
Fexinidazole for Human African Trypanosomiasis, the Fruit of a Successful Public-Private Partnership



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. A Neglected Disease of Neglected Patients
2. The Chemotherapy of Sleeping Sickness: Old Roots but Little Fruit
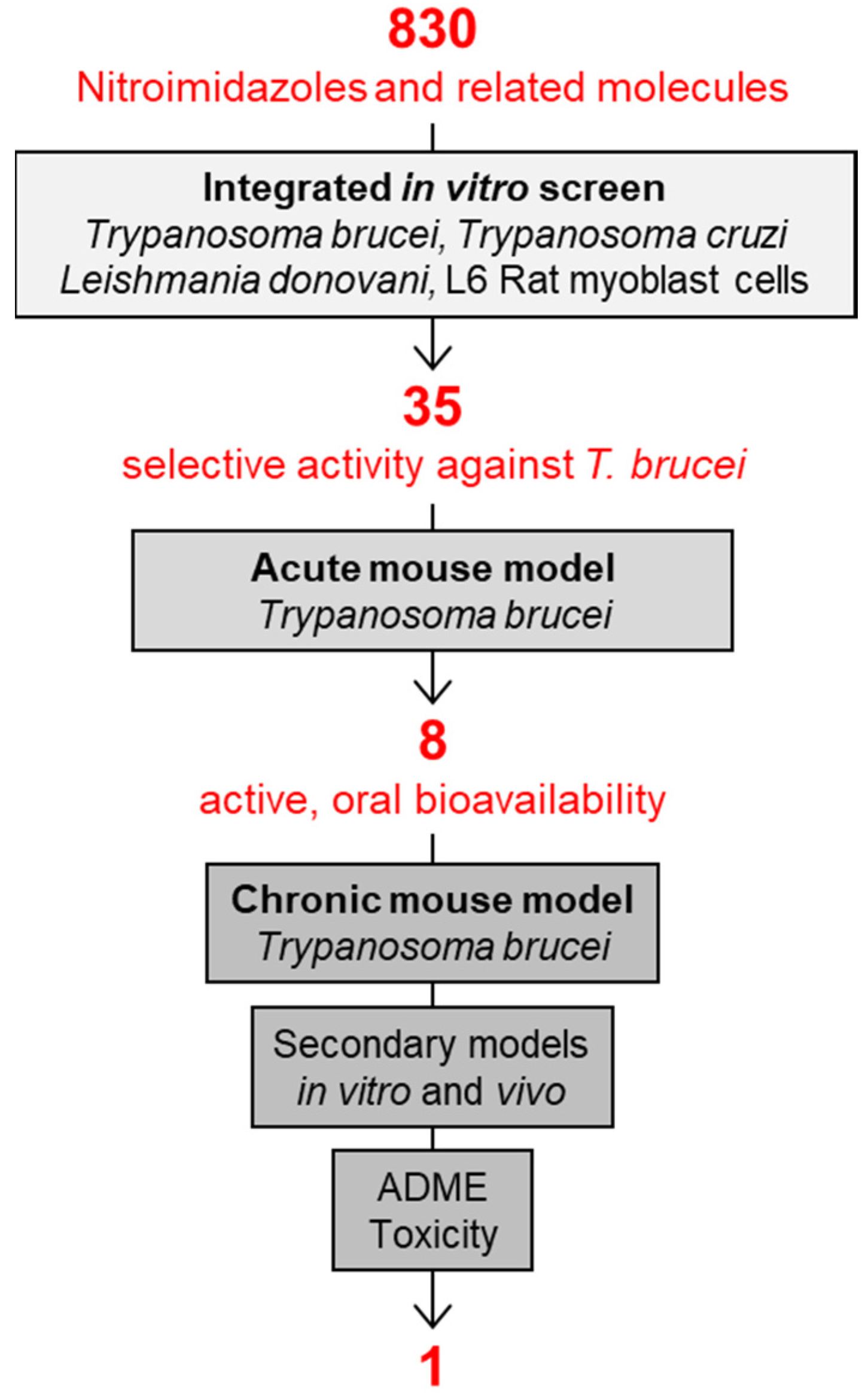
3. Preclinical Development of Fexinidazole
4. Fexinidazole Is a Magic Bomb
5. Clinical Development of Fexinidazole
6. Elements of Success
7. Conclusions: Lessons Learnt
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Barrett, M.P. The fall and rise of sleeping sickness. Lancet 1999, 353, 1113–1114. [Google Scholar] [CrossRef]
- Smith, D.H.; Pepin, J.; Stich, A.H. Human African trypanosomiasis: An emerging public health crisis. Br. Med. Bull. 1998, 54, 341–355. [Google Scholar] [CrossRef]
- Pays, E.; Vanhollebeke, B.; Uzureau, P.; Lecordier, L.; Perez-Morga, D. The molecular arms race between African trypanosomes and humans. Nat. Rev. Microbiol. 2014, 12, 575–584. [Google Scholar] [CrossRef]
- Brun, R.; Blum, J.; Chappuis, F.; Burri, C. Human African trypanosomiasis. Lancet 2010, 375, 148–159. [Google Scholar] [CrossRef]
- Fevre, E.M.; Coleman, P.G.; Odiit, M.; Magona, J.W.; Welburn, S.C.; Woolhouse, M.E. The origins of a new Trypanosoma brucei rhodesiense sleeping sickness outbreak in eastern Uganda. Lancet 2001, 358, 625–628. [Google Scholar] [CrossRef]
- Lemerani, M.; Jumah, F.; Bessell, P.; Bieler, S.; Ndung’u, J.M. Improved Access to Diagnostics for Rhodesian Sleeping Sickness around a Conservation Area in Malawi Results in Earlier Detection of Cases and Reduced Mortality. J. Epidemiol. Glob. Health 2020, 10, 280–287. [Google Scholar] [CrossRef] [PubMed]
- Ehrlich, P. Chemotherapeutische Trypanosomen-Studien. Berl. Klin. Wochenschr. 1907, 11, 310–314. [Google Scholar]
- Wiedemar, N.; Hauser, D.A.; Mäser, P. 100 Years of Suramin. Antimicrob. Agents Chemother. 2020, 64, e01168-19. [Google Scholar] [CrossRef]
- Van Hoof, L.; Henrad, C.; Peel, E. Pentamidine in the prevention and treatment of trypanosomiasis. Trans. R. Soc. Trop. Med. Hyg. 1944, 37, 271–280. [Google Scholar] [CrossRef]
