S. Force-induced conformational changes in PIEZO1. Nature 2019, 573, 230–234. [Google Scholar] [CrossRef]
- Pontes, B.; Monzo, P.; Gauthier, N.C. Membrane tension: A challenging but universal physical parameter in cell biology. Semin. Cell Dev. Biol. 2017, 71, 30–41. [Google Scholar] [CrossRef]
- Tsujita, K.; Satow, R.; Asada, S.; Nakamura, Y.; Arnes, L.; Sako, K.; Fujita, Y.; Fukami, K.; Itoh, T. Homeostatic membrane tension constrains cancer cell dissemination by counteracting BAR protein assembly. Nat. Commun. 2021, 12, 5930. [Google Scholar] [CrossRef]
- Pontes, B.; Monzo, P.; Gole, L.; Le Roux, A.L.; Kosmalska, A.J.; Tam, Z.Y.; Luo, W.; Kan, S.; Viasnoff, V.; Roca-Cusachs, P.; et al. Membrane tension controls adhesion positioning at the leading edge of cells. J. Cell Biol. 2017, 216, 2959–2977. [Google Scholar] [CrossRef]
- Huang, J.; Wu, S.; Barrera, J.; Matthews, K.; Pan, D. The Hippo signaling pathway coordinately regulates cell proliferation and apoptosis by inactivating Yorkie, the Drosophila Homolog of YAP. Cell 2005, 122, 421–434. [Google Scholar] [CrossRef]
- Dupont, S.; Morsut, L.; Aragona, M.; Enzo, E.; Giulitti, S.; Cordenonsi, M.; Zanconato, F.; Le Digabel, J.; Forcato, M.; Bicciato, S.; et al. Role of YAP/TAZ in mechanotransduction. Nature 2011, 474, 179–183. [Google Scholar] [CrossRef] [PubMed]
- Alexander, B.M.; Cloughesy, T.F. Adult Glioblastoma. J. Clin. Oncol. 2017, 35, 2402–2409. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Hegi, M.E.; Mason, W.P.; van den Bent, M.J.; Taphoorn, M.J.; Janzer, R.C.; Ludwin, S.K.; Allgeier, A.; Fisher, B.; Belanger, K.; et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009, 10, 459–466. [Google Scholar] [CrossRef] [PubMed]
- Cruz, J.V.R.; Batista, C.; Afonso, B.H.; Alexandre-Moreira, M.S.; Dubois, L.G.; Pontes, B.; Moura Neto, V.; Mendes, F.A. Obstacles to Glioblastoma Treatment Two Decades after Temozolomide. Cancers 2022, 14, 3203. [Google Scholar] [CrossRef] [PubMed]
- Sorribes, I.C.; Moore, M.N.J.; Byrne, H.M.; Jain, H.V. A Biomechanical Model of Tumor-Induced Intracranial Pressure and Edema in Brain Tissue. Biophys. J. 2019, 116, 1560–1574. [Google Scholar] [CrossRef]
- Reetz, K.; Abbas, Z.; Costa, A.S.; Gras, V.; Tiffin-Richards, F.; Mirzazade, S.; Holschbach, B.; Frank, R.D.; Vassiliadou, A.; Krüger, T.; et al. Increased cerebral water content in hemodialysis patients. PLoS ONE 2015, 10, e0122188. [Google Scholar] [CrossRef]
- Ciasca, G.; Sassun, T.E.; Minelli, E.; Antonelli, M.; Papi, M.; Santoro, A.; Giangaspero, F.; Delfini, R.; De Spirito, M. Nano-mechanical signature of brain tumours. Nanoscale 2016, 8, 19629–19643. [Google Scholar] [CrossRef]
- Hirata, E.; Yukinaga, H.; Kamioka, Y.; Arakawa, Y.; Miyamoto, S.; Okada, T.; Sahai, E.; Matsuda, M. In vivo fluorescence resonance energy transfer imaging reveals differential activation of Rho-family GTPases in glioblastoma cell invasion. J. Cell Sci. 2012, 125, 858–868. [Google Scholar] [CrossRef]
