
A Case of Light Chain Deposition Disease Leading to Acute Liver Failure and Review of Literature
 , ,
, , 
Abstract
1. Introduction
- Heavy chain deposition disease (HCDD).
- Light chain deposition disease (LCDD).
- Heavy and light chain deposition disease (HLCDD).
- Immunoglobulin light chain (AL) amyloidosis.
2. Case Presentation
3. Discussion

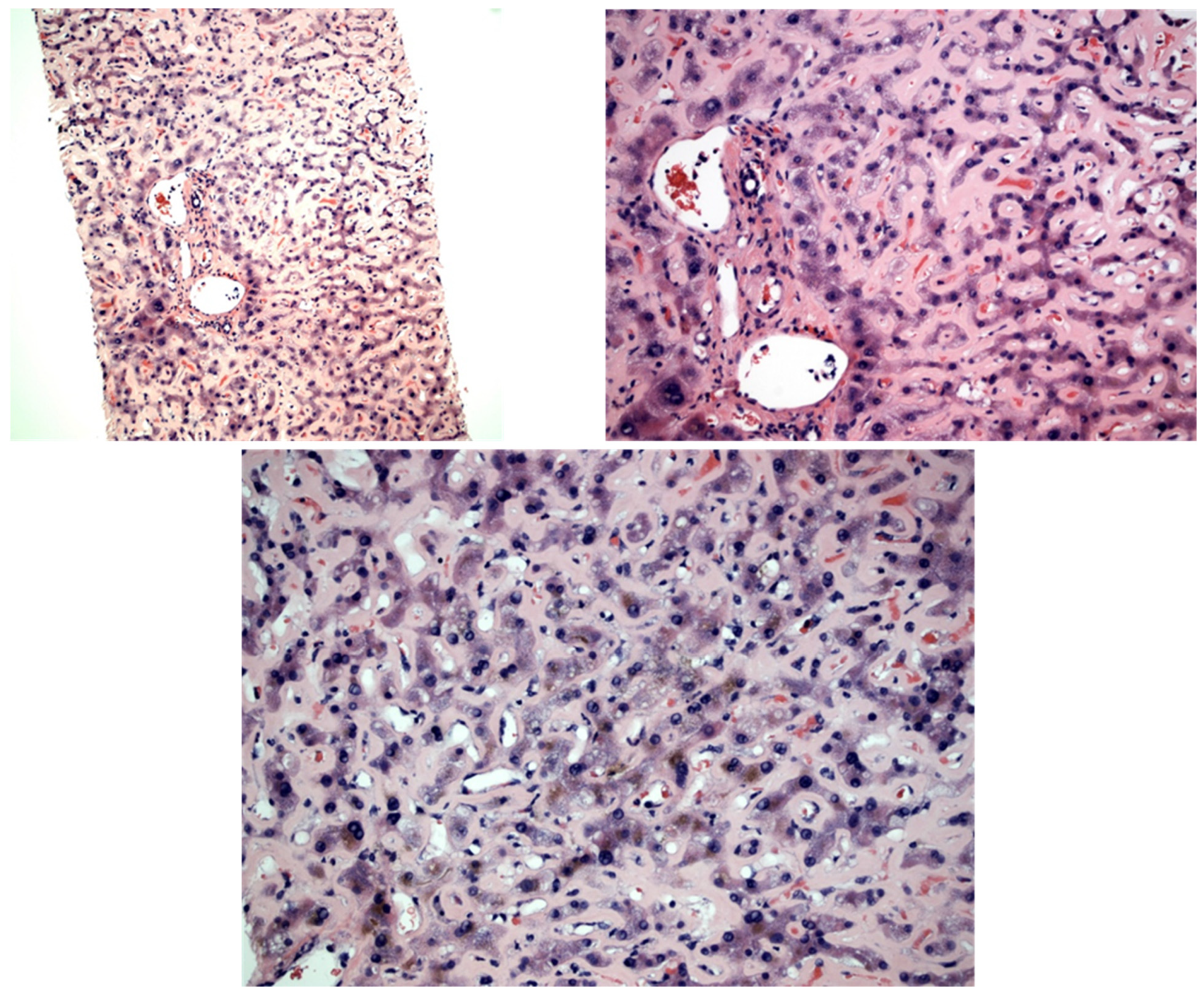{kind=link}
| Year | Age | Gender | Race | Symptoms | Total Bilirubin | Albumin | AST (U/L) | ALT (U/L) | Alkaline Phosphatase (U/L) | INR | Creatinine | Hepatomegaly | Ascites | Encephalopathy | Treatment | Outcome | Lymphoproliferative Disorder | Liver Biopsy | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Our study | 2022 | 83 | F | Caucasian | Generalized edema | 36.45 mg/dL | 3.2 g/dL | 289 | 95 | 1116 | 3.6 | 2.79 mg/dL | No | Yes | Yes | Supportive, CTX offered but declined | Multiorgan failure, circulatory shock; died shortly after | Monoclonal gammopathy of unknown significance | PAS positive, Congo red negative pale pink hyaline material expanding the sinusoid throughout the biopsy |
| Plummer et al. [9] | 2021 | 42 | M | Unknown | Abdominal pain, fatigue, early satiety | 0.9 mg/dL | 2.2 g/dL | 91 | 118 | 990 | Unknown | Unknown | Yes | Yes | Unknown | Bortezomib, cyclophosphamide, and dexamethasone | Discontinued CTX and died shortly thereafter | Unknown | Scattered sinusoidal eosinophilic deposits; lambda restriction on IF |
| Talukdar et al. [8] | 2013 | 55 | M | Unknown | Dyspnea, edema, JVD, muscle wasting | Unknown | 2 g/dL | Unknown | Unknown | 822 | Unknown | 2 mg/dL | Yes | Yes | Unknown | Bortezomib-based induction CTX offered but patient declined | Lost to follow-up | Unknown | Extracellular perisinusoidal deposits compressing bile ductules, hepatocytes and portal tracts; it also showed Kupffer cell hyperplasia |
| Tsushima et al. [19] | 2021 | 64 | F | Japanese | Malaise and anorexia | Unknown | 3.8 g/dL | 13 | 21 | Unknown | Unknown | 6.31 mg/dL | Unknown | Yes | Unknown | Daratumumab, bortezomib and dexamethasone | Responded to treatment | Ig D Multiple myeloma | Hepatocyte atrophy and perisinusoidal space deposits; IF positive for antikappa |
| Mena-Durán et al. [11] | 2011 | 81 | M | Caucasian | Jaundice, anorexia, weight loss, macroglossia | 6.35 mg/dL | 1.9 g/dL | 102 | 33 | 699 | Unknown | 1.3 mg/dL | Yes | Unknown | Yes | High-dose dexamethasone | Died within 3 weeks of hospitalization | IgG Kappa Multiple Myeloma | Perisinusoidal deposits of Congo red negative and eosinophilic material. Kappa chain restriction on IF |
| Cristino et al. [16] | 2017 | 60 | M | Caucasian | Weakness, dyspnea on exertion, early satiety | Elevated | Unknown | Unknown | Unknown | 1377 | Unknown | 2.7 mg/dL | Yes | Unknown | Unknown | Chemotherapy (not specified) | Died 2 months later with infectious complication | No | Sinusoidal deposits of eosinophilic and Congo red negative material. IHC with antikappa Ab inconclusive |
