

Towards Understanding the Key Signature Pathways Associated from Differentially Expressed Gene Analysis in an Indian Prostate Cancer Cohort
 , , ,
, , , 
Abstract
1. Introduction
2. Materials and Methods
2.1. Patients, Clinical Samples, and Criteria
2.2. Tissue Preparation and RNA Sequencing
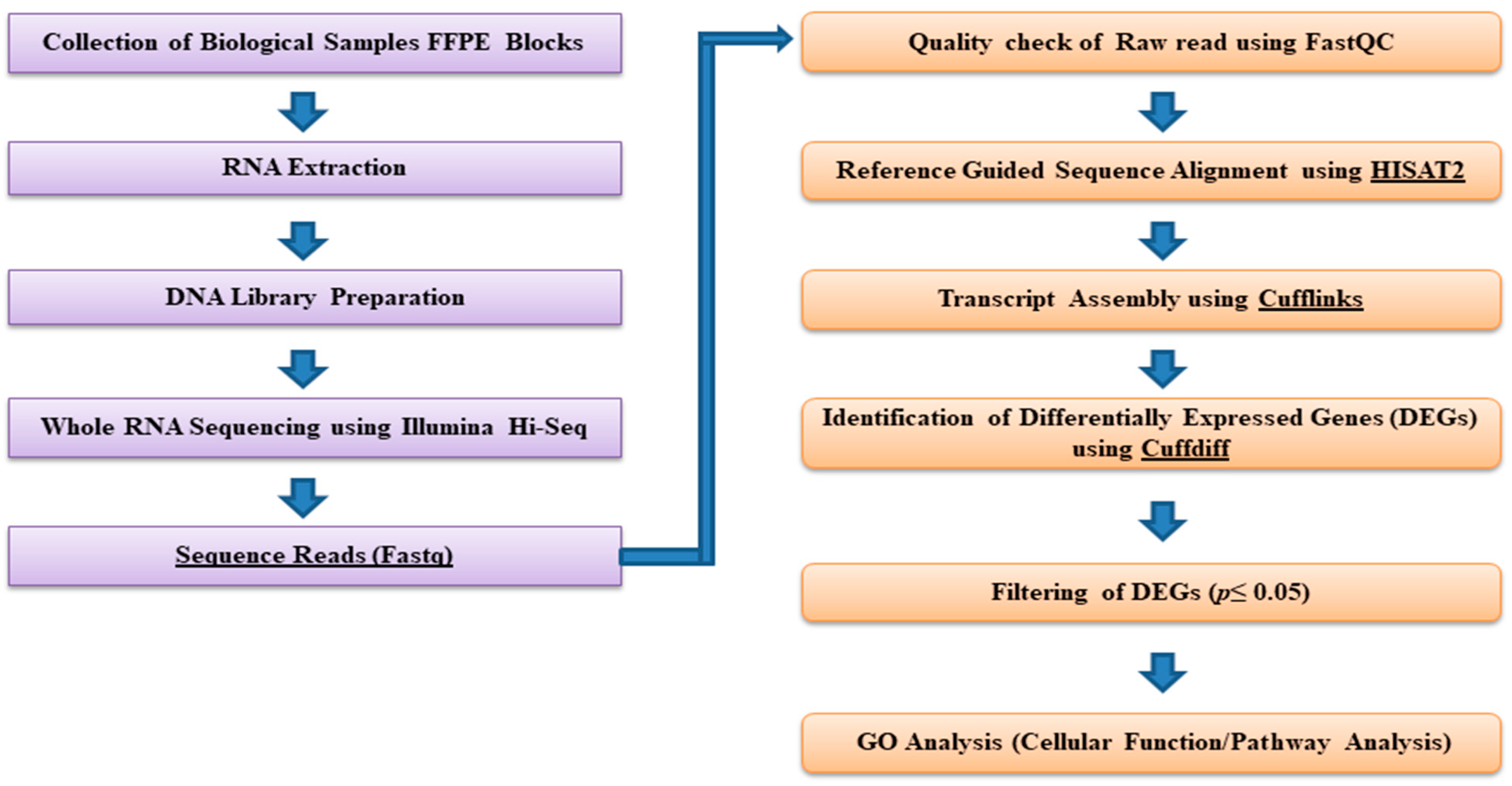
2.3. Bioinformatics and Downstream RNA-Sequencing Analysis
2.4. Interaction Networks, Statistical Analysis, Gene Ontology, and Cbioportal Analyses
3. Results
3.1. Distinct DEGs Were Obtained
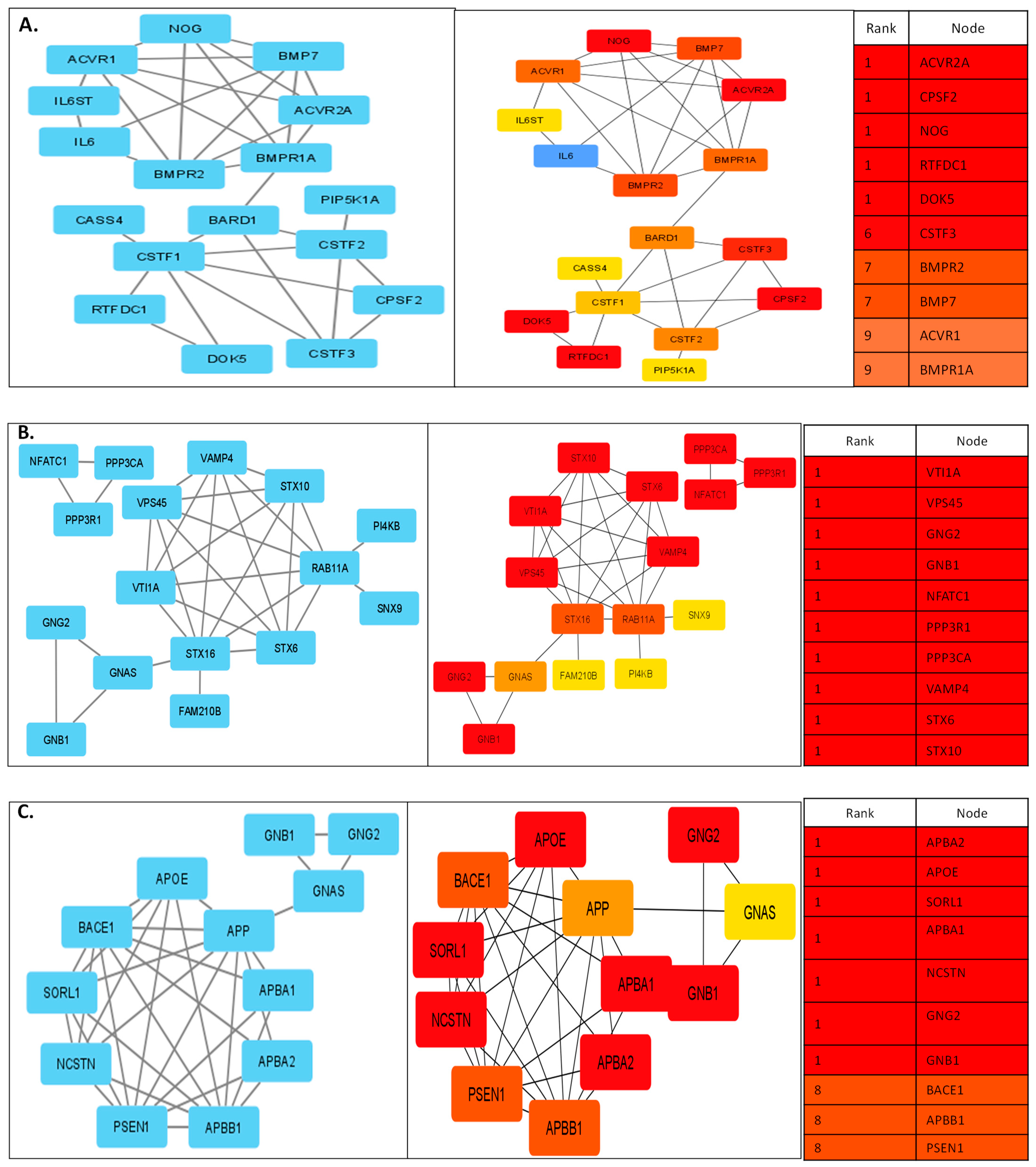
3.2. Protein Interactions Yielded Innate Pathways Responsible for PCa
3.3. Validation of RNA-seq Result Using TCGA Dataset by Cbioportal
3.4. Gene Ontology Yielded Distinct Pathways Regulating Biogenesis
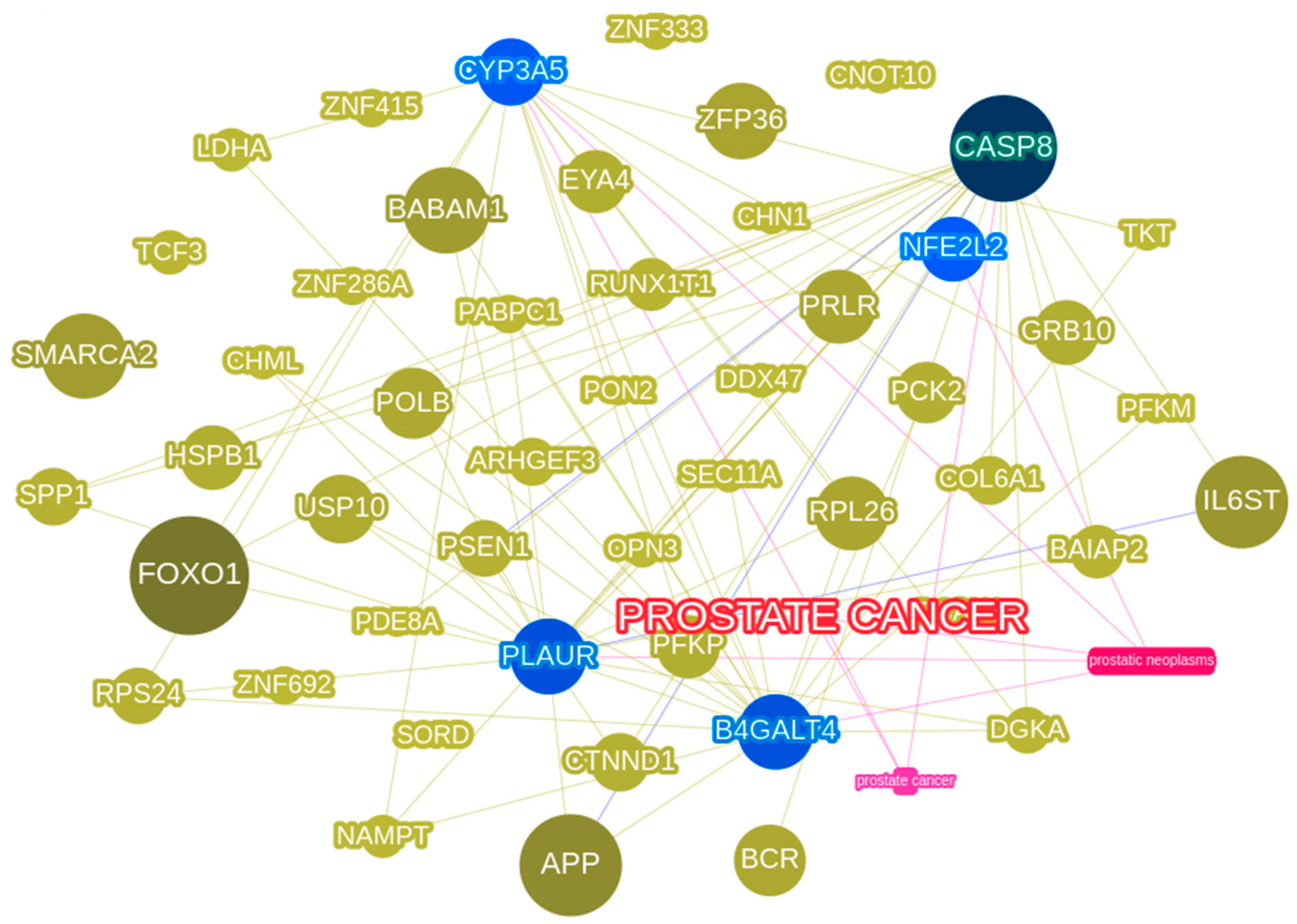
3.5. Phenolyzer Highlights Important DEGs
4. Discussion
4.1. Comparative Analysis of RNA-seq Data with Other Publicly Available Datasets
4.2. A Major Chunk of lncRNAs Are Novel and Regulated in Distinct Pathways
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jemal, A.; Bray, F.; Center, M.M.; Ferlay, J.; Ward, E.; Forman, D. Global cancer statistics. CA Cancer J. Clin. 2011, 61, 69–90. [Google Scholar] [CrossRef] [PubMed]
- Sim, H.G.; Cheng, C.W.S. Changing demography of prostate cancer in Asia. Eur. J. Cancer 2005, 41, 834–845. [Google Scholar] [CrossRef]
- Jain, S.; Saxena, S.; Kumar, A. Epidemiology of prostate cancer in India. Meta Gene 2014, 2, 596–605. [Google Scholar] [CrossRef] [PubMed]
- NCRP. Three-Year Report of the Population Based Cancer Registries-2009–2011; National Centre for Disease Informatics and Research (NCDIR); Indian Council of Medical Research ICMR: Bangalore, India, 2013.