- Farin, A.; Suzuki, S.O.; Weiker, M.; Goldman, J.E.; Bruce, J.N.; Canoll, P. Transplanted glioma cells migrate and proliferate on host brain vasculature: A dynamic analysis. Glia 2006, 53, 799–808. [Google Scholar] [CrossRef]
- Winkler, F.; Kienast, Y.; Fuhrmann, M.; Von Baumgarten, L.; Burgold, S.; Mitteregger, G.; Kretzschmar, H.; Herms, J. Imaging glioma cell invasion in vivo reveals mechanisms of dissemination and peritumoral angiogenesis. Glia 2009, 57, 1306–1315. [Google Scholar] [CrossRef]
- Quail, D.F.; Joyce, J.A. The Microenvironmental Landscape of Brain Tumors. Cancer Cell 2017, 31, 326–341. [Google Scholar] [CrossRef] [PubMed]
- Khoonkari, M.; Liang, D.; Kamperman, M.; Kruyt, F.A.E.; van Rijn, P. Physics of Brain Cancer: Multiscale Alterations of Glioblastoma Cells under Extracellular Matrix Stiffening. Pharmaceutics 2022, 14, 1031. [Google Scholar] [CrossRef] [PubMed]
- Barnes, J.M.; Przybyla, L.; Weaver, V.M. Tissue mechanics regulate brain development, homeostasis and disease. J. Cell Sci. 2017, 130, 71–82. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wang, J. Mechanical tumor microenvironment and transduction: Cytoskeleton mediates cancer cell invasion and metastasis. Int. J. Biol. Sci. 2020, 16, 2014–2028. [Google Scholar] [CrossRef]
- Stewart, D.C.; Rubiano, A.; Dyson, K.; Simmons, C.S. Mechanical characterization of human brain tumors from patients and comparison to potential surgical phantoms. PLoS ONE 2017, 12, e0177561. [Google Scholar] [CrossRef]
- Miroshnikova, Y.A.; Mouw, J.K.; Barnes, J.M.; Pickup, M.W.; Lakins, J.N.; Kim, Y.; Lobo, K.; Persson, A.I.; Reis, G.F.; McKnight, T.R.; et al. Tissue mechanics promote IDH1-dependent HIF1α-tenascin C feedback to regulate glioblastoma aggression. Nat. Cell Biol. 2016, 18, 1336–1345. [Google Scholar] [CrossRef]
- Chen, J.E.; Pedron, S.; Shyu, P.; Hu, Y.; Sarkaria, J.N.; Harley, B.A.C. Influence of Hyaluronic Acid Transitions in Tumor Microenvironment on Glioblastoma Malignancy and Invasive Behavior. Front. Mater. 2018, 5, 39. [Google Scholar] [CrossRef]
- Riegler, J.; Labyed, Y.; Rosenzweig, S.; Javinal, V.; Castiglioni, A.; Dominguez, C.X.; Long, J.E.; Li, Q.; Sandoval, W.; Junttila, M.R.; et al. Tumor Elastography and Its Association with Collagen and the Tumor Microenvironment. Clin. Cancer Res. 2018, 24, 4455–4467. [Google Scholar] [CrossRef]
- Lin, T.C.; Yang, C.H.; Cheng, L.H.; Chang, W.T.; Lin, Y.R.; Cheng, H.C. Fibronectin in Cancer: Friend or Foe. Cells 2019, 9, 27. [Google Scholar] [CrossRef]
- De Oliveira Rosario, L.V.; da Rosa, B.G.; Goncalves, T.L.; Matias, D.I.L.; Freitas, C.; Ferrer, V.P. Glioblastoma Factors Increase the Migration of Human Brain Endothelial Cells In Vitro by Increasing MMP-9/CXCR4 Levels. Anti-Cancer Res. 2020, 40, 2725–2737. [Google Scholar] [CrossRef]
- Beliveau, A.; Thomas, G.; Gong, J.; Wen, Q.; Jain, A. Aligned Nanotopography Promotes a Migratory State in Glioblastoma Multiforme Tumor Cells. Sci. Rep. 2016, 6, 26143. [Google Scholar] [CrossRef]
- Sharma, P.; Sheets, K.; Elankumaran, S.; Nain, A.S. The mechanistic influence of aligned nanofibers on cell shape, migration and blebbing dynamics of glioma cells. Integr. Biol. 2013, 5, 1036–1044. [Google Scholar] [CrossRef]
