
Emerging Trends in Immunotherapy for Cancer


Abstract
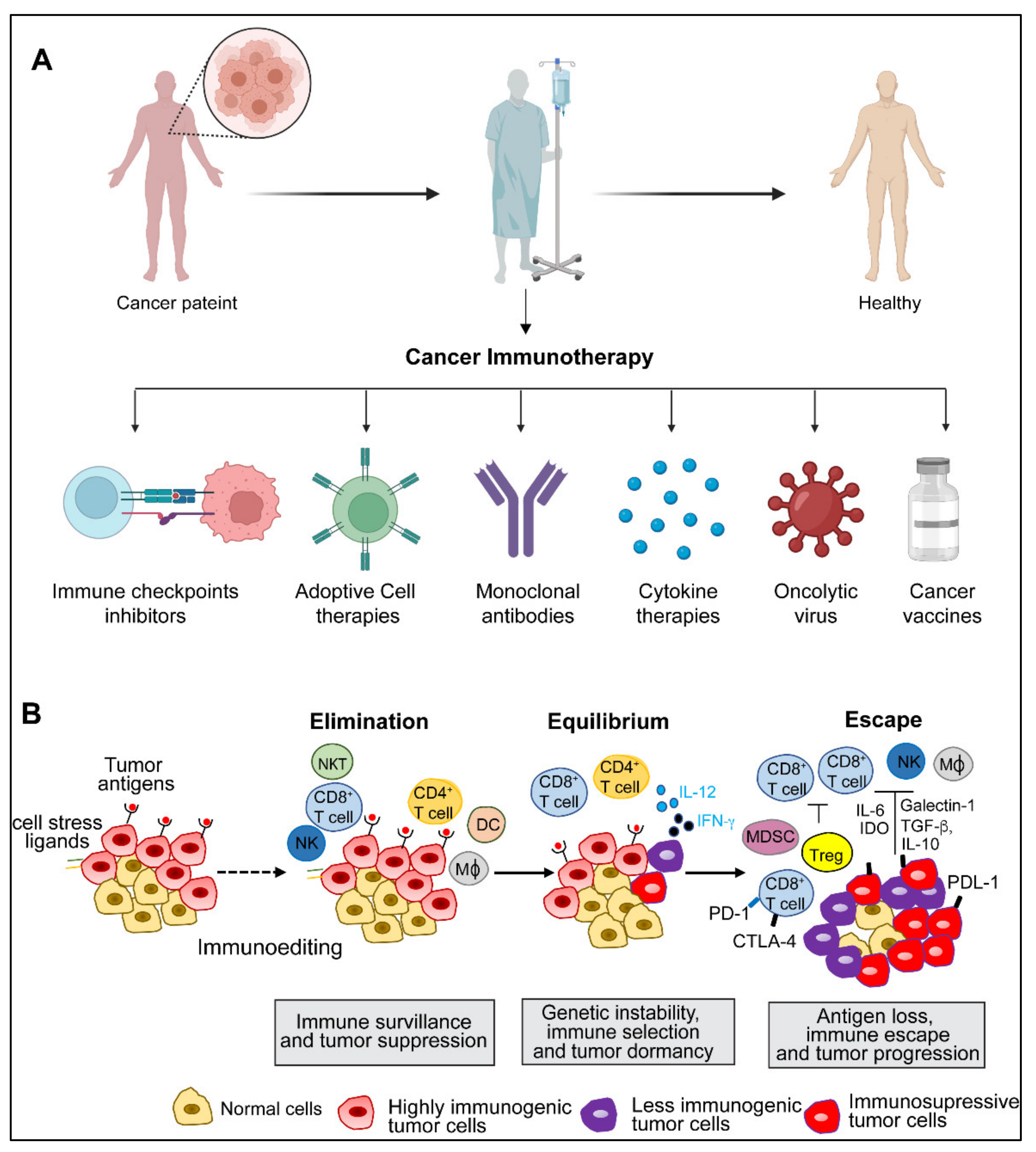
:1. Introduction
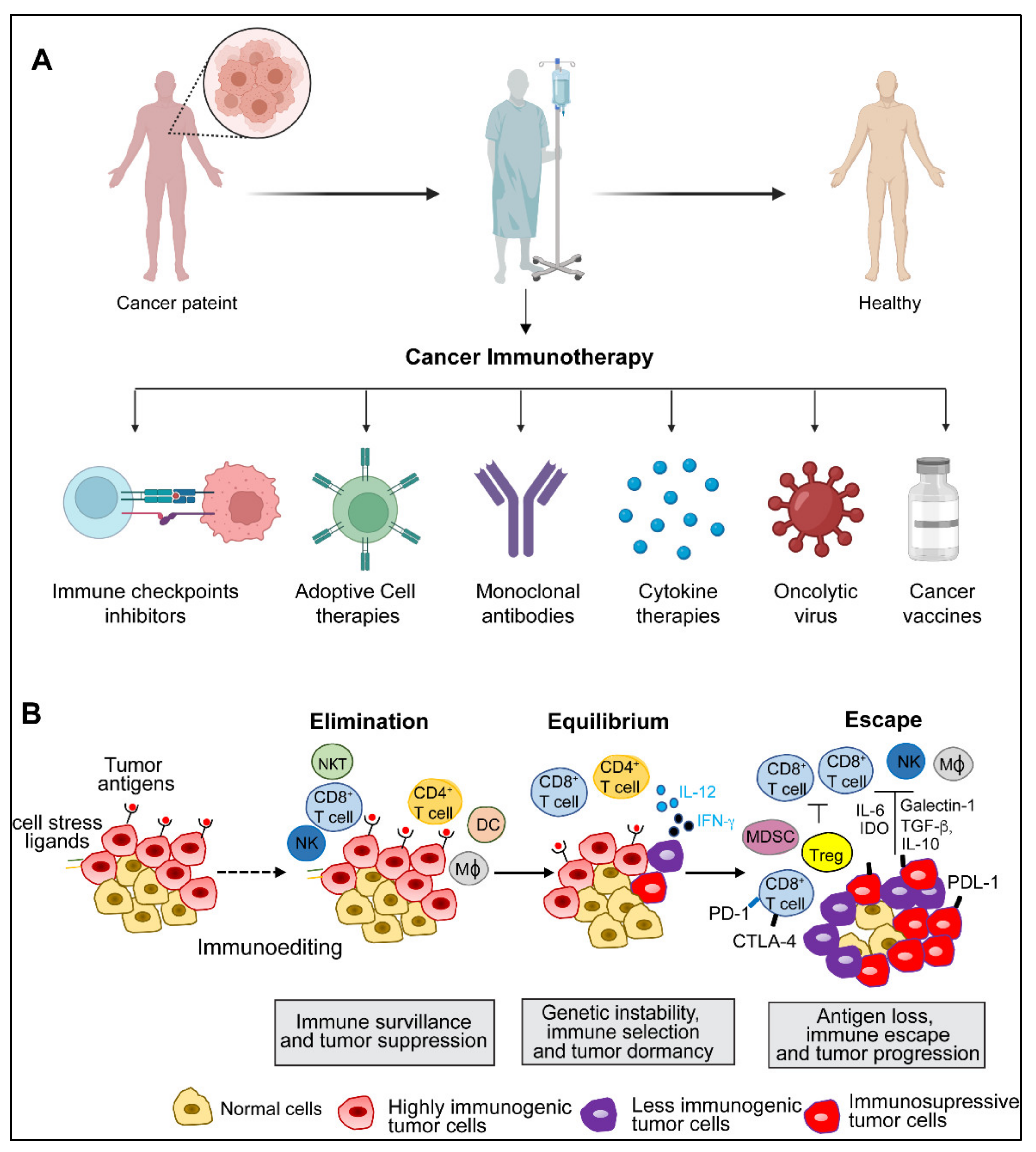
2. Cancer Immunity and Immune Evasion
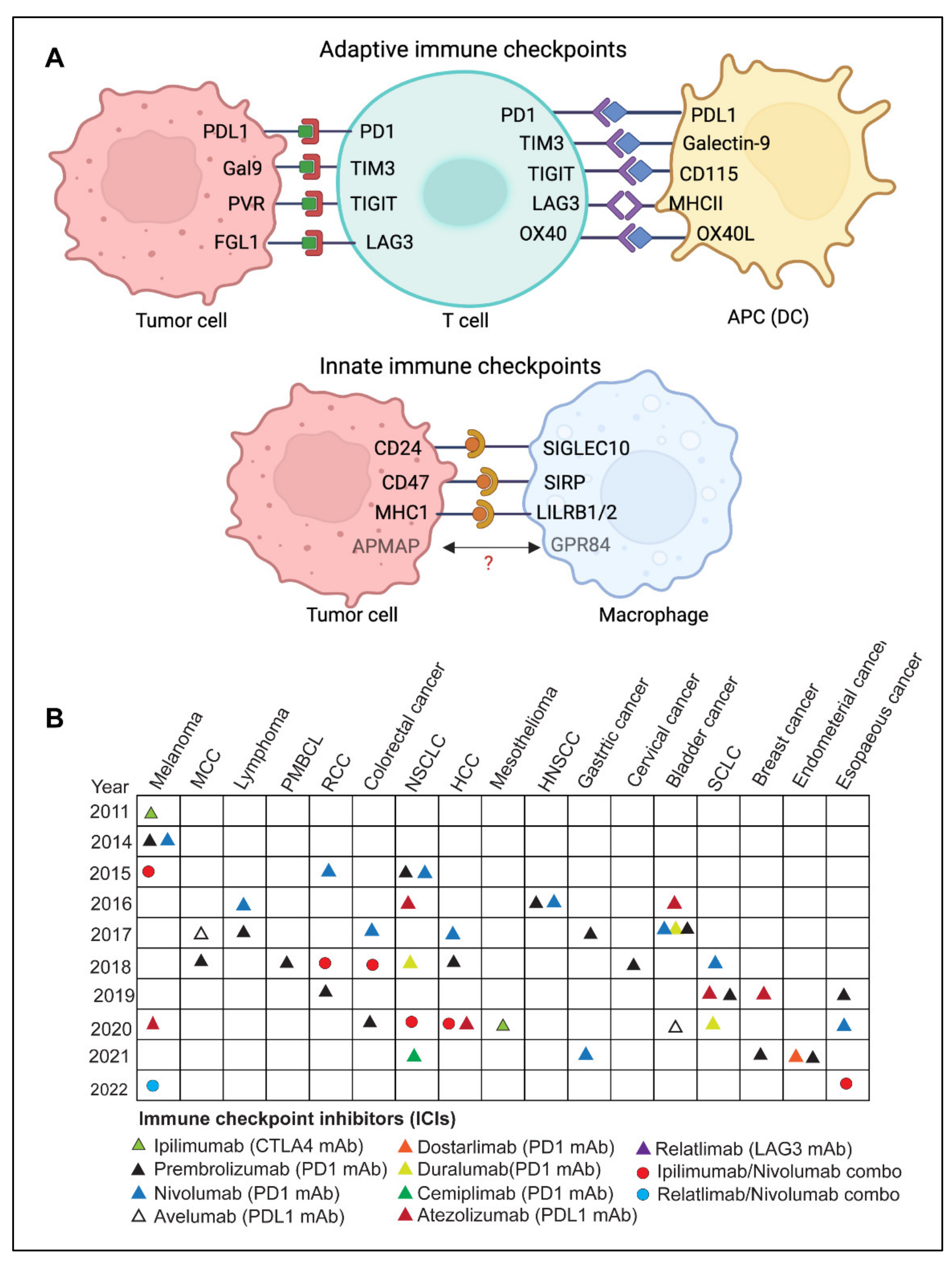
3. Immune Checkpoints
3.1. Adaptive Immune Checkpoints
3.1.1. CTLA-4
3.1.2. PD-1
3.1.3. LAG-3
3.1.4. TIGIT
3.1.5. TIM-3
3.1.6. B7-H3 and B7-H4
3.1.7. VISTA
3.1.8. OX40/OX40L
3.1.9. A2A/B-R and CD73
3.1.10. NKG2A
3.2. Innate Immune Checkpoints
3.2.1. SIRPα-CD47
3.2.2. LILRB1/MHC-I and LILRB2/MHC-I
3.2.3. Siglec10-CD24
3.2.4. APMAP
3.3. Trends in Clinical Trials with Immune Checkpoint Inhibitors
3.4. Limitations and Challenges of ICI Therapy
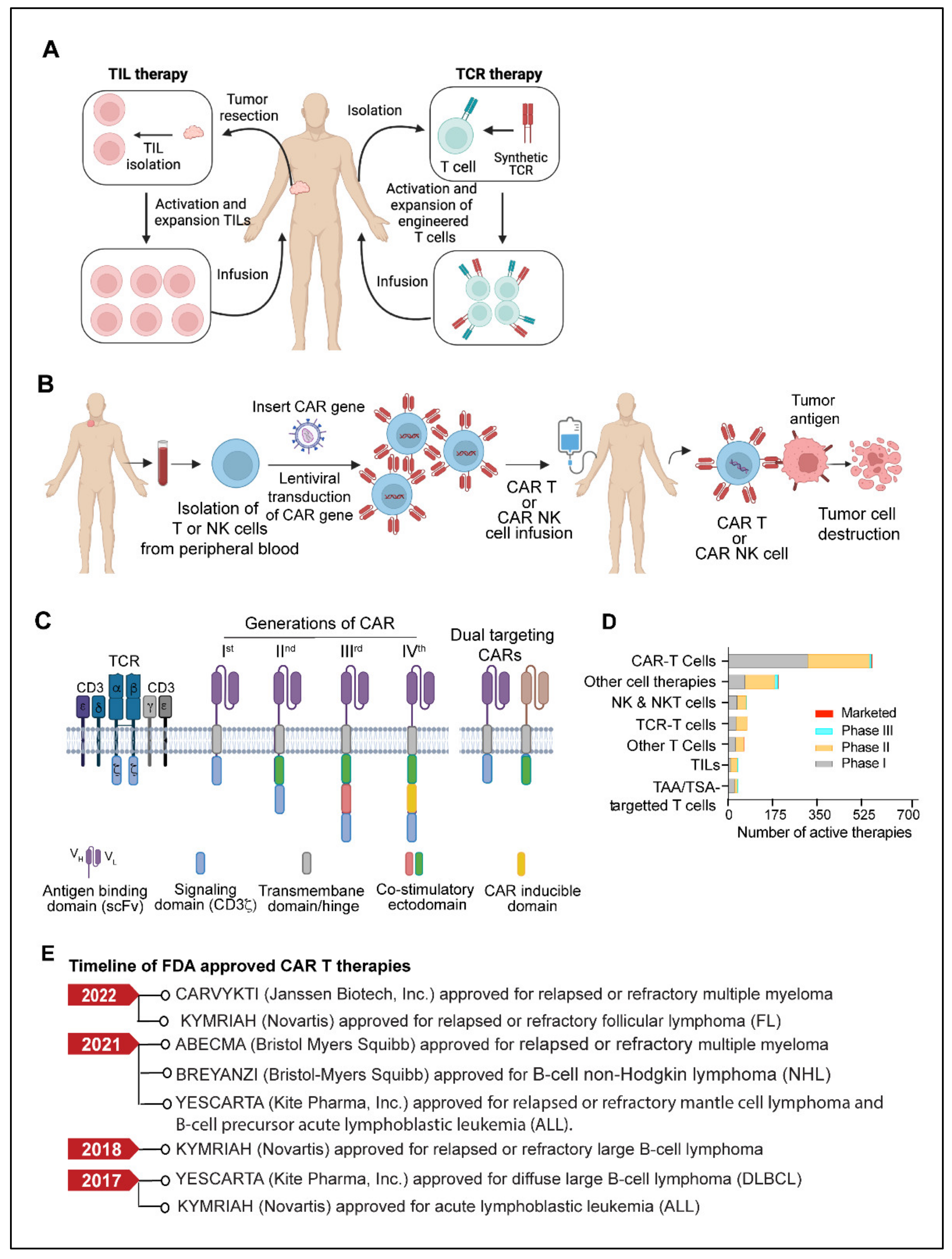
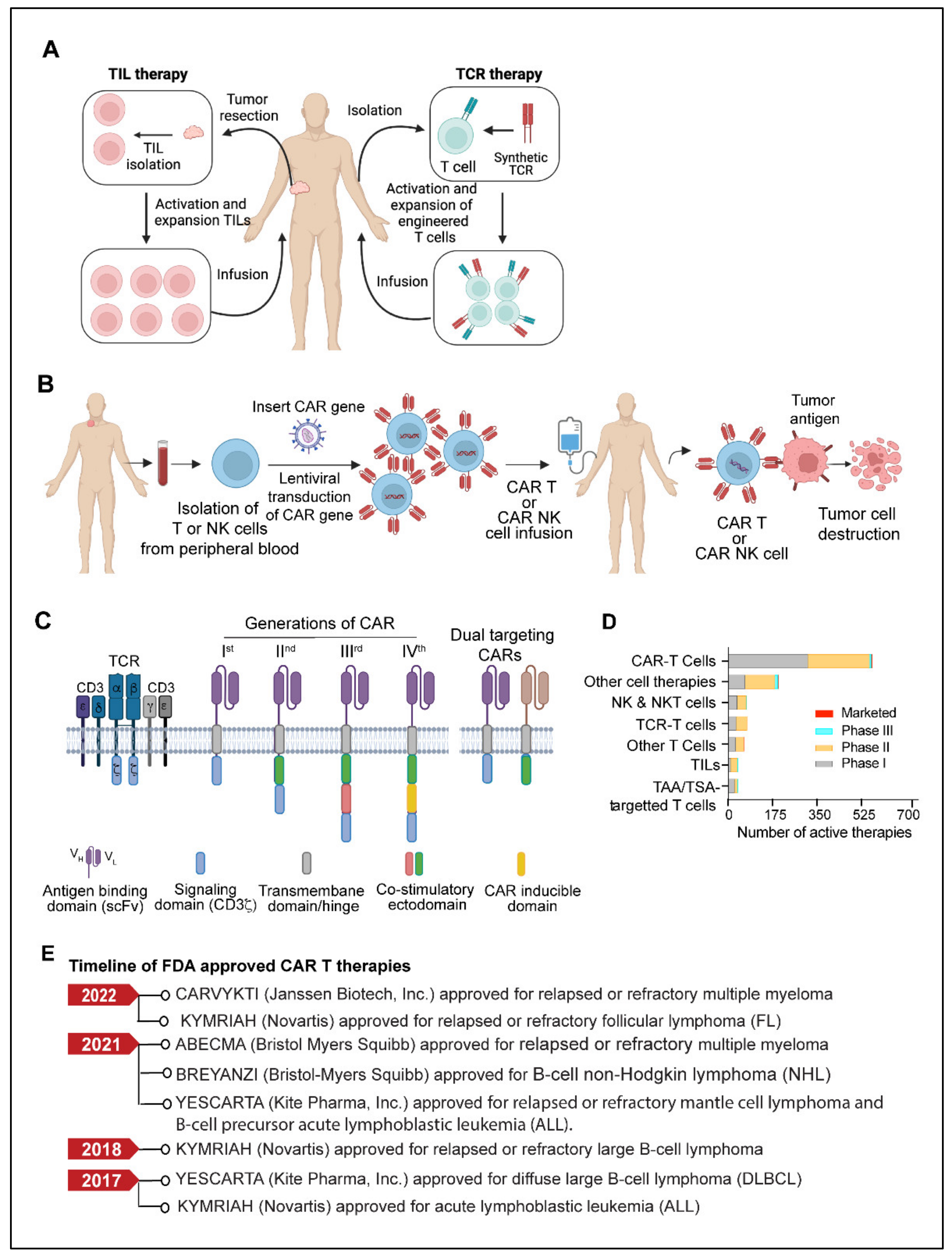
4. Adoptive Cell Therapy
4.1. TILs (Tumor-Infiltrating Lymphocytes)
4.2. TCR (T Cell Receptor) Therapy
4.3. CAR T Cells
4.4. CAR-NK Cells
4.5. Limitations and Challenges of CAR T Therapy
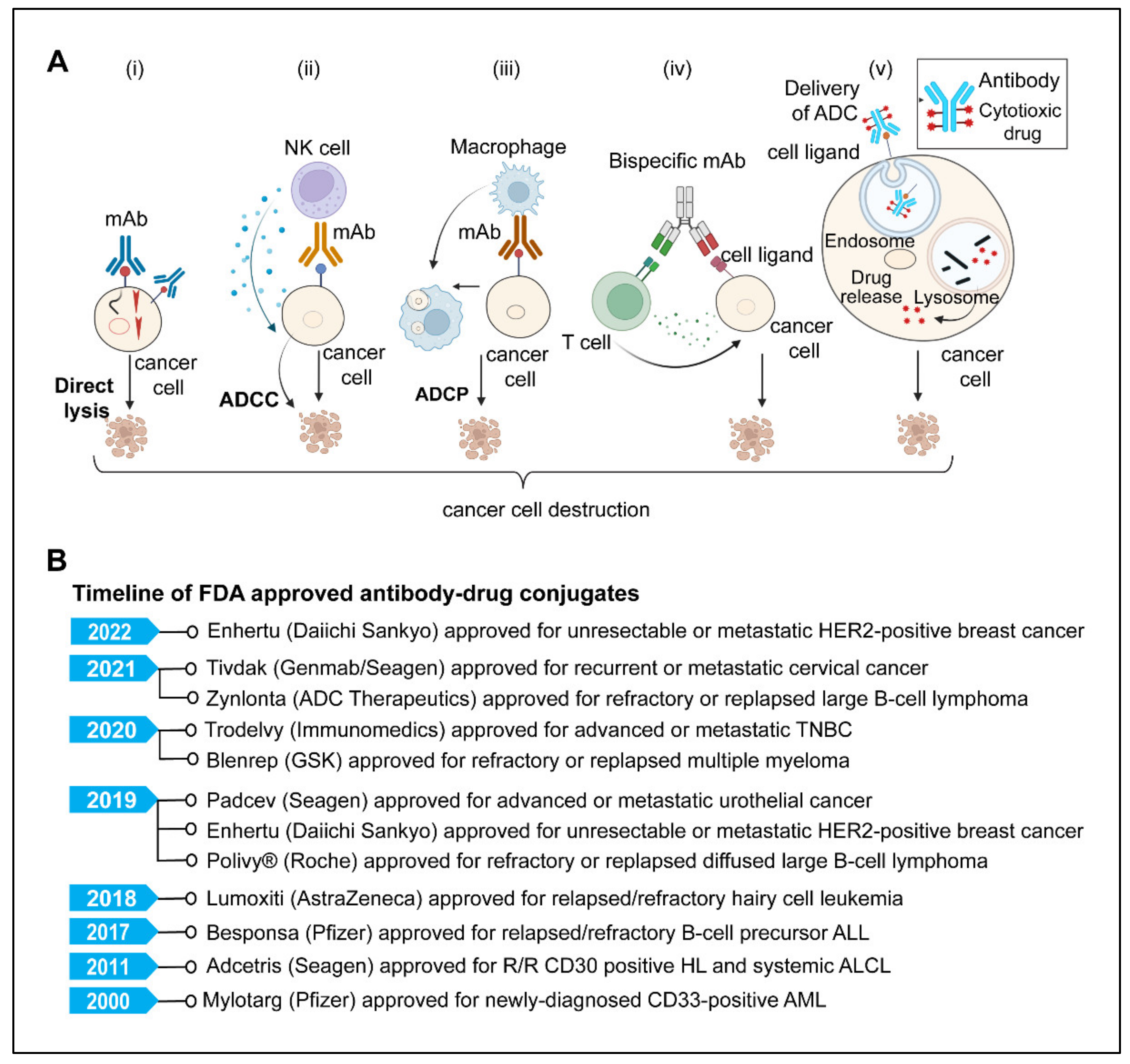
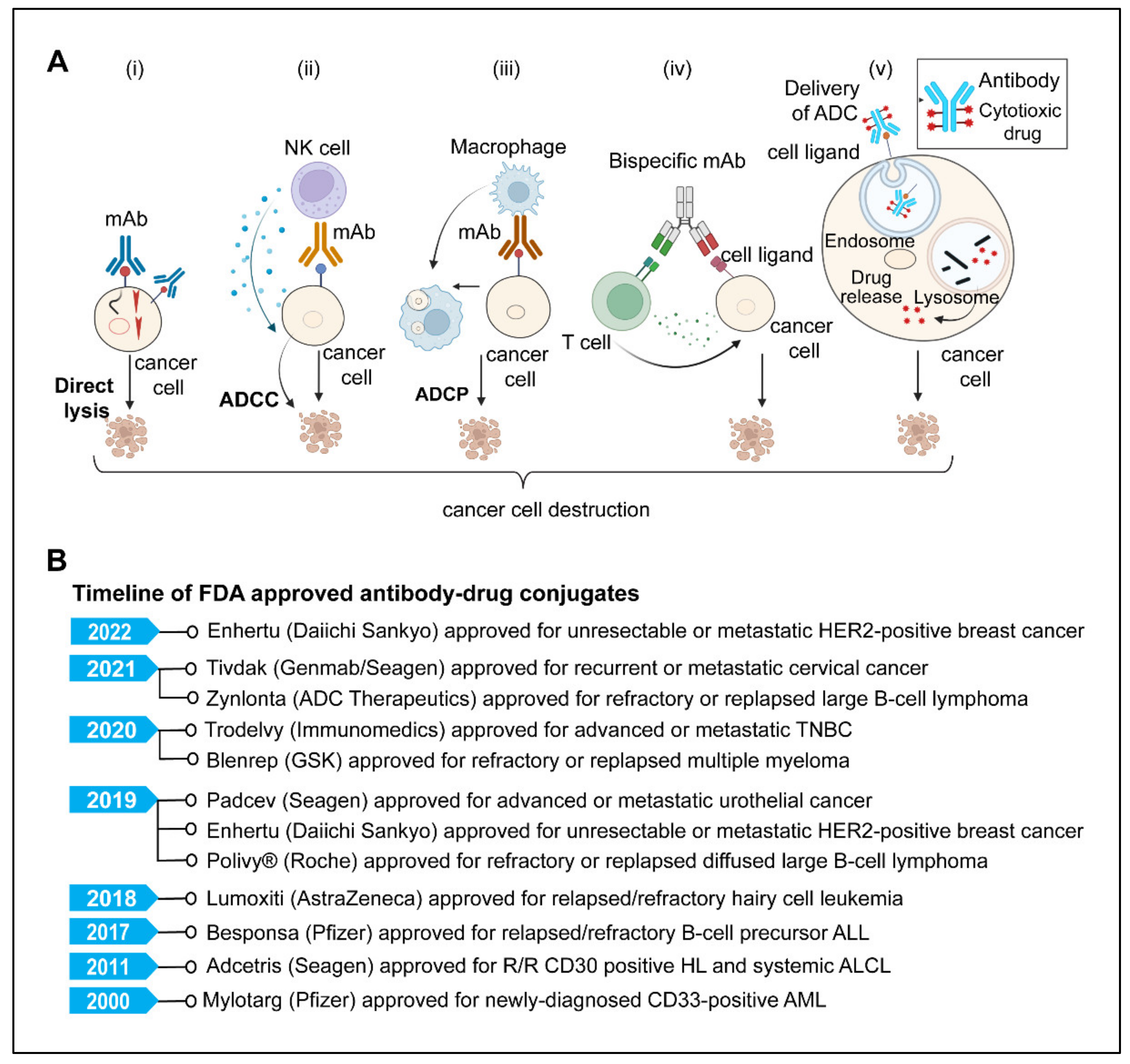
5. Monoclonal Antibodies
5.1. Direct Killing and Immune-Mediated Killing
5.2. mAbs Targeting Angiogenesis
5.3. Antibody-Drug Conjugates
5.4. Antibody Radioimmunoconjugate (RIC)
5.5. Bispecific Antibodies
6. Cytokine Therapies
Limitations and Challenges of Cytokine Therapy
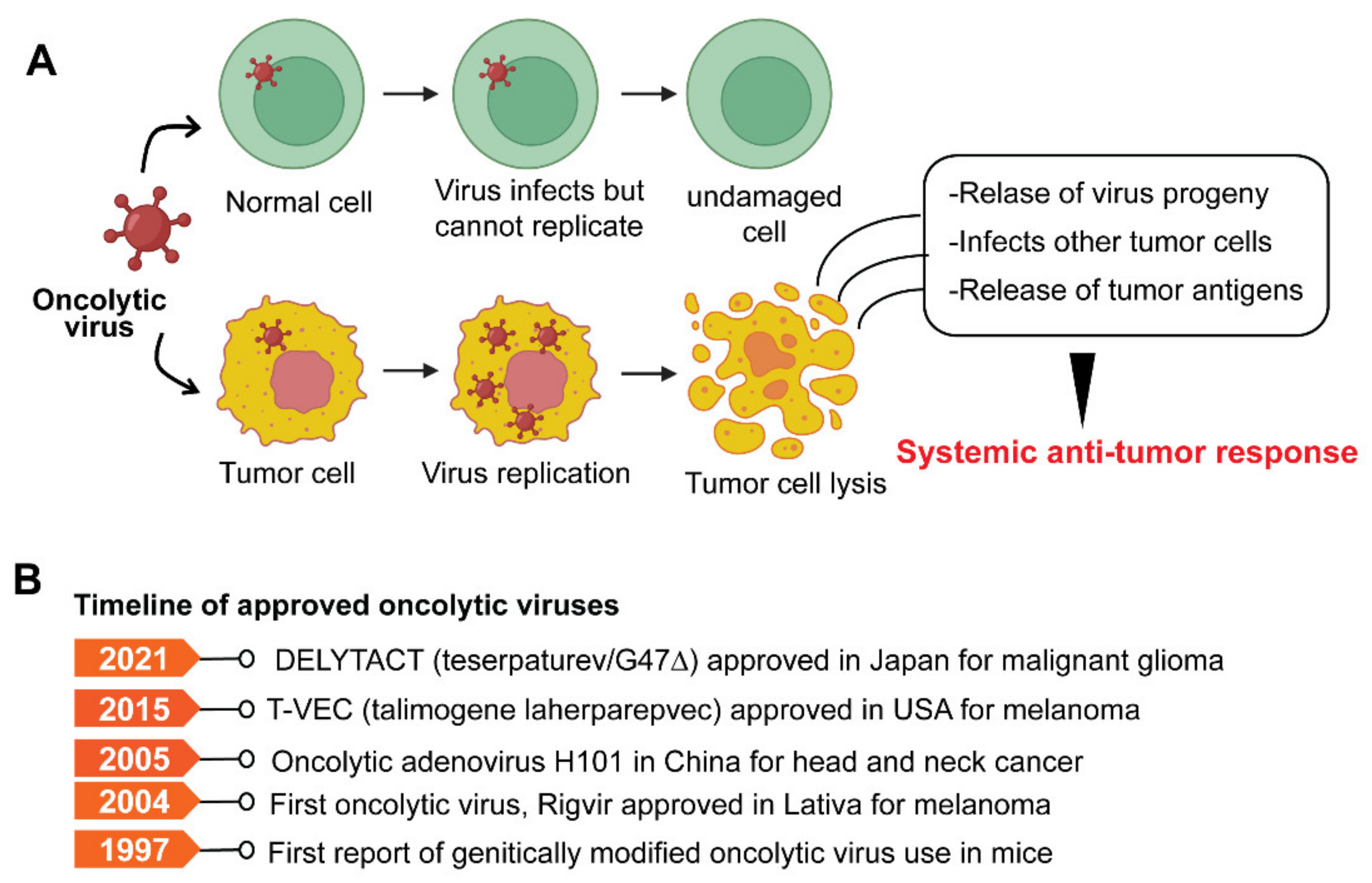
7. Oncolytic Viruses
Limitations and Challenges of OV Therapy
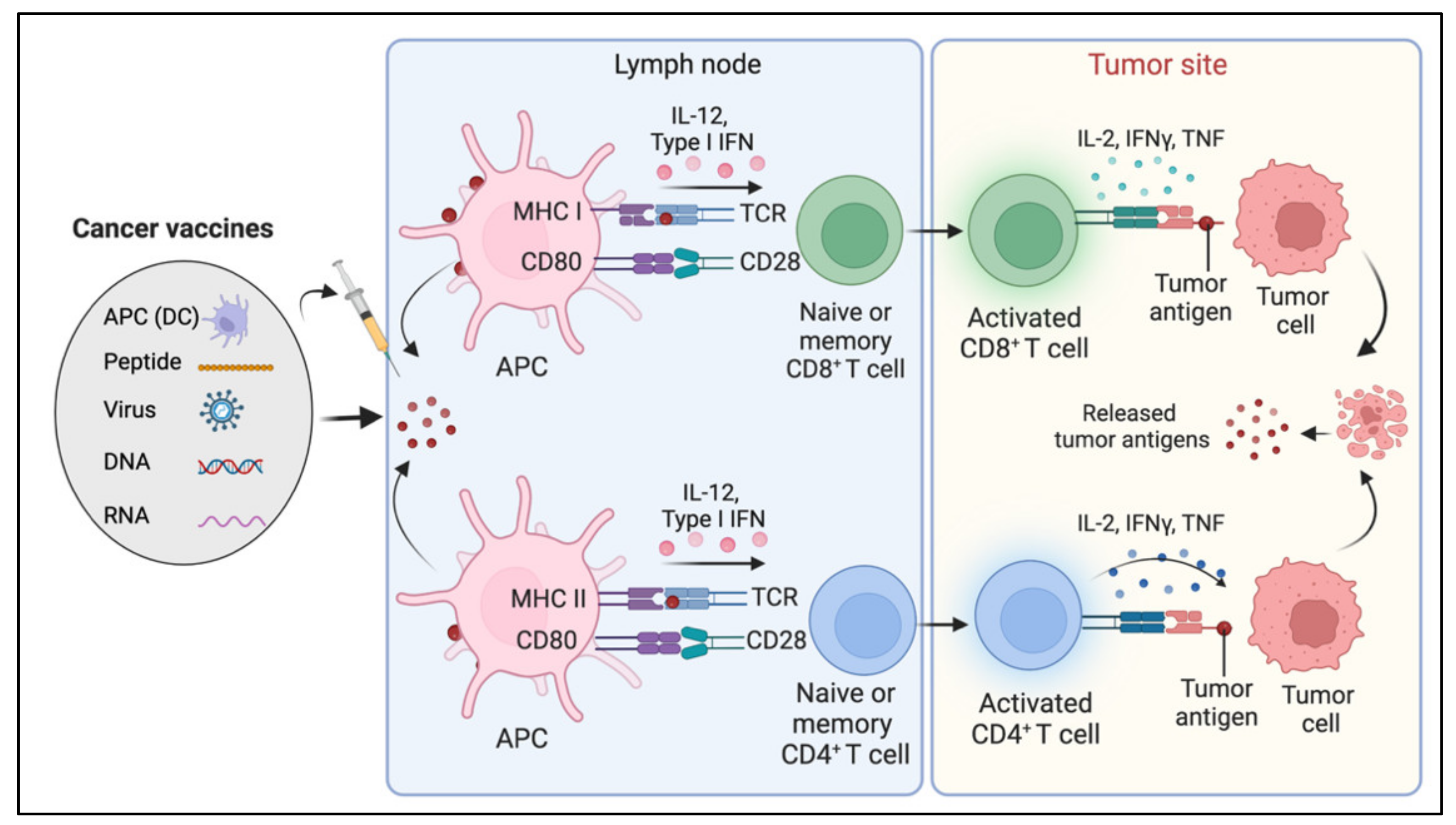
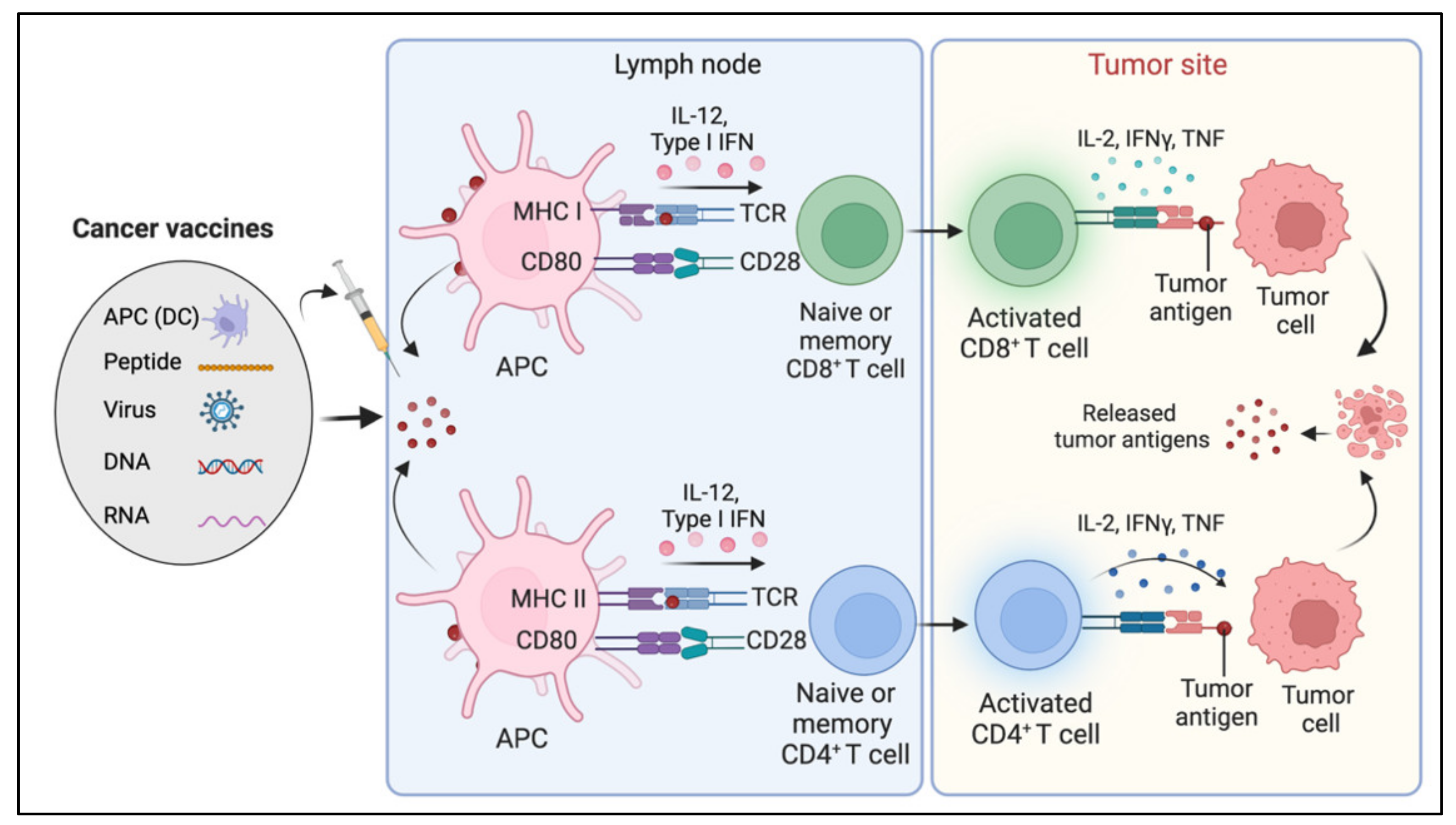
8. Cancer Vaccines
Limitations and Challenges of Cancer Vaccines
9. Conclusions and Future Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- McCarthy, E.F. The toxins of William B. Coley and the treatment of bone and soft-tissue sarcomas. Iowa Orthop. J. 2006, 26, 154–158. [Google Scholar] [PubMed]
- Hayre, D.S. 23. Coley’s toxin and spontaneous tumour regression. Clin. Investig. Med. 2007, 30, 39–40. [Google Scholar] [CrossRef]
- Kramer, M.G.; Masner, M.; Ferreira, F.A.; Hoffman, R.M. Bacterial Therapy of Cancer: Promises, Limitations, and Insights for Future Directions. Front. Microbiol. 2018, 9, 16. [Google Scholar] [CrossRef] [PubMed]
- Oiseth, S.J.; Aziz, M.S. Cancer immunotherapy: A brief review of the history, possibilities, and challenges ahead. J. Cancer Metastasis Treat. 2017, 3, 250. [Google Scholar] [CrossRef]
- Bhatia, A.; Kumar, Y. Cellular and molecular mechanisms in cancer immune escape: A comprehensive review. Expert Rev. Clin. Immunol. 2014, 10, 41–62. [Google Scholar] [CrossRef]
- Dunn, G.P.; Bruce, A.T.; Ikeda, H.; Old, L.J.; Schreiber, R.D. Cancer immunoediting: From immunosurveillance to tumor escape. Nat. Immunol. 2002, 3, 991–998. [Google Scholar] [CrossRef]
- Smyth, M.J.; Dunn, G.P.; Schreiber, R.D. Cancer Immunosurveillance and Immunoediting: The Roles of Immunity in Suppressing Tumor Development and Shaping Tumor Immunogenicity. Adv. Immunol. 2006, 90, 1–50. [Google Scholar]