- Trapnell, C.; Pachter, L.; Salzberg, S.L. TopHat: Discovering splice junctions with RNA-Seq. Bioinformatics 2009, 25, 1105–1111. [Google Scholar] [CrossRef] [PubMed]
- Guan, Y.F.; Li, G.R.; Wang, R.J.; Yi, Y.T.; Yang, L.; Jiang, D.; Zhang, X.P.; Peng, Y. Application of next-generation sequencing in clinical oncology to advance personalized treatment of cancer. Chin. J. Cancer 2012, 31, 463. [Google Scholar] [CrossRef]
- Gupta, A.; Shukla, N.; Nehra, M.; Gupta, S.; Malik, B.; Mishra, A.K.; Vijay, M.; Batra, J.; Lohiya, N.K.; Sharma, D.; et al. A Pilot Study on the Whole Exome Sequencing of Prostate Cancer in the Indian Phenotype Reveals Distinct Polymorphisms. Front. Genet. 2020, 11, 874. [Google Scholar] [CrossRef]
- Stark, R.; Grzelak, M.; Hadfield, J. RNA sequencing: The teenage years. Nat. Rev. Genet. 2019, 20, 631–656. [Google Scholar] [CrossRef]
- Marco-Puche, G.; Lois, S.; Benítez, J.; Trivino, J.C. RNA-Seq Perspectives to Improve Clinical Diagnosis. Front. Genet. 2019, 10, 1152. [Google Scholar] [CrossRef]
- Stupnikov, A.; McInerney, C.E.; Savage, K.I.; McIntosh, S.A.; Emmert-Streib, F.; Kennedy, R.; Salto-Tellez, M.; Prise, K.M.; McArt, D.G. Robustness of differential gene expression analysis of RNA-seq. Comput. Struct. Biotechnol. J. 2021, 19, 3470–3481. [Google Scholar] [CrossRef]
- Kukurba, K.R.; Montgomery, S.B. RNA sequencing and analysis. Cold Spring Harb. Protoc. 2015, 2015, 951–969. [Google Scholar] [CrossRef]
- Xi, X.; Li, T.; Huang, Y.; Sun, J.; Zhu, Y.; Yang, Y.; Lu, Z.J. RNA biomarkers: Frontier of precision medicine for cancer. Non-Coding RNA 2017, 3, 9. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, S.K.; Zetter, B.R. Cancer biomarkers: Knowing the present and predicting the future. Future Oncol. 2005, 1, 37–50. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.L. The biogenesis and emerging roles of circular RNAs. Nat. Rev. Mol. Cell Biol. 2016, 17, 205–211. [Google Scholar] [CrossRef]
- Lopez, J.P.; Diallo, A.; Cruceanu, C.; Fiori, L.M.; Laboissiere, S.; Guillet, I.; Fontaine, J.; Ragoussis, J.; Benes, V.; Turecki, G.; et al. Biomarker discovery: Quantification of microRNAs and other small non-coding RNAs using next generation sequencing. BMC Med. Genom. 2015, 8, 35. [Google Scholar] [CrossRef]
- Krishnan, P.; Damaraju, S. The Challenges and Opportunities in the Clinical Application of Noncoding RNAs: The Road Map for miRNAs and piRNAs in Cancer Diagnostics and Prognostics. Int. J. Genom. 2018, 2018, 5848046. [Google Scholar] [CrossRef]
- Grixti, J.M.; Ayers, D. Long noncoding RNAs and their link to cancer. Non-Coding RNA Res. 2020, 5, 77–82. [Google Scholar] [CrossRef]
- Evans, J.R.; Feng, F.Y.; Chinnaiyan, A.M. The bright side of dark matter: LncRNAs in cancer. J. Clin. Investig. 2016, 126, 2775–2782. [Google Scholar] [CrossRef] [PubMed]
- Arun, G.; Diermeier, S.D.; Spector, D.L. Therapeutic Targeting of Long Non-Coding RNAs in Cancer. Trends Mol. Med. 2018, 24, 257–277. [Google Scholar] [CrossRef]
- Gao, N.; Li, Y.; Li, J.; Gao, Z.; Yang, Z.; Li, Y.; Liu, H.; Fan, T. Long Non-Coding RNAs: The Regulatory Mechanisms, Research Strategies, and Future Directions in Cancers. Front. Oncol. 2020, 10, 598817. [Google Scholar] [CrossRef]
- Winkle, M.; El-Daly, S.M.; Fabbri, M.; Calin, G.A. Noncoding RNA therapeutics—Challenges and potential solutions. Nat. Rev. Drug Discov. 2021, 20, 629–651. [Google Scholar] [CrossRef]
- Kumar, P.; Chakraborty, J.; Sukumar, G.; Dalgard, C.; Chatterjee, R.; Biswas, R. Comparative RNA-seq analysis reveals dys-regulation of major canonical pathways in ERG-inducible LNCaP cell progression model of prostate cancer. Oncotarget 2019, 10, 4290–4306. [Google Scholar] [CrossRef]
- Tripathi, R.; Sharma, P.; Chakraborty, P.; Varadwaj, P. Count-based transcriptome analysis to identify differentially expressed genes for breast cancer. In Proceedings of the 2016 International Conference on Bioinformatics and Systems Biology (BSB), Allahabad, India, 4–6 March 2016; pp. 1–5. [Google Scholar] [CrossRef]
- Kim, S.M.; Rampogu, S.; Vetrivel, P.; Kulkarni, A.M.; Ha, S.E.; Kim, H.H.; Lee, K.W.; Kim, G.S. Transcriptome analysis of sinensetin-treated liver cancer cells guided by biological network analysis. Oncol. Lett. 2021, 21, 355. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Roberts, A.; Goff, L.; Pertea, G.; Kim, D.; Kelley, D.R.; Pimentel, H.; Salzberg, S.L.; Rinn, J.L.; Pachter, L. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 2012, 7, 562–578. [Google Scholar] [CrossRef]
- Gupta, A.; Gupta, S.; Jatawa, S.K.; Kumar, A.; Suravajhala, P. A simplest bioinformatics pipeline for whole transcriptome sequencing: Overview of the processing and steps from raw data to downstream analysis. BioRxiv 2019. [Google Scholar] [CrossRef]
- Ferragina, P.; Manzini, G. Opportunistic data structures with applications. In Proceedings of the Annual Symposium on Foundations of Computer Science—Proceedings, Redondo Beach, CA, USA, 12–14 November 2000. [Google Scholar]