- Simi, A.K.; Pang, M.F.; Nelson, C.M. Extracellular Matrix Stiffness Exists in a Feedback Loop that Drives Tumor Progression. In Biomechanics in Oncology; Advances in Experimental Medicine and Biology; Springer: Cham, Switzerland, 2018; Volume 1092, pp. 57–67. [Google Scholar] [CrossRef]
- Rutka, J.T.; Muller, M.; Hubbard, S.L.; Forsdike, J.; Dirks, P.B.; Jung, S.; Tsugu, A.; Ivanchuk, S.; Costello, P.; Mondal, S.; et al. Astrocytoma adhesion to extracellular matrix: Functional significance of integrin and focal adhesion kinase expression. J. Neuropathol. Exp. Neurol. 1999, 58, 198–209. [Google Scholar] [CrossRef]
- Belot, N.; Rorive, S.; Doyen, I.; Lefranc, F.; Bruyneel, E.; Dedecker, R.; Micik, S.; Brotchi, J.; Decaestecker, C.; Salmon, I.; et al. Molecular characterization of cell substratum attachments in human glial tumors relates to prognostic features. Glia 2001, 36, 375–390. [Google Scholar] [CrossRef]
- Friedlander, D.R.; Zagzag, D.; Shiff, B.; Cohen, H.; Allen, J.C.; Kelly, P.J.; Grumet, M. Migration of brain tumor cells on extracellular matrix proteins in vitro correlates with tumor type and grade and involves αV and β1 integrins. Cancer Res. 1996, 56, 1939–1947. [Google Scholar]
- Paulus, W.; Baur, I.; Schuppan, D.; Roggendorf, W. Characterization of integrin receptors in normal and neoplastic human brain. Am. J. Pathol. 1993, 143, 154–163. [Google Scholar]
- Mavrakis, M.; Juanes, M.A. The compass to follow: Focal adhesion turnover. Curr. Opin. Cell Biol. 2023, 80, 102152. [Google Scholar] [CrossRef]
- Schwarz, U.S.; Gardel, M.L. United we stand: Integrating the actin cytoskeleton and cell-matrix adhesions in cellular mechanotransduction. J. Cell Sci. 2012, 125, 3051–3060. [Google Scholar] [CrossRef]
- Koh, I.; Cha, J.; Park, J.; Choi, J.; Kang, S.G.; Kim, P. The mode and dynamics of glioblastoma cell invasion into a decellularized tissue-derived extracellular matrix-based three-dimensional tumor model. Sci. Rep. 2018, 8, 4608. [Google Scholar] [CrossRef]
- Shen, K.; Kenche, H.; Zhao, H.; Li, J.; Stone, J. The role of extracellular matrix stiffness in regulating cytoskeletal remodeling via vinculin in synthetic smooth muscle cells. Biochem. Biophys. Res. Commun. 2019, 508, 302–307. [Google Scholar] [CrossRef]
- Pietras, A.; Katz, A.M.; Ekström, E.J.; Wee, B.; Halliday, J.J.; Pitter, K.L.; Werbeck, J.L.; Amankulor, N.M.; Huse, J.T.; Holland, E.C. Osteopontin-CD44 signaling in the glioma perivascular niche enhances cancer stem cell phenotypes and promotes aggressive tumor growth. Cell Stem Cell 2014, 14, 357–369. [Google Scholar] [CrossRef] [PubMed]
- Klank, R.L.; Decker Grunke, S.A.; Bangasser, B.L.; Forster, C.L.; Price, M.A.; Odde, T.J.; SantaCruz, K.S.; Rosenfeld, S.S.; Canoll, P.; Turley, E.A.; et al. Biphasic Dependence of Glioma Survival and Cell Migration on CD44 Expression Level. Cell Rep. 2017, 19, 668. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Kumar, S. CD44-mediated adhesion to hyaluronic acid contributes to mechanosensing and invasive motility. Mol. Cancer Res. 2014, 12, 1416–1429. [Google Scholar] [CrossRef] [PubMed]