- Dunn, G.P.; Old, L.J.; Schreiber, R.D. The three Es of cancer immunoediting. Annu. Rev. Immunol. 2004, 22, 329–360. [Google Scholar] [CrossRef]
- Koebel, C.M.; Vermi, W.; Swann, J.B.; Zerafa, N.; Rodig, S.J.; Old, L.J.; Smyth, M.J.; Schreiber, R.D. Adaptive immunity maintains occult cancer in an equilibrium state. Nature 2007, 450, 903–907. [Google Scholar] [CrossRef]
- Zitvogel, L.; Tesniere, A.; Kroemer, G. Cancer despite immunosurveillance: Immunoselection and immunosubversion. Nat. Rev. Immunol. 2006, 6, 715–727. [Google Scholar] [CrossRef]
- Vesely, M.; Schreiber, R.D. Cancer immunoediting: Antigens, mechanisms, and implications to cancer immunotherapy. Ann. N. Y. Acad. Sci. 2013, 1284, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y. Cancer immunotherapy: Harnessing the immune system to battle cancer. J. Clin. Investig. 2015, 125, 3335–3337. [Google Scholar] [CrossRef]
- Lentz, R.W.; Colton, M.D.; Mitra, S.S.; Messersmith, W.A. Innate Immune Checkpoint Inhibitors: The Next Breakthrough in Medical Oncology? Mol. Cancer Ther. 2021, 20, 961–974. [Google Scholar] [CrossRef]
- Ghahremanloo, A.; Soltani, A.; Modaresi, S.M.S.; Hashemy, S.I. Recent advances in the clinical development of immune checkpoint blockade therapy. Cell. Oncol. 2019, 42, 609–626. [Google Scholar] [CrossRef]
- Hargadon, K.M.; Johnson, C.E.; Williams, C.J. Immune checkpoint blockade therapy for cancer: An overview of FDA-approved immune checkpoint inhibitors. Int. Immunopharmacol. 2018, 62, 29–39. [Google Scholar] [CrossRef]
- Nirschl, C.J.; Drake, C.G. Molecular Pathways: Coexpression of Immune Checkpoint Molecules: Signaling Pathways and Implications for Cancer Immunotherapy. Clin. Cancer Res. 2013, 19, 4917–4924. [Google Scholar] [CrossRef]
- Dunn, G.P.; Old, L.J.; Schreiber, R.D. The immunobiology of cancer immunosurveillance and immunoediting. Immunity 2004, 21, 137–148. [Google Scholar] [CrossRef]
- Chen, D.S.; Mellman, I. Elements of cancer immunity and the cancer-immune set point. Nature 2007, 541, 321–330. [Google Scholar] [CrossRef]
- Liu, J.-N.; Kong, X.-S.; Huang, T.; Wang, R.; Li, W.; Chen, Q.-F. Clinical Implications of Aberrant PD-1 and CTLA4 Expression for Cancer Immunity and Prognosis: A Pan-Cancer Study. Front. Immunol. 2020, 11, 2048. [Google Scholar] [CrossRef]
- Dovedi, S.J.; Elder, M.J.; Yang, C.; Sitnikova, S.I.; Irving, L.; Hansen, A.; Hair, J.; Jones, D.C.; Hasani, S.; Wang, B.; et al. Design and Efficacy of a Monovalent Bispecific PD-1/CTLA4 Antibody That Enhances CTLA4 Blockade on PD-1+ Activated T Cells. Cancer Discov. 2021, 11, 1100–1117. [Google Scholar] [CrossRef]