- De Koning, H.P. The Drugs of Sleeping Sickness: Their Mechanisms of Action and Resistance, and a Brief History. Trop. Med. Infect. Dis. 2020, 5, 14. [Google Scholar] [CrossRef] [PubMed]
- Friedheim, E.A.H. Mel B in the treatment of human trypanosomiasis. Am. J. Trop. Med. Hyg. 1949, 29, 173–180. [Google Scholar] [CrossRef] [PubMed]
- Kuzoe, F.A. Current situation of African trypanosomiasis. Acta Trop. 1993, 54, 153–162. [Google Scholar] [CrossRef]
- Barrett, M.P.; Vincent, I.M.; Burchmore, R.J.; Kazibwe, A.J.; Matovu, E. Drug resistance in human African trypanosomiasis. Future Microbiol. 2011, 6, 1037–1047. [Google Scholar] [CrossRef] [PubMed]
- Baker, N.; de Koning, H.P.; Mäser, P.; Horn, D. Drug resistance in African trypanosomiasis: The melarsoprol and pentamidine story. Trends Parasitol. 2013, 29, 110–118. [Google Scholar] [CrossRef] [PubMed]
- Bacchi, C.J.; Nathan, H.C.; Hutner, S.H.; McCann, P.P.; Sjoerdsma, A. Polyamine metabolism: A potential therapeutic target in trypanosomes. Science 1980, 210, 332–334. [Google Scholar] [CrossRef] [PubMed]
- Milord, F.; Pepin, J.; Loko, L.; Ethier, L.; Mpia, B. Efficacy and toxicity of eflornithine for treatment of Trypanosoma brucei gambiense sleeping sickness. Lancet 1992, 340, 652–655. [Google Scholar] [CrossRef]
- Pepin, J.; Khonde, N.; Maiso, F.; Doua, F.; Jaffar, S.; Ngampo, S.; Mpia, B.; Mbulamberi, D.; Kuzoe, F. Short-course eflornithine in Gambian trypanosomiasis: A multicentre randomized controlled trial. Bull. World Health Organ. 2000, 78, 1284–1295. [Google Scholar] [PubMed]
- Barrett, M.P. The rise and fall of sleeping sickness. Lancet 2006, 367, 1377–1378. [Google Scholar] [CrossRef]
- Priotto, G.; Kasparian, S.; Mutombo, W.; Ngouama, D.; Ghorashian, S.; Arnold, U.; Ghabri, S.; Baudin, E.; Buard, V.; Kazadi-Kyanza, S.; et al. Nifurtimox-eflornithine combination therapy for second-stage African Trypanosoma brucei gambiense trypanosomiasis: A multicentre, randomised, phase III, non-inferiority trial. Lancet 2009, 374, 56–64. [Google Scholar] [CrossRef]
- WHO. WHO Model List of Essential Medicines, 16th list March 2009, Geneva; WHO: Geneva, Switzerland, 2009; Available online: http://apps.who.int/iris/handle/10665/70643 (accessed on 6 October 2022).