- Trapnell, C.; Hendrickson, D.G.; Sauvageau, M.; Goff, L.; Rinn, J.L.; Pachter, L. Differential analysis of gene regulation at transcript resolution with RNA-seq. Nat. Biotechnol. 2013, 31, 46–53. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Zhao, J.; Xue, L.; Zhao, T.; Ding, W.; Han, Y.; Ye, H. A comparison of transcriptome analysis methods with reference genome. BMC Genom. 2022, 23, 232. [Google Scholar] [CrossRef]
- Mostafavi, S.; Morris, Q. Combining many interaction networks to predict gene function and analyze gene lists. Proteomics 2012, 12, 1687–1696. [Google Scholar] [CrossRef]
- Warde-Farley, D.; Donaldson, S.L.; Comes, O.; Zuberi, K.; Badrawi, R.; Chao, P.; Franz, M.; Grouios, C.; Kazi, F.; Lopes, C.T.; et al. The GeneMANIA prediction server: Biological network integration for gene prioritization and predicting gene function. Nucleic Acids Res. 2010, 38, W214–W220. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Gable, A.L.; Nastou, K.C.; Lyon, D.; Kirsch, R.; Pyysalo, S.; Doncheva, N.T.; Legeay, M.; Fang, T.; Bork, P.; et al. The STRING database in 2021: Customizable protein-protein networks, and functional characterization of user-uploaded gene/measurement sets. Nucleic Acids Res. 2021, 49, D605–D612. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software Environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Saito, R.; Smoot, M.E.; Ono, K.; Ruscheinski, J.; Wang, P.L.; Lotia, S.; Pico, A.R.; Bader, G.D.; Ideker, T. A travel guide to Cytoscape plugins. Nat. Methods 2012, 9, 1069–1076. [Google Scholar] [CrossRef] [PubMed]
- Chin, C.H.; Chen, S.H.; Wu, H.H.; Ho, C.W.; Ko, M.T.; Lin, C.Y. cytoHubba: Identifying hub objects and sub-networks from complex interactome. BMC Syst. Biol. 2014, 8, S11. [Google Scholar] [CrossRef] [PubMed]
- Mi, H.; Muruganujan, A.; Casagrande, J.T.; Thomas, P.D. Large-scale gene function analysis with the panther classification system. Nat. Protoc. 2013, 8, 1551–1566. [Google Scholar] [CrossRef] [PubMed]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio Cancer Genomics Portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Lichtenberg, T.; Hoadley, K.A.; Poisson, L.M.; Lazar, A.J.; Cherniack, A.D.; Kovatich, A.J.; Benz, C.C.; Levine, D.A.; Lee, A.V.; et al. An Integrated TCGA Pan-Cancer Clinical Data Resource to Drive High-Quality Survival Outcome Analytics. Cell 2018, 173, 400–416.e11. [Google Scholar] [CrossRef]
- Shukla, N.; Siva, N.; Sivakumar, M.; Parveen, R.; Mishra, A.; Shah, A.; Medicherla, K.; Suravajhala, P. Extraction of DNA and RNA from Formalin-fixed Paraffin-embedded Tissue Specimens. Bio-Protocol 2021, 11, e4095. [Google Scholar] [CrossRef]
- Tanaka, T.; Ikari, K.; Furushima, K.; Okada, A.; Tanaka, H.; Furukawa, K.I.; Yoshida, K.; Ikeda, T.; Ikegawa, S.; Hunt, S.C.; et al. Genomewide Linkage and Linkage Disequilibrium Analyses Identify COL6A1, on Chromosome 21, as the Locus for Ossification of the Posterior Longitudinal Ligament of the Spine. Am. J. Hum. Genet. 2003, 73, 812–822. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, Z.; Yang, X.; Lu, W.; Chen, Y.; Lin, Y.; Wang, J.; Lin, S.; Yun, J.P. H3K27 acetylation activated-COL6A1 promotes osteosarcoma lung metastasis by repressing STAT1 and activating pulmonary cancer-associated fibroblasts. Theranostics 2021, 11, 1473–1492. [Google Scholar] [CrossRef]
- Owusu-Ansah, K.G.; Song, G.; Chen, R.; Edoo, M.I.A.; Li, J.; Chen, B.; Wu, J.; Zhou, L.; Xie, H.; Jiang, D.; et al. COL6A1 promotes metastasis and predicts poor prognosis in patients with pancreatic cancer. Int. J. Oncol. 2019, 55, 391–404. [Google Scholar] [CrossRef]
- Li, N.; Liu, M.; Cao, X.; Li, W.; Li, Y.; Zhao, Z. Identification of differentially expressed genes using microarray analysis and COL6A1 induction of bone metastasis in non-small cell lung cancer. Oncol. Lett. 2021, 22, 12954. [Google Scholar] [CrossRef]
- Lin, Z.; Zhu, G.; Tang, D.; Bu, J.; Zou, J. High expression of COL6A1 correlates with poor prognosis in patients with breast cancer. Int. J. Clin. Exp. Med. 2018, 11, 12157–12164. [Google Scholar]
- Lu, Q. δ-Catenin dysregulation in cancer: Interactions with E-cadherin and beyond. J. Pathol. 2010, 222, 119–123. [Google Scholar] [CrossRef] [PubMed]
- Tang, B.; Tang, F.; Wang, Z.; Qi, G.; Liang, X.; Li, B.; Yuan, S.; Liu, J.; Yu, S.; He, S. Overexpression of CTNND1 in hepatocellular carcinoma promotes carcinous characters through activation of Wnt/β-catenin signaling. J. Exp. Clin. Cancer Res. 2016, 35, 82. [Google Scholar] [CrossRef]
- Liao, X.H.; Zhang, Y.; Dong, W.J.; Shao, Z.M.; Li, D.Q. Chromatin remodeling protein MORC2 promotes breast cancer invasion and metastasis through a PRD domain-mediated interaction with CTNND1. Oncotarget 2017, 8, 97941–97954. [Google Scholar] [CrossRef] [PubMed]
- Burnell, S.E.A.; Spencer-Harty, S.; Howarth, S.; Bodger, O.; Kynaston, H.; Morgan, C.; Doak, S.H. STEAP2 Knockdown Reduces the Invasive Potential of Prostate Cancer Cells. Sci. Rep. 2018, 8, 6252. [Google Scholar] [CrossRef] [PubMed]