- Buchbinder, E.I.; Desai, A. CTLA-4 and PD-1 pathways similarities, differences, and implications of their inhibition. Am. J. Clin. Oncol. Cancer Clin. Trials 2016, 39, 98–106. [Google Scholar]
- Takahashi, T.; Tagami, T.; Yamazaki, S.; Uede, T.; Shimizu, J.; Sakaguchi, N.; Mak, T.W.; Sakaguchi, S. Immunologic Self-Tolerance Maintained by Cd25+Cd4+Regulatory T Cells Constitutively Expressing Cytotoxic T Lymphocyte–Associated Antigen 4. J. Exp. Med. 2000, 192, 303–310. [Google Scholar] [CrossRef] [PubMed]
- Schadendorf, D.; Hodi, F.S.; Robert, C.; Weber, J.S.; Margolin, K.; Hamid, O.; Patt, D.; Chen, T.-T.; Berman, D.M.; Wolchok, J.D. Pooled Analysis of Long-Term Survival Data From Phase II and Phase III Trials of Ipilimumab in Unresectable or Metastatic Melanoma. J. Clin. Oncol. 2015, 33, 1889–1894. [Google Scholar] [CrossRef] [PubMed]
- Postow, M.A.; Chesney, J.; Pavlick, A.C.; Robert, C.; Grossmann, K.; McDermott, D.; Linette, G.P.; Meyer, N.; Giguere, J.K.; Agarwala, S.S.; et al. Nivolumab and Ipilimumab versus Ipilimumab in Untreated Melanoma. N. Engl. J. Med. 2015, 372, 2006–2017. [Google Scholar] [CrossRef]
- Larkin, J.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.-J.; Cowey, C.L.; Lao, C.D.; Schadendorf, D.; Dummer, R.; Smylie, M.; Rutkowski, P.; et al. Combined Nivolumab and Ipilimumab or Monotherapy in Untreated Melanoma. N. Engl. J. Med. 2015, 373, 23–34. [Google Scholar] [CrossRef]
- Wolchok, J.D.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.-J.; Rutkowski, P.; Lao, C.D.; Cowey, C.L.; Schadendorf, D.; Wagstaff, J.; Dummer, R.; et al. Long-Term Outcomes With Nivolumab Plus Ipilimumab or Nivolumab Alone Versus Ipilimumab in Patients With Advanced Melanoma. J. Clin. Oncol. 2022, 40, 127–137. [Google Scholar] [CrossRef]
- Kamatham, S.; Shahjehan, F.; Kasi, P.M. Immune Checkpoint Inhibitors in Metastatic Colorectal Cancer: Current Status, Recent Advances, and Future Directions. Curr. Colorectal Cancer Rep. 2019, 15, 112–121. [Google Scholar] [CrossRef]
- Morse, M.A.; Hochster, H.; Benson, A. Perspectives on Treatment of Metastatic Colorectal Cancer with Immune Checkpoint Inhibitor Therapy. Oncologist 2019, 25, 33–45. [Google Scholar] [CrossRef]
- Saung, M.T.; Pelosof, L.; Casak, S.; Donoghue, M.; Lemery, S.; Yuan, M.; Rodriguez, L.; Schotland, P.; Chuk, M.; Davis, G.; et al. FDA Approval Summary: Nivolumab Plus Ipilimumab for the Treatment of Patients with Hepatocellular Carcinoma Previously Treated with Sorafenib. Oncologist 2021, 26, 797–806. [Google Scholar] [CrossRef]
- Baas, P.; Scherpereel, A.; Nowak, A.K.; Fujimoto, N.; Peters, S.; Tsao, A.S.; Mansfield, A.S.; Popat, S.; Jahan, T.; Antonia, S.; et al. First-line nivolumab plus ipilimumab in unresectable malignant pleural mesothelioma (CheckMate 743): A multicentre, randomised, open-label, phase 3 trial. Lancet 2021, 397, 375–386. [Google Scholar] [CrossRef]