| Grembiale et al. [20] | 2020 | 70 | M | Unknown | Fatigue and weight loss | 4.8 mg/dL | Unknown | 647 | 485 | Unknown | Unknown | 6.7 mg/dL | Yes | Unknown | Unknown | Dexamethasone andbortezomib | Complete recovery in 6 months | Unknown | Congo red and PAS negative, eosinophilic deposits; kappa LC predominant on IF |
| Kwon JH et al. [12] | 2011 | 62 | M | Mongolian | Generalized edema, hepatomegaly | 0.4 mg/dL | 2 g/dL | 76 | 53 | 276 | Unknown | 1.7 mg/dL | Unknown | Yes | Unknown | Peg-IFN + ribavirin for coexisting chronic HCV | Died 2 months later | Multiple myeloma | Sinusoidal expansion with eosinophilic, Congo red negative deposits; IHC stains show deposits composed of kappa LC |
| Michopulous S et al. [10] | 2002 | 36 | M | Unknown | Jaundice | 7.8 mg/dL | Unknown | 89 | 100 | 2315 | Unknown | Unknown | Yes | Unknown | Unknown | Unknown | Rapid decline with multiorgan failure and death | Multiple myeloma | Slightly eosinophilic, Congo red negative, PAS positive perisinusoidal deposits; positive for kappa LC on IF |
| Kumar PN et al. [13] | 2012 | 66 | M | Unknown | Jaundice and pruritis | 11.3 mg/dL | 2.7 g/dL | 177 | 75 | 1965 | Unknown | Unknown | Yes | Unknown | Unknown | Unknown | Progressive worsening and loss to follow-up | Unknown | Congo red negative, PAS positive, eosinophilic perisinusoidal deposits compressing hepatocytes; bile ductules |
| Pelletier et al. [21] | 1988 | 63 | F | Unknown | Weight loss | 44 µmol/L | 47 IU/L | Unknown | Unknown | 469 | Unknown | 71 µmol/L | Yes | Unknown | Yes | Cyclophosphamide, melphalan andprednisolone × 6 months | Died in 9 months | Unknown | Deposition of Congo red negative, antilamba LC positive material along the space of Disse outlining the sinusoids |
| Girelli et al. [14] | 1998 | 59 | F | Caucasian | Fatigue, anorexia, epigastric pain, weight loss | Unknown | Unknown | Unknown | Unknown | 632 | Unknown | Unknown | Yes | Unknown | Unknown | Received at different center | Lost to follow-up | Unknown | Slightly eosinophilic, PAS positive, Congo red negative, staining positively with antikappa antiserum in the perisinusoidal area |
| Faa G et al. [15] | 1989 | 67 | M | Unknown | Jaundice | 11.4 mg/dL | 2.12 g/dL | 94 | 87 | 3905 | Unknown | Unknown | Yes | Unknown | Unknown | CTX (not specified) following 32 mg methylprednisolone | Improved | Unknown | Congo red negative, PAS positive, eosinophilic deposits in the space of Disse, making sinusoidal appear thickened; Congo red positive deposits were seen in arterial wall and connective tissue; incubation with antilamba LC antiserum showed intense staining and specific staining with antikappa |