- Grimm, J.; Sachs, M.; Britsch, S.; Di Cesare, S.; Schwarz-Romond, T.; Alitalo, K.; Birchmeier, W. Novel p62dok family members, dok-4 and dok-5, are substrates of the c-Ret receptor tyrosine kinase and mediate neuronal differentiation. J. Cell Biol. 2001, 154, 345–354. [Google Scholar] [CrossRef] [PubMed]
- Luo, F.; Wang, Z.; Chen, S.; Luo, Z.; Wang, G.; Yang, H.; Tang, L. DOK5 as a Prognostic Biomarker of Gastric Cancer Immunoinvasion: A Bioinformatics Analysis. Biomed. Res. Int. 2022, 2022, 9914778. [Google Scholar] [CrossRef]
- Amaral, P.P.; Neyt, C.; Wilkins, S.J.; Askarian-Amiri, M.E.; Sunkin, S.M.; Perkins, A.C.; Mattick, J.S. Complex architecture and regulated expression of the Sox2ot locus during vertebrate development. RNA 2009, 15, 2013–2027. [Google Scholar] [CrossRef]
- Hou, Z.; Zhao, W.; Zhou, J.; Shen, L.; Zhan, P.; Xu, C.; Chang, C.; Bi, H.; Zou, J.; Yao, X.; et al. A long noncoding RNA Sox2ot regulates lung cancer cell proliferation and is a prognostic indicator of poor survival. Int. J. Biochem. Cell Biol. 2014, 53, 380–388. [Google Scholar] [CrossRef]
- Qu, F.; Cao, P. Long noncoding RNA SOX2OT contributes to gastric cancer progression by sponging miR-194-5p from AKT2. Exp. Cell Res. 2018, 369, 187–196. [Google Scholar] [CrossRef]
- Shahryari, A.; Rafiee, M.R.; Fouani, Y.; Oliae, N.A.; Samaei, N.M.; Shafiee, M.; Semnani, S.; Vasei, M.; Mowla, S.J. Two novel splice variants of SOX2OT, SOX2OT-S1, and SOX2OT-S2 are coupregulated with SOX2 and OCT4 in esophageal squamous cell carcinoma. Stem Cells 2014, 32, 126–134. [Google Scholar] [CrossRef]
- Askarian-Amiri, M.E.; Seyfoddin, V.; Smart, C.E.; Wang, J.; Kim, J.E.; Hansji, H.; Baguley, B.C.; Finlay, G.J.; Leung, E.Y. Emerging role of long non-coding RNA SOX2OT in SOX2 regulation in breast cancer. PLoS ONE 2014, 9, e102140. [Google Scholar] [CrossRef]
- Sun, J.; Wei, X.; Xu, L. Upregulation of lncRna Sox2ot indicates a poor prognosis for patients with hepatocellular carcinoma and promotes cell invasion. Oncol. Lett. 2018, 16, 1189–1195. [Google Scholar] [CrossRef] [PubMed]
- Han, L.; Zhang, W.; Zhang, B.; Zhan, L. Long non-coding RNA SOX2OT promotes cell proliferation and motility in human ovarian cancer. Exp. Ther. Med. 2018, 15, 2182–2188. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhang, X.F.; Wang, D.Y.; Zhu, Y.; Chen, L.; Zhang, J.J. Long noncoding RNA SOX2OT promotes pancreatic cancer cell migration and invasion through destabilizing FUS protein via ubiquitination. Cell Death Discov. 2021, 7, 261. [Google Scholar] [CrossRef]
- Wang, Y.; Wu, N.; Luo, X.; Zhang, X.; Liao, Q.; Wang, J. SOX2OT, a novel tumor-related long non-coding RNA. Biomed. Pharmacother. 2020, 123, 109725. [Google Scholar] [CrossRef]
- Liu, B.; Zhou, J.; Wang, C.; Chi, Y.; Wei, Q.; Fu, Z.; Lian, C.; Huang, Q.; Liao, C.; Yang, Z.; et al. LncRNA SOX2OT promotes temozolomide resistance by elevating SOX2 expression via ALKBH5-mediated epigenetic regulation in glioblastoma. Cell Death Dis. 2020, 11, 384. [Google Scholar] [CrossRef]
- Li, S.; Zhang, Q.; Liu, W.; Zhao, C. Silencing of FTX suppresses pancreatic cancer cell proliferation and invasion by upregulating miR-513b-5p. BMC Cancer 2021, 21, 290. [Google Scholar] [CrossRef]
- Chen, H.; Liu, T.; Ouyang, H.; Lin, S.; Zhong, H.; Zhang, H.; Yang, Y. Upregulation of ftx promotes osteosarcoma tumorigenesis by increasing sox4 expression via mir-214-5p. Onco. Targets. Ther. 2020, 2020, 7125–7136. [Google Scholar] [CrossRef]
- Guo, X.B.; Hua, Z.; Li, C.; Peng, L.P.; Wang, J.S.; Wang, B.; Zhi, Q.M. Biological significance of long non-coding RNA FTX expression in human colorectal cancer. Int. J. Clin. Exp. Med. 2015, 8, 15591–15600. [Google Scholar]
- Chen, W.; Li, Y.; Guo, L.; Zhang, C.; Tang, S. Long non-coding RNA FTX predicts a poor prognosis of human cancers: A meta-analysis. Biosci. Rep. 2021, 41, BSR20203995. [Google Scholar] [CrossRef]
- Huo, X.; Wang, H.; Huo, B.; Wang, L.; Yang, K.; Wang, J.; Wang, L.; Wang, H. FTX contributes to cell proliferation and migration in lung adenocarcinoma via targeting miR-335-5p/NUCB2 axis. Cancer Cell Int. 2020, 20, 89. [Google Scholar] [CrossRef]
- Zhang, F.; Wang, X.S.; Tang, B.; Li, P.A.; Wen, Y.; Yu, P.W. Long non-coding RNA FTX promotes gastric cancer progression by targeting miR-215. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 3037–3048. [Google Scholar] [CrossRef]
- Azuaje, F.J. Selecting biologically informative genes in co-expression networks with a centrality score. Biol. Direct 2014, 9, 12. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.J.; Li, J.P.; Zeng, S.H.; Han, M.; Liu, S.L.; Zou, X. DZIP1 expression as a prognostic marker in gastric cancer: A bioinformatics-based analysis. Pharmgenom. Pers. Med. 2021, 2021, 1151–1168. [Google Scholar] [CrossRef] [PubMed]