- Vellanki, P.J.; Mulkey, F.; Jaigirdar, A.A.; Rodriguez, L.; Wang, Y.; Xu, Y.; Zhao, H.; Liu, J.; Howe, G.; Wang, J.; et al. FDA approval summary: Nivolumab with ipilimumab and chemotherapy for metastatic non–small cell lung cancer, A collaborative project orbis review. Clin. Cancer Res. 2021, 27, 3522–3527. [Google Scholar] [CrossRef]
- Gao, X.; McDermott, D.F. Ipilimumab in combination with nivolumab for the treatment of renal cell carcinoma. Expert Opin. Biol. Ther. 2018, 18, 947–957. [Google Scholar] [CrossRef]
- Sharpe, A.H.; Pauken, K.E. The diverse functions of the PD1 inhibitory pathway. Nat. Rev. Immunol. 2018, 18, 153–167. [Google Scholar] [CrossRef]
- Mallett, G.; Laurence, A.; Amarnath, S. Programmed cell death-1 receptor (Pd-1)-mediated regulation of innate lymphoid cells. Int. J. Mol. Sci. 2019, 20, 2836. [Google Scholar] [CrossRef]
- Upadhaya, S.; Neftelinov, S.T.; Hodge, J.; Campbell, J. Challenges and opportunities in the PD1/PDL1 inhibitor clinical trial landscape. Nat. Rev. Drug Discov. 2022, 21, 482–483. [Google Scholar] [CrossRef]
- Kraehenbuehl, L.; Weng, C.H.; Eghbali, S.; Wolchok, J.D.; Merghoub, T. Enhancing immunotherapy in cancer by targeting emerging immunomodulatory pathways. Nat. Rev. Clin. Oncol. 2022, 19, 37–50. [Google Scholar] [CrossRef]
- Triebel, F.; Jitsukawa, S.; Baixeras, E.; Roman-Roman, S.; Genevee, C.; Viegas-Pequignot, E.; Hercend, T. LAG-3, a novel lymphocyte activation gene closely related to CD4. J. Exp. Med. 1990, 171, 1393–1405. [Google Scholar] [CrossRef]
- Huard, B.; Tournier, M.; Triebel, F. LAG-3 does not define a specific mode of natural killing in human. Immunol. Lett. 1998, 61, 109–112. [Google Scholar] [CrossRef]
- Goldberg, M.V.; Drake, C.G. LAG-3 in cancer immunotherapy. Curr. Top Microbiol. Immunol. 2010, 344, 269–278. [Google Scholar] [CrossRef]
- Shi, A.-P.; Tang, X.-Y.; Xiong, Y.-L.; Zheng, K.-F.; Liu, Y.-J.; Shi, X.-G.; Lv, Y.; Jiang, T.; Ma, N.; Zhao, J.-B. Immune Checkpoint LAG3 and Its Ligand FGL1 in Cancer. Front. Immunol. 2022, 12, 785091. [Google Scholar] [CrossRef]
- Wang, J.; Sanmamed, M.F.; Datar, I.; Su, T.T.; Ji, L.; Sun, J.; Chen, L.; Chen, Y.; Zhu, G.; Yin, W.; et al. Fibrinogen-like Protein 1 Is a Major Immune Inhibitory Ligand of LAG-3. Cell 2019, 176, 334–347.e12. [Google Scholar] [CrossRef] [PubMed]
- Souri, Z.; Wierenga, A.P.A.; Kroes, W.G.M.; van der Velden, P.A.; Verdijk, R.M.; Eikmans, M.; Luyten, G.P.M.; Jager, M.J. LAG3 and Its Ligands Show Increased Expression in High-Risk Uveal Melanoma. Cancers 2021, 13, 4445. [Google Scholar] [CrossRef] [PubMed]