- Wenzler, T.; Boykin, D.W.; Ismail, M.A.; Hall, J.E.; Tidwell, R.R.; Brun, R. New treatment option for second-stage African sleeping sickness: In vitro and in vivo efficacy of aza analogs of DB289. Antimicrob. Agents Chemother. 2009, 53, 4185–4192. [Google Scholar] [CrossRef] [PubMed]
- Burri, C.; Yeramian, P.D.; Allen, J.L.; Merolle, A.; Serge, K.K.; Mpanya, A.; Lutumba, P.; Mesu, V.K.; Bilenge, C.M.; Lubaki, J.P.; et al. Efficacy, Safety, and Dose of Pafuramidine, a New Oral Drug for Treatment of First Stage Sleeping Sickness, in a Phase 2a Clinical Study and Phase 2b Randomized Clinical Studies. PLoS Negl. Trop. Dis. 2016, 10, e0004362. [Google Scholar] [CrossRef]
- Pohlig, G.; Bernhard, S.C.; Blum, J.; Burri, C.; Mpanya, A.; Lubaki, J.P.; Mpoto, A.M.; Munungu, B.F.; N’Tombe, P.M.; Deo Manesa, K.G.; et al. Efficacy and Safety of Pafuramidine versus Pentamidine Maleate for Treatment of First Stage Sleeping Sickness in a Randomized, Comparator-Controlled, International Phase 3 Clinical Trial. PLoS Negl. Trop. Dis. 2016, 10, e0004363. [Google Scholar] [CrossRef] [PubMed]
- Kaminsky, R.; Brun, R. In vitro assays to determine drug sensitivities of African trypanosomes: A review. Acta Trop. 1993, 54, 279–289. [Google Scholar] [CrossRef]
- Opperdoes, F.R. Biochemical peculiarities of trypanosomes, African and South American. Br. Med. Bull. 1985, 41, 130–136. [Google Scholar] [CrossRef]
- Berriman, M.; Ghedin, E.; Hertz-Fowler, C.; Blandin, G.; Renauld, H.; Bartholomeu, D.C.; Lennard, N.J.; Caler, E.; Hamlin, N.E.; Haas, B.; et al. The genome of the African trypanosome Trypanosoma brucei. Science 2005, 309, 416–422. [Google Scholar] [CrossRef] [PubMed]
- Franco, J.R.; Simarro, P.P.; Diarra, A.; Jannin, J.G. Epidemiology of human African trypanosomiasis. Clin. Epidemiol. 2014, 6, 257–275. [Google Scholar] [CrossRef]
- WHO. Accelerating Work to Overcome the Global Impact of Neglected Tropical Diseases: A Roadmap for Implementation, Geneva. 2012. Available online: https://apps.who.int/iris/handle/10665/70809 (accessed on 6 October 2022).