- Piao, X.M.; Hwang, B.; Jeong, P.; Byun, Y.J.; Kang, H.W.; Seo, S.P.; Kim, W.T.; Lee, J.Y.; Ha, Y.S.; Lee, Y.S.; et al. Collagen type VIα1 and 2 repress the proliferation, migration and invasion of bladder cancer cells. Int. J. Oncol. 2021, 59, 37. [Google Scholar] [CrossRef] [PubMed]
- Singhal, S.K.; Sens, D.; Sens, M.A.; Byun, J.; Yancey, R.; Caban, A.; Boisvert, H.; Hennek, S.; Bobrow, M.; Ahmed, S.; et al. Subcellular partitioning of Kaiso (ZBTB33) as a biomarker to predict overall breast cancer survival. J. Clin. Oncol. 2020, 38, 3534. [Google Scholar] [CrossRef]
- Wang, Q.; Yao, J.; Jin, Q.; Wang, X.; Zhu, H.; Huang, F.; Wang, W.; Qiang, J.; Ni, Q. LAMP1 expression is associated withpoor prognosis in breast cancer. Oncol. Lett. 2017, 14, 4729–4735. [Google Scholar] [CrossRef]
- Alessandrini, F.; Pezzè, L.; Ciribilli, Y. LAMPs: Shedding light on cancer biology. Semin. Oncol. 2017, 44, 239–253. [Google Scholar] [CrossRef]
- Sun, R.; Yang, L.; Wang, Y.; Zhang, Y.; Ke, J.; Zhao, D. DNAJB11 predicts a poor prognosis and is associated with immune infiltration in thyroid carcinoma: A bioinformatics analysis. J. Int. Med. Res. 2021, 49. [Google Scholar] [CrossRef]
- Assenov, Y.; Ramírez, F.; Schelhorn, S.E.S.E.; Lengauer, T.; Albrecht, M. Computing topological parameters of biological networks. Bioinformatics 2008, 24, 282–284. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Robinson, P.N.; Wang, K. Phenolyzer: Phenotype-based prioritization of candidate genes for human diseases. Nat. Methods 2015, 12, 841–843. [Google Scholar] [CrossRef] [PubMed]
- Klokk, T.I.; Kilander, A.; Xi, Z.; Wæhre, H.; Risberg, B.; Danielsen, H.E.; Saatcioglu, F. Kallikrein 4 is a proliferative factor that is overexpressed in prostate cancer. Cancer Res. 2007, 67, 5221–5230. [Google Scholar] [CrossRef] [PubMed]
- Carleton, N.M.; Zhu, G.; Gorbounov, M.; Miller, M.C.; Pienta, K.J.; Resar, L.M.S.; Veltri, R.W. PBOV1 as a potential biomarker for more advanced prostate cancer based on protein and digital histomorphometric analysis. Prostate 2018, 78, 547–559. [Google Scholar] [CrossRef] [PubMed]
- Du, J.X.; Luo, Y.H.; Zhang, S.J.; Wang, B.; Chen, C.; Zhu, G.Q.; Zhu, P.; Cai, C.Z.; Wan, J.L.; Cai, J.L.; et al. Splicing factor SRSF1 promotes breast cancer progression via oncogenic splice switching of PTPMT1. J. Exp. Clin. Cancer Res. 2021, 40, 171. [Google Scholar] [CrossRef]
- Pandey, P.; Sliker, B.; Peters, H.L.; Tuli, A.; Herskovitz, J.; Smits, K.; Purohit, A.; Singh, R.K.; Dong, J.; Batra, S.K.; et al. Amyloid precursor protein and amyloid precursor-like protein 2 in cancer. Oncotarget 2016, 7, 19430–19444. [Google Scholar] [CrossRef] [PubMed]
- Kantidze, O.L.; Kamalyukova, I.M.; Razin, S.V. Association of the mammalian transcriptional regulator kaiso with centrosomes and the midbody. Cell Cycle 2009, 8, 2303–2304. [Google Scholar] [CrossRef] [PubMed]
- Schackmann, R.C.J.; Tenhagen, M.; van de Ven, R.A.H.; Derksen, P.W.B. P120-catenin in cancer—Mechanisms, models and opportunities for intervention. J. Cell Sci. 2013, 126, 3515–3525. [Google Scholar] [CrossRef]
- Singh, K.K.; Chaubey, G.; Chen, J.Y.; Suravajhala, P. Decoding SARS-CoV-2 hijacking of host mitochondria in COVID-19 pathogenesis. Am. J. Physiol.-Cell Physiol. 2020, 319, C258–C267. [Google Scholar] [CrossRef] [PubMed]
- Shukla, N.; Prasad, A.; Kanga, U.; Suravajhala, R.; Nigam, V.K.; Kishor, P.B.K.; Polavarapu, R.; Chaubey, G.; Singh, K.K.; Suravajhala, P. SARS-CoV-2 transgressing LncRNAs uncovers the known unknowns. Physiol. Genom. 2021, 53, 433–440. [Google Scholar] [CrossRef]
- Stamey, T.A.; Yang, N.; Hay, A.R.; McNeal, J.E.; Freiha, F.S.; Redwine, E. Prostate-Specific Antigen as a Serum Marker for Adenocarcinoma of the Prostate. N. Engl. J. Med. 1987, 317, 909–916. [Google Scholar] [CrossRef] [PubMed]
- Ahn, J.; Berndt, S.I.; Wacholder, S.; Kraft, P.; Kibel, A.S.; Yeager, M.; Albanes, D.; Giovannucci, E.; Stampfer, M.J.; Virtamo, J.; et al. Variation in KLK genes, prostate-specific antigen and risk of prostate cancer. Nat. Genet. 2008, 40, 1032–1034. [Google Scholar] [CrossRef] [PubMed]
- Shang, Z.; Niu, Y.; Cai, Q.; Chen, J.; Tian, J.; Yeh, S.; Lai, K.P.; Chang, C. Human kallikrein 2 (KLK2) promotes prostate cancer cell growth via function as a modulator to promote the ARA70-enhanced androgen receptor transactivation. Tumor Biol. 2014, 35, 1881–1890. [Google Scholar] [CrossRef] [PubMed]
- Meng, J.; Wang, J. Role of SNARE proteins in tumourigenesis and their potential as targets for novel anti-cancer therapeutics. Biochim. Biophys. Acta-Rev. Cancer 2015, 1856, 1–12. [Google Scholar] [CrossRef]
- Han, J.; Pluhackova, K.; Böckmann, R.A. The multifaceted role of SNARE proteins in membrane fusion. Front. Physiol. 2017, 8, 5. [Google Scholar] [CrossRef]
- Riggs, K.A.; Hasan, N.; Humphrey, D.; Raleigh, C.; Nevitt, C.; Corbin, D.; Hu, C. Regulation of integrin endocytic recycling and chemotactic cell migration by syntaxin 6 and VAMP3 interaction. J. Cell Sci. 2012, 125, 3827–3839. [Google Scholar] [CrossRef]