- Kwiatkowska, A.; Symons, M. Signaling Determinants of Glioma Cell Invasion. In Glioma Signaling; Advances in Experimental Medicine and Biology; Springer: Cham, Switzerland, 2020; Volume 1202, pp. 129–149. [Google Scholar] [CrossRef]
- Chen, X.; Wanggou, S.; Bodalia, A.; Zhu, M.; Dong, W.; Fan, J.J.; Yin, W.C.; Min, H.K.; Hu, M.; Draghici, D.; et al. A Feed-forward Mechanism Mediated by Mechanosensitive Ion Channel PIEZO1 and Tissue Mechanics Promotes Glioma Aggression. Neuron 2018, 100, 799–815.e7. [Google Scholar] [CrossRef] [PubMed]
- Momin, A.; Bahrampour, S.; Min, H.K.; Chen, X.; Wang, X.; Sun, Y.; Huang, X. Channeling Force in the Brain: Mechanosensitive Ion Channels Choreograph Mechanics and Malignancies. Trends Pharmacol. Sci. 2021, 42, 367–384. [Google Scholar] [CrossRef]
- Creasy, C.L.; Chernoff, J. Cloning and characterization of a human protein kinase with homology to Ste20. J. Biol. Chem. 1995, 270, 21695–21700. [Google Scholar] [CrossRef]
- Wu, S.; Huang, J.; Dong, J.; Pan, D. hippo encodes a Ste-20 family protein kinase that restricts cell proliferation and promotes apoptosis in conjunction with salvador and warts. Cell 2003, 114, 445–456. [Google Scholar] [CrossRef]
- Udan, R.S.; Kango-Singh, M.; Nolo, R.; Tao, C.; Halder, G. Hippo promotes proliferation arrest and apoptosis in the Salvador/Warts pathway. Nat. Cell Biol. 2003, 5, 914–920. [Google Scholar] [CrossRef]
- Vassilev, A.; Kaneko, K.J.; Shu, H.; Zhao, Y.; DePamphilis, M.L. TEAD/TEF transcription factors utilize the activation domain of YAP65, a Src/Yes-associated protein localized in the cytoplasm. Genes Dev. 2001, 15, 1229–1241. [Google Scholar] [CrossRef]
- Totaro, A.; Panciera, T.; Piccolo, S. YAP/TAZ upstream signals and downstream responses. Nat. Cell Biol. 2018, 20, 888–899. [Google Scholar] [CrossRef]
- Wilson, K.E.; Yang, N.; Mussell, A.L.; Zhang, J. The Regulatory Role of KIBRA and PTPN14 in Hippo Signaling and Beyond. Genes 2016, 7, 23. [Google Scholar] [CrossRef]
- Yu, J.; Zheng, Y.; Dong, J.; Klusza, S.; Deng, W.M.; Pan, D. Kibra functions as a tumor suppressor protein that regulates Hippo signaling in conjunction with Merlin and Expanded. Dev. Cell 2010, 18, 288–299. [Google Scholar] [CrossRef]
- Xiao, L.; Chen, Y.; Ji, M.; Dong, J. KIBRA regulates Hippo signaling activity via interactions with large tumor suppressor kinases. J. Biol. Chem. 2011, 286, 7788–7796. [Google Scholar] [CrossRef]
- Couzens, A.L.; Knight, J.D.; Kean, M.J.; Teo, G.; Weiss, A.; Dunham, W.H.; Lin, Z.Y.; Bagshaw, R.D.; Sicheri, F.; Pawson, T.; et al. Protein interaction network of the mammalian Hippo pathway reveals mechanisms of kinase-phosphatase interactions. Sci. Signal. 2013, 6, rs15. [Google Scholar] [CrossRef]
- Meng, Z.; Moroishi, T.; Mottier-Pavie, V.; Plouffe, S.W.; Hansen, C.G.; Hong, A.W.; Park, H.W.; Mo, J.S.; Lu, W.; Lu, S.; et al. MAP4K family kinases act in parallel to MST1/2 to activate LATS1/2 in the Hippo pathway. Nat. Commun. 2015, 6, 8357. [Google Scholar] [CrossRef]