- Matsuzaki, J.; Gnjatic, S.; Mhawech-Fauceglia, P.; Beck, A.; Miller, A.; Tsuji, T.; Eppolito, C.; Qian, F.; Lele, S.; Shrikant, P.; et al. Tumor-infiltrating NY-ESO-1–specific CD8 + T cells are negatively regulated by LAG-3 and PD-1 in human ovarian cancer. Proc. Natl. Acad. Sci. USA 2010, 107, 7875–7880. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.-Z.; Kim, H.J.; Villasboas, J.C.; Chen, Y.-P.; Price-Troska, T.; Jalali, S.; Wilson, M.; Novak, A.J.; Ansell, S.M. Expression of LAG-3 defines exhaustion of intratumoral PD-1+ T cells and correlates with poor outcome in follicular lymphoma. Oncotarget 2017, 8, 61425–61439. [Google Scholar] [CrossRef] [PubMed]
- Deng, W.-W.; Mao, L.; Yu, G.-T.; Bu, L.-L.; Ma, S.-R.; Liu, B.; Gutkind, J.S.; Kulkarni, A.B.; Zhang, W.-F.; Sun, Z.-J. LAG-3 confers poor prognosis and its blockade reshapes antitumor response in head and neck squamous cell carcinoma. OncoImmunology 2016, 5, e1239005. [Google Scholar] [CrossRef] [PubMed]
- Maruhashi, T.; Sugiura, D.; Okazaki, I.M.; Okazaki, T. LAG-3: From molecular functions to clinical applications. J. Immunother. Cancer 2020, 8, e001014. [Google Scholar] [CrossRef]
- Andrews, L.P.; Cillo, A.R.; Karapetyan, L.; Kirkwood, J.M.; Workman, C.J.; Vignali, D.A. Molecular Pathways and Mechanisms of LAG-3 in Cancer Therapy. Clin. Cancer Res. 2022, OF1–OF10. [Google Scholar] [CrossRef]
- Lichtenegger, F.S.; Rothe, M.; Schnorfeil, F.M.; Deiser, K.; Krupka, C.; Augsberger, C.; Schlüter, M.; Neitz, J.; Subklewe, M. Targeting LAG-3 and PD-1 to Enhance T Cell Activation by Antigen-Presenting Cells. Front. Immunol. 2018, 9, 385. [Google Scholar] [CrossRef]
- Wierz, M.; Pierson, S.; Guyonnet, L.; Viry, E.; Lequeux, A.; Oudin, A.; Niclou, S.P.; Ollert, M.; Berchem, G.; Janji, B.; et al. Dual PD1/LAG3 immune checkpoint blockade limits tumor development in a murine model of chronic lymphocytic leukemia. Blood 2018, 131, 1617–1621. [Google Scholar] [CrossRef]
- Qi, Y.; Chen, L.; Liu, Q.; Kong, X.; Fang, Y.; Wang, J. Research Progress Concerning Dual Blockade of Lymphocyte-Activation Gene 3 and Programmed Death-1/Programmed Death-1 Ligand-1 Blockade in Cancer Immunotherapy: Preclinical and Clinical Evidence of This Potentially More Effective Immunotherapy Strategy. Front. Immunol. 2021, 11, 563258. [Google Scholar] [CrossRef]
- Andrews, L.P.; Yano, H.; Vignali, D.A.A. Inhibitory receptors and ligands beyond PD-1, PD-L1 and CTLA-4: Breakthroughs or backups. Nat. Immunol. 2019, 20, 1425–1434. [Google Scholar] [CrossRef]
- Lipson, E.J.; Tawbi, H.A.-H.; Schadendorf, D.; Ascierto, P.A.; Matamala, L.; Gutiérrez, E.C.; Rutkowski, P.; Gogas, H.; Lao, C.D.; de Menezes, J.J.; et al. Relatlimab (RELA) plus nivolumab (NIVO) versus NIVO in first-line advanced melanoma: Primary phase III results from RELATIVITY-047 (CA224-047). J. Clin. Oncol. 2021, 39, 9503. [Google Scholar] [CrossRef]
- Paik, J. Nivolumab Plus Relatlimab: First Approval. Drugs 2022, 82, 925–931. [Google Scholar] [CrossRef]