- Yan, R. Physiological functions of the β-site amyloid precursor protein cleaving enzyme 1 and 2. Front. Mol. Neurosci. 2017, 10, 97. [Google Scholar]
- Farris, F.; Matafora, V.; Bachi, A. The emerging role of β-secretases in cancer. J. Exp. Clin. Cancer Res. 2021, 40, 147. [Google Scholar] [CrossRef]
- Ren, S.; Peng, Z.; Mao, J.H.; Yu, Y.; Yin, C.; Gao, X.; Cui, Z.; Zhang, J.; Yi, K.; Xu, W.; et al. RNA-seq analysis of prostate cancer in the Chinese population identifies recurrent gene fusions, cancer-associated long noncoding RNAs and aberrant alternative splicings. Cell Res. 2012, 22, 806–821. [Google Scholar] [CrossRef]
- Guo, H.; Ahmed, M.; Zhang, F.; Yao, C.Q.; Li, S.; Liang, Y.; Hua, J.; Soares, F.; Sun, Y.; Langstein, J.; et al. Modulation of long noncoding RNAs by risk SNPs underlying genetic predispositions to prostate cancer. Nat. Genet. 2016, 48, 1142–1150. [Google Scholar] [CrossRef]
- Ji, P.; Diederichs, S.; Wang, W.; Böing, S.; Metzger, R.; Schneider, P.M.; Tidow, N.; Brandt, B.; Buerger, H.; Bulk, E.; et al. MALAT-1, a novel noncoding RNA, and thymosin β4 predict metastasis and survival in early-stage non-small cell lung cancer. Oncogene 2003, 22, 8031–8041. [Google Scholar] [CrossRef]
- Ren, S.; Liu, Y.; Xu, W.; Sun, Y.; Lu, J.; Wang, F.; Wei, M.; Shen, J.; Hou, J.; Gao, X.; et al. Long noncoding RNA MALAT-1 is a new potential therapeutic target for castration resistant prostate cancer. J. Urol. 2013, 190, 2278–2287. [Google Scholar] [CrossRef] [PubMed]
- Crea, F.; Watahiki, A.; Quagliata, L.; Xue, H.; Pikor, L.; Parolia, A.; Wang, Y.; Lin, D.; Lam, W.L.; Farrar, W.L.; et al. Identification of a long non-coding RNA as a novel biomarker and potential therapeutic target for metastatic prostate cancer. Oncotarget 2014, 5, 764–774. [Google Scholar] [CrossRef] [PubMed]
- Prensner, J.R.; Zhao, S.; Erho, N.; Schipper, M.; Iyer, M.K.; Dhanasekaran, S.M.; Magi-Galluzzi, C.; Mehra, R.; Sahu, A.; Siddiqui, J.; et al. RNA biomarkers associated with metastatic progression in prostate cancer: A multi-institutional high-throughput analysis of SChLAP1. Lancet Oncol. 2014, 15, 1469–1480. [Google Scholar] [CrossRef]
- Kong, D.; Zhao, Q.; Liu, W.; Wang, F. Identification of crucial miRNAs and IncRNAs for ossification of ligamentum flavum. Mol. Med. Rep. 2019, 20, 1683–1699. [Google Scholar] [CrossRef] [PubMed]
- Rajagopal, S.; Sharma, A.; Simlot, A.; Mathur, P.; Mehta, S.; Mehta, S.; Naravula, J.; Medicherla, K.M.; Kumar, A.; Kanga, U.; et al. Inferring bona fide Differentially Expressed Genes and Their Variants Associated with Vitamin K Deficiency Using a Systems Genetics Approach. Genes 2022, 13, 2078. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample ID | Condition | Gleason Score (Primary + Secondary) |
|---|---|---|
| 69/19 | Adenocarcinoma | 3 + 4 |
| 1631/H19 | Adenocarcinoma | 4 + 4 |
| 4226/H19 | Adenocarcinoma | 3 + 4 |
| 5110/H20 | Adenocarcinoma | 4 + 4 |
| 1374/19 | Benign Nodular Prostatic Hyperplasia | <6 |
| 1266/19 | Benign Nodular Prostatic Hyperplasia | <6 |
| Inclusion Criteria | Exclusion Criteria | |
|---|---|---|
| Malignant (PCa) | Age > 55 years PSA > 4 ng/mL Gleason score > 6 Non-diabetic or any other co-morbidity | Smoking Familial history of BPH |
| Normal (BPH) | Age > 55 years PSA < 4 ng/mL Gleason score < 6 | Those with Associated diseases/phenotypes or any urological diseases. |
| Gene Id | Locus | log2 Fold | p-Value | Gene Name |
|---|---|---|---|---|
| CUFF.1000 | chr1:12314982–12315401 | 3.1420 | 0.0254 | VPS13D |
| CUFF.100001 | chr20:53952153–53952369 | 2.9034 | 0.0366 | BCAS1 |
| CUFF.10004 | chr1:151206377–151206638 | 4.6543 | 0.02435 | PIP5K1A |
| CUFF.100042 | chr20:54490801–54490981 | 8.8744 | 0.0245 | DOK5 |
| CUFF.100060 | chr20:55553908–55554084 | 7.1775 | 0.0213 | LINC01440 |
| CUFF.134059 | chr5:55937823–55938153 | 4.6464 | 0.0334 | IL6ST |
| CUFF.100106 | chr20:57168914–57169100 | 2.8468 | 0.02305 | BMP7 |
| CUFF.100077 | chr20:56403670–56403826 | 2.7896 | 0.011 | CSTF1 |
| CUFF.100080 | chr20:56412720–56412894 | 3.2345 | 0.02435 | CASS4 |
| CUFF.100086 | chr20:56487111–56487297 | 3.1269 | 0.02305 | RTF2 |
| CUFF.102595 | chr21:41275383–41275947 | 3.4149 | 0.0307 | BACE2 |
| CUFF.109200 | chr3:43335859–43336390 | 3.4173 | 0.0307 | SNRK |
| CUFF.109337 | chr3:44926095–44926812 | 4.0190 | 0.02435 | ZDHHC3 |
| CUFF.119448 | chr3:181441034–181441358 | 3.3563 | 0.0131 | SOX2OT |
| CUFF.126886 | chr4:101280781–101281251 | 3.0588 | 0.0234 | PPP3CA |
| CUFF.131979 | chr5:13991457–13991753 | 3.1847 | 0.04515 | DNAH5 |
| CUFF.13343 | chr1:201898030–201898394 | 4.054158 | 0.02435 | LMOD1 |
| CUFF.141632 | chr5:173034434–173034832 | 4.094679 | 0.02855 | ATP6VOE1 |
| CUFF.151326 | chr6:157942571–157943399 | 4.254992 | 0.004 | SNX9 |
| CUFF.157342 | chr7:90232992–90235254 | 3.248 | 0.03545 | STEAP2 |
| CUFF.100128 | chr20:57648598–57649155 | 4.119627 | 0.00535 | PMEPA1 |
| CUFF.10017 | chr1:151326615–151327193 | 3.3082 | 0.0272 | PI4KB |