- Panciera, T.; Azzolin, L.; Cordenonsi, M.; Piccolo, S. Mechanobiology of YAP and TAZ in physiology and disease. Nat. Rev. Mol. Cell Biol. 2017, 18, 758–770. [Google Scholar] [CrossRef]
- Aragona, M.; Panciera, T.; Manfrin, A.; Giulitti, S.; Michielin, F.; Elvassore, N.; Dupont, S.; Piccolo, S. A mechanical checkpoint controls multicellular growth through YAP/TAZ regulation by actin-processing factors. Cell 2013, 154, 1047–1059. [Google Scholar] [CrossRef]
- Gaspar, P.; Tapon, N. Sensing the local environment: Actin architecture and Hippo signalling. Curr. Opin. Cell Biol. 2014, 31, 74–83. [Google Scholar] [CrossRef]
- Neto, F.; Klaus-Bergmann, A.; Ong, Y.T.; Alt, S.; Vion, A.C.; Szymborska, A.; Carvalho, J.R.; Hollfinger, I.; Bartels-Klein, E.; Franco, C.A.; et al. YAP and TAZ regulate adherens junction dynamics and endothelial cell distribution during vascular development. eLife 2018, 7, e31037. [Google Scholar] [CrossRef]
- Elbediwy, A.; Vincent-Mistiaen, Z.I.; Spencer-Dene, B.; Stone, R.K.; Boeing, S.; Wculek, S.K.; Cordero, J.; Tan, E.H.; Ridgway, R.; Brunton, V.G.; et al. Integrin signalling regulates YAP and TAZ to control skin homeostasis. Development 2016, 143, 1674–1687. [Google Scholar] [CrossRef]
- Meng, Z.; Qiu, Y.; Lin, K.C.; Kumar, A.; Placone, J.K.; Fang, C.; Wang, K.C.; Lu, S.; Pan, M.; Hong, A.W.; et al. RAP2 mediates mechanoresponses of the Hippo pathway. Nature 2018, 560, 655–660. [Google Scholar] [CrossRef]
- Elosegui-Artola, A.; Andreu, I.; Beedle, A.E.M.; Lezamiz, A.; Uroz, M.; Kosmalska, A.J.; Oria, R.; Kechagia, J.Z.; Rico-Lastres, P.; Le Roux, A.L.; et al. Force Triggers YAP Nuclear Entry by Regulating Transport across Nuclear Pores. Cell 2017, 171, 1397–1410.e14. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Wei, X.; Li, W.; Udan, R.S.; Yang, Q.; Kim, J.; Xie, J.; Ikenoue, T.; Yu, J.; Li, L.; et al. Inactivation of YAP oncoprotein by the Hippo pathway is involved in cell contact inhibition and tissue growth control. Genes Dev. 2007, 21, 2747–2761. [Google Scholar] [CrossRef] [PubMed]
- Barry, E.R.; Simov, V.; Valtingojer, I.; Venier, O. Recent Therapeutic Approaches to Modulate the Hippo Pathway in Oncology and Regenerative Medicine. Cells 2021, 10, 2715. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.; Hu, J.W.; He, X.R.; Jin, W.L.; He, X.Y. Statins: A repurposed drug to fight cancer. J. Exp. Clin. Cancer Res. 2021, 40, 241. [Google Scholar] [CrossRef]
- Shorstova, T.; Foulkes, W.D.; Witcher, M. Achieving clinical success with BET inhibitors as anti-cancer agents. Br. J. Cancer 2021, 124, 1478–1490. [Google Scholar] [CrossRef]
- Luo, J.; Deng, L.; Zou, H.; Guo, Y.; Tong, T.; Huang, M.; Ling, G.; Li, P. New insights into the ambivalent role of YAP/TAZ in human cancers. J. Exp. Clin. Cancer Res. 2023, 42, 130. [Google Scholar] [CrossRef]
- Zanconato, F.; Battilana, G.; Cordenonsi, M.; Piccolo, S. YAP/TAZ as therapeutic targets in cancer. Curr. Opin. Pharmacol. 2016, 29, 26–33. [Google Scholar] [CrossRef]