| Ichikawa et al. [23] | 2007 | 62 | F | Unknown | Facial edema and lumbar pain | 3.2 mg/dL | Unknown | Unknown | Unknown | Unknown | Unknown | 3.9 mg/dL | Unknown | Unknown | Unknown | Dexamethasone, plasma exchange and dialysis | Death from disseminated intravascular coagulation | Multiple myeloma | Amyloid-like deposits in perisinusoidal space, Congo red negative, IgA and kappa positive on immunohistochemical stain |
| Bedossa P et al. [17] | 1988 | 54 | M | Unknown | Hepatomegaly and portal hypertension | Unknown | Unknown | Unknown | Unknown | Very elevated | Unknown | Unknown | Yes | Unknown | Unknown | Unknown | Hepatic failure, renal failure | Unknown | PAS positive, Congo red negative, amorphous ribbon-like material, green with Masson’s trichrome and red after picrosirius |
| Bedossa P et al. [17] | 1988 | 69 | M | Unknown | Hepatomegaly and splenomegaly | Unknown | Unknown | Unknown | Unknown | Elevated | Unknown | Unknown | Yes | Unknown | Unknown | Unknown | Renal failure, portal hypertension | Myeloma | PAS positive, Congo red negative, amorphous ribbon-like material, green with Masson’s trichrome and red after picrosirius |
| Bedossa P et al. [17] | 1988 | 60 | F | Unknown | Hepatomegaly | Elevated | Unknown | Unknown | Unknown | Very elevated | Unknown | Unknown | Yes | Unknown | Unknown | Unknown | Portal hypertension, renal abnormalities | Unknown | PAS positive, Congo red apple green birefringence, amorphous ribbon-like material expanding the sinusoid, green with Masson’s trichrome and red after picrosirius |
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Randall, R.E.; Williamson, W.C., Jr.; Mullinax, F.; Tung, M.Y.; Still, W.J. Manifestations of systemic light chain deposition. Am. J. Med. 1976, 60, 293–299. [Google Scholar] [CrossRef] [PubMed]
- Pozzi, C.; D’Amico, M.; Fogazzi, G.B.; Curioni, S.; Ferrario, F.; Pasquali, S.; Quattrocchio, G.; Rollino, C.; Segagni, S.; Locatelli, F. Light chain deposition disease with renal involvement: Clinical characteristics and prognostic factors. Am. J. Kidney Dis. 2003, 42, 1154–1163. [Google Scholar] [CrossRef] [PubMed]
- Kanzaki, G.; Okabayashi, Y.; Nagahama, K.; Ohashi, R.; Tsuboi, N.; Yokoo, T.; Shimizu, A. Monoclonal Immunoglobulin Deposition Disease and Related Diseases. J. Nippon Med. Sch. 2019, 86, 2–9. [Google Scholar] [CrossRef]
- Mohan, M.; Buros, A.; Mathur, P.; Gokden, N.; Singh, M.; Susanibar, S.; Jo Kamimoto, J.; Hoque, S.; Radhakrishnan, M.; Matin, A.; et al. Clinical characteristics and prognostic factors in multiple myeloma patients with light chain deposition disease. Am. J. Hematol. 2017, 92, 739–745. [Google Scholar] [CrossRef] [PubMed]
- Kourelis, T.V.; Nasr, S.H.; Dispenzieri, A.; Kumar, S.K.; Gertz, M.A.; Fervenza, F.C.; Buadi, F.K.; Lacy, M.Q.; Erickson, S.B.; Cosio, F.G.; et al. Outcomes of patients with renal monoclonal immunoglobulin deposition disease. Am. J. Hematol. 2016, 91, 1123–1128. [Google Scholar] [CrossRef] [PubMed]
- Merlini, G.; Palladini, G. Enlightening light chain deposition disease. Blood 2015, 126, 2770–2771. [Google Scholar] [CrossRef]