- Agocs, G.R.; Assarzadegan, N.; Kirsch, R.; Dawson, H.; Galván, J.; Lugli, A.; Zlobec, I.; Berger, M. LAG-3 Expression Predicts Outcome in Stage II Colon Cancer. J. Pers. Med. 2021, 11, 749. [Google Scholar] [CrossRef]
- Qin, S.; Xu, L.; Yi, M.; Yu, S.; Wu, K.; Luo, S. Novel immune checkpoint targets: Moving beyond PD-1 and CTLA-4. Mol. Cancer 2009, 18, 155. [Google Scholar] [CrossRef]
- Chauvin, J.M.; Zarour, H.M. TIGIT in cancer immunotherapy. J. Immunother. Cancer 2020, 8, e000957. [Google Scholar] [CrossRef]
- Harjunpää, H.; Guillerey, C. TIGIT as an emerging immune checkpoint. Clin. Exp. Immunol. 2020, 200, 108–119. [Google Scholar] [CrossRef]
- Starbuck, K.; Morrell, K.; Odunsi, K.; Zsiros, E.; Szender, J.; Frederick, P.; Lele, S.; Akers, S.; Eng, K. TIGIT ligands CD155, CD112, and galectin-9 are associated with immune infiltration and increased overall survival in ovarian cancer. Gynecol. Oncol. 2018, 149, 56. [Google Scholar] [CrossRef]
- Lozano, E.; Mena, M.-P.; Díaz, T.; Martin-Antonio, B.; León, S.; Rodríguez-Lobato, L.-G.; Oliver-Caldés, A.; Cibeira, M.T.; Bladé, J.; Prat, A.; et al. Nectin-2 Expression on Malignant Plasma Cells Is Associated with Better Response to TIGIT Blockade in Multiple Myeloma. Clin. Cancer Res. 2020, 26, 4688–4698. [Google Scholar] [CrossRef]
- Yu, X.; Harden, K.; Gonzalez, L.C.; Francesco, M.; Chiang, E.; Irving, B.; Tom, I.; Ivelja, S.; Refino, C.J.; Clark, H.; et al. The surface protein TIGIT suppresses T cell activation by promoting the generation of mature immunoregulatory dendritic cells. Nat. Immunol. 2008, 10, 48–57. [Google Scholar] [CrossRef]
- Stanietsky, N.; Simic, H.; Arapovic, J.; Toporik, A.; Levy, O.; Novik, A.; Levine, Z.; Beiman, M.; Dassa, L.; Achdout, H.; et al. The interaction of TIGIT with PVR and PVRL2 inhibits human NK cell cytotoxicity. Proc. Natl. Acad. Sci. USA 2009, 106, 17858–17863. [Google Scholar] [CrossRef] [PubMed]
- Lozano, E.; Dominguez-Villar, M.; Kuchroo, V.; Hafler, D.A. The TIGIT/CD226 Axis Regulates Human T Cell Function. J. Immunol. 2012, 188, 3869–3875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnston, R.J.; Comps-Agrar, L.; Hackney, J.; Yu, X.; Huseni, M.; Yang, Y.; Park, S.; Javinal, V.; Chiu, H.; Irving, B.; et al. The Immunoreceptor TIGIT Regulates Antitumor and Antiviral CD8 + T Cell Effector Function. Cancer Cell 2014, 26, 923–937. [Google Scholar] [CrossRef] [PubMed]
- McNulty, R. FDA Grants Tiragolumab Breakthrough Therapy Designation for PD-L1–High NSCLC. Evid.-Based Oncol. 2021, 27, SP58. [Google Scholar]
- Anderson, A.C.; Joller, N.; Kuchroo, V.K. Lag-3, Tim-3, and TIGIT: Co-inhibitory Receptors with Specialized Functions in Immune Regulation. Immunity 2016, 44, 989–1004. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Cai, P.; Li, L.; Shi, L.; Chang, P.; Liang, T.; Yang, Q.; Liu, Y.; Wang, L.; Hu, L. Co-expression of TIM-3 and CEACAM1 promotes T cell exhaustion in colorectal cancer patients. Int. Immunopharmacol. 2017, 43, 210–218. [Google Scholar] [CrossRef]