- Zanconato, F.; Cordenonsi, M.; Piccolo, S. YAP/TAZ at the Roots of Cancer. Cancer Cell 2016, 29, 783–803. [Google Scholar] [CrossRef]
- Orr, B.A.; Bai, H.; Odia, Y.; Jain, D.; Anders, R.A.; Eberhart, C.G. Yes-associated protein 1 is widely expressed in human brain tumors and promotes glioblastoma growth. J. Neuropathol. Exp. Neurol. 2011, 70, 568–577. [Google Scholar] [CrossRef]
- Fernandez, L.A.; Northcott, P.A.; Dalton, J.; Fraga, C.; Ellison, D.; Angers, S.; Taylor, M.D.; Kenney, A.M. YAP1 is amplified and up-regulated in hedgehog-associated medulloblastomas and mediates Sonic hedgehog-driven neural precursor proliferation. Genes Dev. 2009, 23, 2729–2741. [Google Scholar] [CrossRef]
- Li, W.; Dong, S.; Wei, W.; Wang, G.; Zhang, A.; Pu, P.; Jia, Z. The role of transcriptional coactivator TAZ in gliomas. Oncotarget 2016, 7, 82686–82699. [Google Scholar] [CrossRef][Green Version]
- Xu, Y.; Stamenkovic, I.; Yu, Q. CD44 attenuates activation of the hippo signaling pathway and is a prime therapeutic target for glioblastoma. Cancer Res. 2010, 70, 2455–2464. [Google Scholar] [CrossRef]
- Lau, Y.K.; Murray, L.B.; Houshmandi, S.S.; Xu, Y.; Gutmann, D.H.; Yu, Q. Merlin is a potent inhibitor of glioma growth. Cancer Res. 2008, 68, 5733–5742. [Google Scholar] [CrossRef]
- Chao, Y.; Wang, Y.; Liu, X.; Ma, P.; Shi, Y.; Gao, J.; Shi, Q.; Hu, J.; Yu, R.; Zhou, X. Mst1 regulates glioma cell proliferation via the AKT/mTOR signaling pathway. J. Neuro-Oncol. 2015, 121, 279–288. [Google Scholar] [CrossRef]
- Quick, Q.A.; Gewirtz, D.A. An accelerated senescence response to radiation in wild-type p53 glioblastoma multiforme cells. J. Neurosurg. 2006, 105, 111–118. [Google Scholar] [CrossRef]
- Zhang, L.; Cheng, F.; Wei, Y.; Zhang, L.; Guo, D.; Wang, B.; Li, W. Inhibition of TAZ contributes radiation-induced senescence and growth arrest in glioma cells. Oncogene 2019, 38, 2788–2799. [Google Scholar] [CrossRef]
- Minata, M.; Audia, A.; Shi, J.; Lu, S.; Bernstock, J.; Pavlyukov, M.S.; Das, A.; Kim, S.H.; Shin, Y.J.; Lee, Y.; et al. Phenotypic Plasticity of Invasive Edge Glioma Stem-like Cells in Response to Ionizing Radiation. Cell Rep. 2019, 26, 1893–1905.e7. [Google Scholar] [CrossRef]
- Verhaak, R.G.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef]
- Bhat, K.P.; Salazar, K.L.; Balasubramaniyan, V.; Wani, K.; Heathcock, L.; Hollingsworth, F.; James, J.D.; Gumin, J.; Diefes, K.L.; Kim, S.H.; et al. The transcriptional coactivator TAZ regulates mesenchymal differentiation in malignant glioma. Genes Dev. 2011, 25, 2594–2609. [Google Scholar] [CrossRef]
- Castellan, M.; Guarnieri, A.; Fujimura, A.; Zanconato, F.; Battilana, G.; Panciera, T.; Sladitschek, H.L.; Contessotto, P.; Citron, A.; Grilli, A.; et al. Single-cell analyses reveal YAP/TAZ as regulators of stemness and cell plasticity in glioblastoma. Nat. Cancer 2021, 2, 174–188. [Google Scholar] [CrossRef] [PubMed]
- Pobbati, A.V.; Kumar, R.; Rubin, B.P.; Hong, W. Therapeutic targeting of TEAD transcription factors in cancer. Trends Biochem. Sci. 2023, 48, 450–462. [Google Scholar] [CrossRef] [PubMed]