- Confalonieri, R.; Barbiano di Belgiojoso, G.; Banfi, G.; Ferrario, F.; Bertani, T.; Pozzi, C.; Casanova, S.; Lupo, A.; De Ferrari, G.; Minetti, L. Light chain nephropathy: Histological and clinical aspects in 15 cases. Nephrol. Dial. Transplant. 1988, 3, 150–156. [Google Scholar]
- Talukdar, A.; Mukherjee, K.; Khanra, D.; Saha, M. Portal hypertension related to light chain deposition disease of liver: An enlightening experience. BMJ Case Rep. 2013, 2013, bcr2013009553. [Google Scholar] [CrossRef]
- Plummer, M.D.; Regenstein, F. Light Chain Deposition Disease: An Unusual Cause of Portal Hypertension. Hepatology 2021, 74, 3546–3548. [Google Scholar] [CrossRef]
- Michopoulos, S.; Petraki, K.; Petraki, C.; Dimopoulos, M.A. Light chain deposition disease of the liver without renal involvement in a patient with multiple myeloma related to liver failure and rapid fatal outcome. Dig. Dis. Sci. 2002, 47, 730–734. [Google Scholar] [CrossRef]
- Mena-Durán, A.; Muñoz Vicente, E.; Pareja Llorens, G.; Sanchis Cervera, J. Liver failure caused by light chain deposition disease associated with multiple myeloma. Intern. Med. 2012, 51, 773–776. [Google Scholar] [CrossRef] [PubMed]
- Kwon, J.H.; Jeong, S.H.; Kim, J.W.; Bang, S.M.; Kim, H.; Kim, Y.H.; Song, S.H. Case report: A case of light chain deposition disease involving liver and stomach with chronic hepatitis C virus infection and hepatocellular carcinoma. J. Med. Virol. 2011, 83, 810–814. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.N. Light chain deposition disease presenting as cholestatic jaundice: A case report. Oman Med. J. 2012, 27, 56–59. [Google Scholar] [CrossRef] [PubMed]
- Girelli, C.M.; Lodi, G.; Rocca, F. κ light chain deposition disease of the liver. Eur. J. Gastroenterol. Hepatol. 1998, 10. [Google Scholar] [CrossRef] [PubMed]
- Faa, G.; Van Eyken, P.; De Vos, R.; Fevery, J.; Van Damme, B.; De Groote, J.; Desmet, V.J. Light chain deposition disease of the liver associated with AL-type amyloidosis and severe cholestasis. J. Hepatol. 1991, 12, 75–82. [Google Scholar] [CrossRef]
- Cristino, A.; Pais, C.; Silva, R.; Carrola, P. Light-Chain Deposition Disease with Prominent Hepatic Involvement. Eur. J. Case Rep. Intern. Med. 2017, 4, 000545. [Google Scholar] [CrossRef]
- Bedossa, P.; Fabre, M.; Paraf, F.; Martin, E.; Lemaigre, G. Light chain deposition disease with liver dysfunction. Hum. Pathol. 1988, 19, 1008–1014. [Google Scholar] [CrossRef]
- Walsh, S.B.; Unwin, R.J. Renal tubular disorders. Clin. Med. 2012, 12, 476–479. [Google Scholar] [CrossRef]
- Tsushima, T.; Suzuki, T.; Terao, T.; Miura, D.; Narita, K.; Takeuchi, M.; Shimuzu, A.; Matsue, K. Light chain deposition disease involving kidney and liver in a patient with IgD myeloma. BMC Nephrol. 2021, 22, 40. [Google Scholar] [CrossRef]
- Grembiale, A.; Garlatti, E.; Ermacora, A.; Grazioli, S.; Balbi, M.; Tonizzo, M. An Unusual Case of Cholestatic Hepatitis due to Light-Chain Deposition Disease. Case Rep. Oncol. 2020, 13, 1343–1348. [Google Scholar] [CrossRef]