- Steinmann, P.; Stone, C.M.; Sutherland, C.S.; Tanner, M.; Tediosi, F. Contemporary and emerging strategies for eliminating human African trypanosomiasis due to Trypanosoma brucei gambiense: Review. Trop. Med. Int. Health 2015, 20, 707–718. [Google Scholar] [CrossRef]
- Raether, W.; Seidenath, H. The activity of fexinidazole (HOE 239) against experimental infections with Trypanosoma cruzi, trichomonads and Entamoeba histolytica. Ann. Trop. Med. Parasitol. 1983, 77, 13–26. [Google Scholar] [CrossRef] [PubMed]
- Winkelmann, E.; Raether, W. Chemotherapeutically acitve nitro compounds. 4. 5-Nitroimidazoles (Part III). Arzneimittelforschung 1978, 28, 739–749. [Google Scholar] [PubMed]
- Imran, M.; Khan, S.A.; Alshammari, M.K.; Alqahtani, A.M.; Alanazi, T.A.; Kamal, M.; Jawaid, T.; Ghoneim, M.M.; Alshehri, S.; Shakeel, F. Discovery, Development, Inventions and Patent Review of Fexinidazole: The First All-Oral Therapy for Human African Trypanosomiasis. Pharmaceuticals 2022, 15, 128. [Google Scholar] [CrossRef]
- Kaiser, M.; Bray, M.A.; Cal, M.; Bourdin Trunz, B.; Torreele, E.; Brun, R. Antitrypanosomal activity of fexinidazole, a new oral nitroimidazole drug candidate for treatment of sleeping sickness. Antimicrob. Agents Chemother. 2011, 55, 5602–5608. [Google Scholar] [CrossRef] [PubMed]
- Torreele, E.; Bourdin Trunz, B.; Tweats, D.; Kaiser, M.; Brun, R.; Mazue, G.; Bray, M.A.; Pecoul, B. Fexinidazole--a new oral nitroimidazole drug candidate entering clinical development for the treatment of sleeping sickness. PLoS Negl. Trop. Dis. 2011, 4, e923. [Google Scholar] [CrossRef]
- Jennings, F.W.; Urquhart, G.M. The use of the 2 substituted 5-nitroimidazole, Fexinidazole (Hoe 239) in the treatment of chronic T. brucei infections in mice. Z. Parasitenkd 1983, 69, 577–581. [Google Scholar]
- Tweats, D.; Bourdin Trunz, B.; Torreele, E. Genotoxicity profile of fexinidazole—A drug candidate in clinical development for human African trypanomiasis (sleeping sickness). Mutagenesis 2012, 27, 523–532. [Google Scholar] [CrossRef] [PubMed]
- Wittlin, S.; Mäser, P. From Magic Bullet to Magic Bomb: Reductive Bioactivation of Antiparasitic Agents. ACS Infect. Dis. 2021, 7, 2777–2786. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, S.R.; Taylor, M.C.; Horn, D.; Kelly, J.M.; Cheeseman, I. A mechanism for cross-resistance to nifurtimox and benznidazole in trypanosomes. Proc. Natl. Acad. Sci. USA 2008, 105, 5022–5027. [Google Scholar] [CrossRef] [PubMed]
- Patterson, S.; Wyllie, S. Nitro drugs for the treatment of trypanosomatid diseases: Past, present, and future prospects. Trends Parasitol. 2014, 30, 289–298. [Google Scholar] [CrossRef]
- Mesu, V.; Kalonji, W.M.; Bardonneau, C.; Mordt, O.V.; Blesson, S.; Simon, F.; Delhomme, S.; Bernhard, S.; Kuziena, W.; Lubaki, J.F.; et al. Oral fexinidazole for late-stage African Trypanosoma brucei gambiense trypanosomiasis: A pivotal multicentre, randomised, non-inferiority trial. Lancet 2018, 391, 144–154. [Google Scholar] [CrossRef]
- Kande Betu Kumesu, V.; Mutombo Kalonji, W.; Bardonneau, C.; Valverde Mordt, O.; Ngolo Tete, D.; Blesson, S.; Simon, F.; Delhomme, S.; Bernhard, S.; Nganzobo Ngima, P.; et al. Safety and efficacy of oral fexinidazole in children with gambiense human African trypanosomiasis: A multicentre, single-arm, open-label, phase 2-3 trial. Lancet Glob. Health 2022, 10, e1665–e1674. [Google Scholar] [CrossRef]
- Kande Betu Ku Mesu, V.; Mutombo Kalonji, W.; Bardonneau, C.; Valverde Mordt, O.; Ngolo Tete, D.; Blesson, S.; Simon, F.; Delhomme, S.; Bernhard, S.; Mahenzi Mbembo, H.; et al. Oral fexinidazole for stage 1 or early stage 2 African Trypanosoma brucei gambiense trypanosomiasis: A prospective, multicentre, open-label, cohort study. Lancet Glob. Health 2021, 9, e999–e1008. [Google Scholar] [CrossRef]
- Watson, J.A.; Strub-Wourgraft, N.; Tarral, A.; Ribeiro, I.; Tarning, J.; White, N.J. Pharmacokinetic-Pharmacodynamic Assessment of the Hepatic and Bone Marrow Toxicities of the New Trypanoside Fexinidazole. Antimicrob. Agents Chemother. 2019, 63, e02515-18. [Google Scholar] [CrossRef] [PubMed]
- WHO. WHO Interim Guidelines for the Treatment of Gambiense Human African Trypanosomiasis, Geneva. 2019. Available online: https://www.who.int/publications/i/item/9789241550567 (accessed on 6 October 2022).
- DNDi. Fexinidazole for T. b. gambiense. Available online: https://dndi.org/research-development/portfolio/fexinidazole/ (accessed on 16 August 2022).