- Beeghly, G.F.; Amofa, K.Y.; Fischbach, C.; Kumar, S. Regulation of Tumor Invasion by the Physical Microenvironment: Lessons from Breast and Brain Cancer. Annu. Rev. Biomed. Eng. 2022, 24, 29–59. [Google Scholar] [CrossRef]
- Parkins, C.C.; McAbee, J.H.; Ruff, L.; Wendler, A.; Mair, R.; Gilbertson, R.J.; Watts, C.; Scherman, O.A. Mechanically matching the rheological properties of brain tissue for drug-delivery in human glioblastoma models. Biomaterials 2021, 276, 120919. [Google Scholar] [CrossRef] [PubMed]
- Sivakumar, H.; Strowd, R.; Skardal, A. Exploration of Dynamic Elastic Modulus Changes on Glioblastoma Cell Populations with Aberrant EGFR Expression as a Potential Therapeutic Intervention Using a Tunable Hyaluronic Acid Hydrogel Platform. Gels 2017, 3, 28. [Google Scholar] [CrossRef]
- Brusatin, G.; Panciera, T.; Gandin, A.; Citron, A.; Piccolo, S. Biomaterials and engineered microenvironments to control YAP/TAZ-dependent cell behaviour. Nat. Mater. 2018, 17, 1063–1075. [Google Scholar] [CrossRef]
- Wolf, K.J.; Chen, J.; Coombes, J.; Aghi, M.K.; Kumar, S. Dissecting and rebuilding the glioblastoma microenvironment with engineered materials. Nat. Rev. Mater. 2019, 4, 651–668. [Google Scholar] [CrossRef]
- Baruffaldi, D.; Palmara, G.; Pirri, C.; Frascella, F. 3D Cell Culture: Recent Development in Materials with Tunable Stiffness. ACS Appl. Bio Mater. 2021, 4, 2233–2250. [Google Scholar] [CrossRef]
- El Kheir, W.; Marcos, B.; Virgilio, N.; Paquette, B.; Faucheux, N.; Lauzon, M.A. Drug Delivery Systems in the Development of Novel Strategies for Glioblastoma Treatment. Pharmaceutics 2022, 14, 1189. [Google Scholar] [CrossRef]
- Stylianopoulos, T.; Munn, L.L.; Jain, R.K. Reengineering the Physical Microenvironment of Tumors to Improve Drug Delivery and Efficacy: From Mathematical Modeling to Bench to Bedside. Trends Cancer 2018, 4, 292–319. [Google Scholar] [CrossRef]
- Ruiz-Molina, D.; Mao, X.; Alfonso-Triguero, P.; Lorenzo, J.; Bruna, J.; Yuste, V.J.; Candiota, A.P.; Novio, F. Advances in Preclinical/Clinical Glioblastoma Treatment: Can Nanoparticles Be of Help? Cancers 2022, 14, 4960. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pontes, B.; Mendes, F.A. Mechanical Properties of Glioblastoma: Perspectives for YAP/TAZ Signaling Pathway and Beyond. Diseases 2023, 11, 86. https://doi.org/10.3390/diseases11020086
Pontes B, Mendes FA. Mechanical Properties of Glioblastoma: Perspectives for YAP/TAZ Signaling Pathway and Beyond. Diseases. 2023; 11(2):86. https://doi.org/10.3390/diseases11020086
Chicago/Turabian StylePontes, Bruno, and Fabio A. Mendes. 2023. "Mechanical Properties of Glioblastoma: Perspectives for YAP/TAZ Signaling Pathway and Beyond" Diseases 11, no. 2: 86. https://doi.org/10.3390/diseases11020086
APA StylePontes, B., & Mendes, F. A. (2023). Mechanical Properties of Glioblastoma: Perspectives for YAP/TAZ Signaling Pathway and Beyond. Diseases, 11(2), 86. https://doi.org/10.3390/diseases11020086