- Pelletier, G.; Fabre, M.; Attali, P.; Ladouch-Badre, A.; Ink, O.; Martin, E.; Etienne, J.P. Light chain deposition disease presenting with hepatomegaly: An association with amyloid-like fibrils. Postgrad. Med. J. 1988, 64, 804–808. [Google Scholar] [CrossRef] [PubMed]
- Stravitz, R.T.; Lee, W.M. Acute liver failure. Lancet 2019, 394, 869–881. [Google Scholar] [CrossRef] [PubMed]
- Ichikawa, A.; Hashimoto, A.; Shimizu, A.; Shizuku, J.; Abe, Y.; Wakai, S.; Ogura, M.; Ogawa, T.; Nitta, K. A case of acute renal and liver dysfunction with light chain deposition disease. Jpn. J. Nephrol. Nihon Jinzo Gakkai Shi 2007, 49, 459–463. [Google Scholar]
- Tan, C.R.C.; Abdul-Majeed, S.; Cael, B.; Barta, S.K. Clinical Pharmacokinetics and Pharmacodynamics of Bortezomib. Clin. Pharmacokinet. 2019, 58, 157–168. [Google Scholar] [CrossRef] [PubMed]
- Joly, F.; Cohen, C.; Javaugue, V.; Bender, S.; Belmouaz, M.; Arnulf, B.; Knebelmann, B.; Nouvier, M.; Audard, V.; Provot, F.; et al. Randall-type monoclonal immunoglobulin deposition disease: Novel insights from a nationwide cohort study. Blood 2019, 133, 576–587. [Google Scholar] [CrossRef]
- Milani, P.; Basset, M.; Curci, P.; Foli, A.; Rizzi, R.; Nuvolone, M.; Guido, R.; Gesualdo, L.; Specchia, G.; Merlini, G.; et al. Daratumumab in light chain deposition disease: Rapid and profound hematologic response preserves kidney function. Blood Adv. 2020, 4, 1321–1324. [Google Scholar] [CrossRef]
- Casiraghi, M.A.; De Paoli, A.; Assi, A.; Palladini, G.; Lavazza, M.T.; Beretta, A.; Gualdoni, G.; Beretta, R. Hepatic amyloidosis with light chain deposition disease. A rare association. Dig. Liver Dis. 2000, 32, 795–798. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gandhi, M.; Pasha, S.B.; Reznicek, E.; Pasha, S.R.; Ertugrul, H.; Araslanova, A.; Yin, F.; Tahan, V. A Case of Light Chain Deposition Disease Leading to Acute Liver Failure and Review of Literature. Diseases 2023, 11, 24. https://doi.org/10.3390/diseases11010024
Gandhi M, Pasha SB, Reznicek E, Pasha SR, Ertugrul H, Araslanova A, Yin F, Tahan V. A Case of Light Chain Deposition Disease Leading to Acute Liver Failure and Review of Literature. Diseases. 2023; 11(1):24. https://doi.org/10.3390/diseases11010024
Chicago/Turabian StyleGandhi, Mustafa, Syed Bilal Pasha, Emily Reznicek, Syed Raheel Pasha, Hamza Ertugrul, Adel Araslanova, Feng Yin, and Veysel Tahan. 2023. "A Case of Light Chain Deposition Disease Leading to Acute Liver Failure and Review of Literature" Diseases 11, no. 1: 24. https://doi.org/10.3390/diseases11010024
APA StyleGandhi, M., Pasha, S. B., Reznicek, E., Pasha, S. R., Ertugrul, H., Araslanova, A., Yin, F., & Tahan, V. (2023). A Case of Light Chain Deposition Disease Leading to Acute Liver Failure and Review of Literature. Diseases, 11(1), 24. https://doi.org/10.3390/diseases11010024