- Vischer, N.; Pfeiffer, C.; Joller, A.; Klingmann, I.; Ka, A.; Kpormegbe, S.K.; Burri, C. The Good Clinical Practice guideline and its interpretation—Perceptions of clinical trial teams in sub-Saharan Africa. Trop. Med. Int. Health 2016, 21, 1040–1048. [Google Scholar] [CrossRef] [PubMed]
- Vischer, N.; Pfeiffer, C.; Kealy, J.; Burri, C. Increasing protocol suitability for clinical trials in sub-Saharan Africa: A mixed methods study. Glob. Health Res. Policy 2017, 2, 11. [Google Scholar] [CrossRef] [PubMed]
- Vischer, N.; Pfeiffer, C.; Limacher, M.; Burri, C. “You can save time if...”—A qualitative study on internal factors slowing down clinical trials in Sub-Saharan Africa. PLoS ONE 2017, 12, e0173796. [Google Scholar] [CrossRef] [PubMed]
- Cecchi, G.; Paone, M.; Franco, J.R.; Fevre, E.M.; Diarra, A.; Ruiz, J.A.; Mattioli, R.C.; Simarro, P.P. Towards the Atlas of human African trypanosomiasis. Int. J. Health Geogr. 2009, 8, 15. [Google Scholar] [CrossRef] [PubMed]
- Kuwawenaruwa, A.; Wyss, K.; Wiedenmayer, K.; Metta, E.; Tediosi, F. The effects of medicines availability and stock-outs on household’s utilization of healthcare services in Dodoma region, Tanzania. Health Policy Plan 2020, 35, 323–333. [Google Scholar] [CrossRef] [PubMed]
- CARAMAL Consortium. Community access to rectal artesunate for malaria (CARAMAL): A large-scale observational implementation study in the Democratic Republic of the Congo, Nigeria and Uganda. PLoS Global Pub. Health 2022, 2, e0000464. [Google Scholar] [CrossRef]
- Franco, J.R.; Cecchi, G.; Priotto, G.; Paone, M.; Diarra, A.; Grout, L.; Simarro, P.P.; Zhao, W.; Argaw, D. Monitoring the elimination of human African trypanosomiasis at continental and country level: Update to 2018. PLoS Negl. Trop. Dis. 2020, 14, e0008261. [Google Scholar] [CrossRef] [PubMed]
- Franco, J.R.; Cecchi, G.; Paone, M.; Diarra, A.; Grout, L.; Kadima Ebeja, A.; Simarro, P.P.; Zhao, W.; Argaw, D. The elimination of human African trypanosomiasis: Achievements in relation to WHO road map targets for 2020. PLoS Negl. Trop. Dis. 2022, 16, e0010047. [Google Scholar] [CrossRef] [PubMed]
- DNDi. A Doctor’s Dream: A Pill for Sleeping Sickness, Geneva. 2018. Available online: https://m.youtube.com/watch?v=Tk31iucWYdE (accessed on 6 October 2022).
- White, N.J.; Nosten, F.H. SERCAP: Is the perfect the enemy of the good? Malar. J. 2021, 20, 281. [Google Scholar] [CrossRef] [PubMed]
- Trunz, B.B.; Jedrysiak, R.; Tweats, D.; Brun, R.; Kaiser, M.; Suwinski, J.; Torreele, E. 1-Aryl-4-nitro-1H-imidazoles, a new promising series for the treatment of human African trypanosomiasis. Eur. J. Med. Chem. 2011, 46, 1524–1535. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bernhard, S.; Kaiser, M.; Burri, C.; Mäser, P. Fexinidazole for Human African Trypanosomiasis, the Fruit of a Successful Public-Private Partnership. Diseases 2022, 10, 90. https://doi.org/10.3390/diseases10040090
Bernhard S, Kaiser M, Burri C, Mäser P. Fexinidazole for Human African Trypanosomiasis, the Fruit of a Successful Public-Private Partnership. Diseases. 2022; 10(4):90. https://doi.org/10.3390/diseases10040090
Chicago/Turabian StyleBernhard, Sonja, Marcel Kaiser, Christian Burri, and Pascal Mäser. 2022. "Fexinidazole for Human African Trypanosomiasis, the Fruit of a Successful Public-Private Partnership" Diseases 10, no. 4: 90. https://doi.org/10.3390/diseases10040090
APA StyleBernhard, S., Kaiser, M., Burri, C., & Mäser, P. (2022). Fexinidazole for Human African Trypanosomiasis, the Fruit of a Successful Public-Private Partnership. Diseases, 10(4), 90. https://doi.org/10.3390/diseases10040090