- Zhu, C.; Anderson, A.C.; Schubart, A.; Xiong, H.; Imitola, J.; Khoury, S.; Zheng, X.X.; Strom, T.B.; Kuchroo, V.K. The Tim-3 ligand galectin-9 negatively regulates T helper type 1 immunity. Nat. Immunol. 2005, 6, 1245–1252. [Google Scholar] [CrossRef]
- Freeman, G.J.; Casasnovas, J.M.; Umetsu, D.T.; Dekruyff, R.H. TIM genes: A family of cell surface phosphatidylserine receptors that regulate innate and adaptive immunity. Immunol. Rev. 2010, 235, 172–189. [Google Scholar] [CrossRef]
- Chiba, S.; Baghdadi, M.; Akiba, H.; Yoshiyama, H.; Kinoshita, I.; Dosaka-Akita, H.; Fujioka, Y.; Ohba, Y.; Gorman, J.V.; Colgan, J.D.; et al. Tumor-infiltrating DCs suppress nucleic acid–mediated innate immune responses through interactions between the receptor TIM-3 and the alarmin HMGB1. Nat. Immunol. 2012, 13, 832–842. [Google Scholar] [CrossRef]
- Huang, Y.-H.; Zhu, C.; Kondo, Y.; Anderson, A.C.; Gandhi, A.; Russell, A.F.; Dougan, S.K.; Petersen, B.-S.; Melum, E.; Pertel, T.; et al. CEACAM1 regulates TIM-3-mediated tolerance and exhaustion. Nature 2015, 517, 386–390. [Google Scholar] [CrossRef]
- Anderson, A.C. Tim-3: An Emerging Target in the Cancer Immunotherapy Landscape. Cancer Immunol. Res. 2014, 2, 393–398. [Google Scholar] [CrossRef]
- Zhou, Q.; Munger, M.; Veenstra, R.G.; Weigel, B.J.; Hirashima, M.; Munn, D.; Murphy, W.J.; Azuma, M.; Anderson, A.C.; Kuchroo, V.K.; et al. Coexpression of Tim-3 and PD-1 identifies a CD8+ T-cell exhaustion phenotype in mice with disseminated acute myelogenous leukemia. Blood 2011, 117, 4501–4510. [Google Scholar] [CrossRef]
- Fourcade, J.; Sun, Z.; Pagliano, O.; Chauvin, J.-M.; Sander, C.; Janjic, B.; Tarhini, A.A.; Tawbi, H.A.; Kirkwood, J.M.; Moschos, S.; et al. PD-1 and Tim-3 Regulate the Expansion of Tumor Antigen–Specific CD8+ T Cells Induced by Melanoma Vaccines. Cancer Res. 2014, 74, 1045–1055. [Google Scholar] [CrossRef] [Green Version]
- Jin, H.-T.; Anderson, A.C.; Tan, W.G.; West, E.E.; Ha, S.-J.; Araki, K.; Freeman, G.J.; Kuchroo, V.K.; Ahmed, R. Cooperation of Tim-3 and PD-1 in CD8 T-cell exhaustion during chronic viral infection. Proc. Natl. Acad. Sci. USA 2010, 107, 14733–14738. [Google Scholar] [CrossRef]
- Picarda, E.; Ohaegbulam, K.C.; Zang, X. Molecular Pathways: Targeting B7-H3 (CD276) for Human Cancer Immunotherapy. Clin. Cancer Res. 2016, 22, 3425–3431. [Google Scholar] [CrossRef]
- Yang, S.; Wei, W.; Zhao, Q. B7-H3, a checkpoint molecule, as a target for cancer immunotherapy. Int. J. Biol. Sci. 2020, 16, 1767–1773. [Google Scholar] [CrossRef]
- Podojil, J.R.; Miller, S.D. Potential targeting of B7-H4 for the treatment of cancer. Immunol. Rev. 2017, 276, 40–51. [Google Scholar] [CrossRef]