| CUFF.100074 | chr20:56367973–56368588 | 4.4636 | 0.0028 | FAM21OB |
| CUFF.100241 | chr20:58895335–58895591 | 2.7206 | 0.0227 | GNAS |
| CUFF.100175 | chr20:58033916–58034202 | 5.059 | 0.04605 | STX16 |
| CUFF.100056 | chr20:55074569–55074768 | 2.743 | 0.00795 | RPL12P4 |
| CUFF.100291 | chr20:59967544–59968164 | 3.415 | 0.04675 | CADHERIN 26 |
| CUFF.100698 | chr21:14084939–14085128 | 3.123919 | 0.0416 | ENSG00000224905 |
| CUFF.10107 | chr1:152032497–152032839 | 4.3215 | 0.0145 | ENSG00000229021 |
| CUFF.101153 | chr21:18857730–18858445 | 2.8043 | 0.04035 | PPIAP22 |
| CUFF.101277 | chr21:25880415–25881777 | 5.6321 | 0.03945 | APP |
| CUFF.101659 | chr21:28672882–28673291 | 3.3034 | 0.00035 | ENSG00000232855 |
| CUFF.101853 | chr21:31210389–31210838 | 3.2157 | 0.0433 | TIAM1 |
| CUFF.102028 | chr21:33551193–33553146 | 5.4886 | 0.02575 | SON |
| CUFF.102981 | chr21:46003384–46005044 | 7.4652 | 0.0004 | COL6A1 |
| CUFF.115488 | chr3:126861928–126865263 | 3. 9928 | 0.04385 | CHCM1/CHCHD6 |
| CUFF.101498 | chr8:100713839–100714204 | 3.23209 | 0.03765 | PABPC1 |
| CUFF.110085 | chrX:74232840–74233182 | 3.23919 | 0.03765 | FTX |
| CUFF.103580 | chr9:14087901–14088194 | 3.42888 | 0.04005 | NFIB |
| CUFF.2892 | chr1:52416916–52417541 | 3.13921 | 0.0423 | TUT4 |
| CUFF.41953 | chr17:48060789–48061102 | 4.18946 | 0.04285 | NFE2L2 |
| CUFF.36847 | chr16:1699878–1700192 | 3.07352 | 0.044 | HN1L/JPT2 |
| CUFF.22947 | chr12:52949238–52950383 | 3.04539 | 0.04445 | KRT18 |
| CUFF.15787 | chr11:6483526–6483970 | 3.31699 | 0.04535 | TIMM10b |
| CUFF.3812 | chr1:70224556–70224999 | 3.31713 | 0.04535 | SRSF11 |
| CUFF.20358 | chr11:123057501–123061280 | 4.93401 | 0.0459 | HSPA8 |
| CUFF.30773 | chr14:58259607–58260234 | 2.96918 | 0.046 | PSMA3 |
| CUFF.39071 | chr16:71729367–71729916 | 2.97406 | 0.046 | AP1G1 |
| CUFF.14995 | chr10:118005601–118006215 | 3.17633 | 0.049 | RAB11FIP2 |
| CUFF.17593 | chr11:61130859–61131197 | 2.90619 | 0.0491 | VPS37C |
| CUFF.11546 | chr10:34656519–34656811 | 3.1245 | 0.00075 | PARD3 |
| CUFF.31039 | chr14:63395971–63396275 | 2.8375 | 0.00075 | PPP2R5E |
| CUFF.110851 | chrX:111699308–111699657 | 3.8764 | 0.00085 | ALG3 |
| CUFF.25323 | chr12:111257228–111257830 | 3.8654 | 0.00095 | CUX2 |
| CUFF.107185 | chr9:127397206–127397465 | 3.2564 | 0.00105 | SLC2A8 |
| CUFF.107177 | chr9:127325581–127325969 | 4.3465 | 0.0007 | GARNL3 |
| CUFF.17462 | chr11:57782675–57782947 | 4.93401 | 0.0003 | CTNND1 |
| CUFF.13238 | chr10:78033882–78040677 | 6.18617 | 0.0252 | RPS24 |
| CUFF.100491 | chr8:71098236–71098451 | 7.9462 | 0.00075 | ENST00000647843.1 |
| CUFF.55161 | chr15:44826300–44826876 | 5.03278 | 0.0428 | ENST00000558419.1 |
| CUFF.10014 | chr1:151287575–151287979 | 3.9867 | 0.01955 | ZNF687 |
| CUFF.100478 | chr20:63890280–63890538 | 4.8968 | 0.0235 | TPD52L2 |
| CUFF.100559 | chr21:6986631–6987286 | 2.9645 | 0.0209 | ENST00000623165.3 |
| CUFF.103593 | chr22:22900959–22901440 | 4.6579 | 0.01695 | IGLC2 |
| CUFF.100039 | chr8:60846260–60846506 | 3.8965 | 0.03515 | CHD7 |
| Down-regulated | ||||
| CUFF.148166 | chr6:109038801–109039233 | −2.85649 | 0.0181 | SESN1 |
| CUFF.27895 | chr13:71866547–71867205 | −4.47892 | 0.03305 | DACH1 |
| CUFF.28742 | chr13:102161569–102161764 | −3.74264 | 0.00095 | FGF14 |
| CUFF.30017 | chr14:37573929–37574220 | −2.98346 | 0.001 | MIPOL1 |
| CUFF.23189 | chr12:56596124–56596418 | −5.36542 | 0.0008 | RBMS2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shukla, N.; Kour, B.; Sharma, D.; Vijayvargiya, M.; Sadasukhi, T.C.; Medicherla, K.M.; Malik, B.; Bissa, B.; Vuree, S.; Lohiya, N.K.; et al. Towards Understanding the Key Signature Pathways Associated from Differentially Expressed Gene Analysis in an Indian Prostate Cancer Cohort. Diseases 2023, 11, 72. https://doi.org/10.3390/diseases11020072
Shukla N, Kour B, Sharma D, Vijayvargiya M, Sadasukhi TC, Medicherla KM, Malik B, Bissa B, Vuree S, Lohiya NK, et al. Towards Understanding the Key Signature Pathways Associated from Differentially Expressed Gene Analysis in an Indian Prostate Cancer Cohort. Diseases. 2023; 11(2):72. https://doi.org/10.3390/diseases11020072
Chicago/Turabian StyleShukla, Nidhi, Bhumandeep Kour, Devendra Sharma, Maneesh Vijayvargiya, T. C. Sadasukhi, Krishna Mohan Medicherla, Babita Malik, Bhawana Bissa, Sugunakar Vuree, Nirmal Kumar Lohiya, and et al. 2023. "Towards Understanding the Key Signature Pathways Associated from Differentially Expressed Gene Analysis in an Indian Prostate Cancer Cohort" Diseases 11, no. 2: 72. https://doi.org/10.3390/diseases11020072
APA StyleShukla, N., Kour, B., Sharma, D., Vijayvargiya, M., Sadasukhi, T. C., Medicherla, K. M., Malik, B., Bissa, B., Vuree, S., Lohiya, N. K., & Suravajhala, P. (2023). Towards Understanding the Key Signature Pathways Associated from Differentially Expressed Gene Analysis in an Indian Prostate Cancer Cohort. Diseases, 11(2), 72. https://doi.org/10.3390/diseases11020072