- Powderly, J.D.; Jang, S.; Lohr, J.; Spira, A.I.; Bohac, G.C.; Sharma, M. Preliminary dose escalation results from a phase I/II, first-in-human study of MGC018 (anti-B7-H3 antibody-drug conjugate) in patients with advanced solid tumors. J. Clin. Oncol. 2020, 38, 3071. [Google Scholar] [CrossRef]
- Im, E.; Sim, D.Y.; Lee, H.-J.; Park, J.E.; Park, W.Y.; Ko, S.; Kim, B.; Shim, B.S.; Kim, S.-H. Immune functions as a ligand or a receptor, cancer prognosis potential, clinical implication of VISTA in cancer immunotherapy. Semin. Cancer Biol. 2021. [Google Scholar] [CrossRef]
- Tagliamento, M.; Bironzo, P.; Novello, S. New emerging targets in cancer immunotherapy: The role of VISTA. ESMO Open 2020, 4, e000683. [Google Scholar] [CrossRef]
- ElTanbouly, M.A.; Schaafsma, E.; Noelle, R.J.; Lines, J.L. VISTA: Coming of age as a multi-lineage immune checkpoint. Clin. Exp. Immunol. 2020, 200, 120–130. [Google Scholar] [CrossRef] [PubMed]
- Yuan, L.; Tatineni, J.; Mahoney, K.M.; Freeman, G.J. VISTA: A Mediator of Quiescence and a Promising Target in Cancer Immunotherapy. Trends Immunol. 2021, 42, 209–227. [Google Scholar] [CrossRef] [PubMed]
- ElTanbouly, M.A.; Zhao, Y.; Nowak, E.; Li, J.; Schaafsma, E.; Le Mercier, I.; Ceeraz, S.; Lines, J.L.; Peng, C.; Carriere, C.; et al. VISTA is a checkpoint regulator for naïve T cell quiescence and peripheral tolerance. Science 2020, 367, eaay0524. [Google Scholar] [CrossRef] [PubMed]
- Johnston, R.J.; Su, L.J.; Pinckney, J.; Critton, D.; Boyer, E.; Krishnakumar, A.; Corbett, M.; Rankin, A.L.; DiBella, R.; Campbell, L.; et al. VISTA is an acidic pH-selective ligand for PSGL-1. Nature 2019, 574, 565–570. [Google Scholar] [CrossRef]
- Liu, J.; Yuan, Y.; Chen, W.; Putra, J.; Suriawinata, A.A.; Schenk, A.D.; Miller, H.E.; Guleria, I.; Barth, R.J.; Huang, Y.H.; et al. Immune-checkpoint proteins VISTA and PD-1 nonredundantly regulate murine T-cell responses. Proc. Natl. Acad. Sci. USA 2015, 112, 6682–6687. [Google Scholar] [CrossRef]
- Wang, L.; Le Mercier, I.; Putra, J.; Chen, W.; Liu, J.; Schenk, A.D.; Nowak, E.C.; Suriawinata, A.A.; Li, J.; Noelle, R.J. Disruption of the immune-checkpoint VISTA gene imparts a proinflammatory phenotype with predisposition to the development of autoimmunity. Proc. Natl. Acad. Sci. USA 2014, 111, 14846–14851. [Google Scholar] [CrossRef]
- Le Mercier, I.; Chen, W.; Lines, J.L.; Day, M.; Li, J.; Sergent, P.; Noelle, R.J.; Wang, L. VISTA Regulates the Development of Protective Antitumor Immunity. Cancer Res. 2014, 74, 1933–1944. [Google Scholar] [CrossRef]
- Han, X.; Vesely, M.D.; Yang, W.; Sanmamed, M.F.; Badri, T.; Alawa, J.; López-Giráldez, F.; Gaule, P.; Lee, S.W.; Zhang, J.-P.; et al. PD-1H (VISTA)–mediated suppression of autoimmunity in systemic and cutaneous lupus erythematosus. Sci. Transl. Med. 2019, 11, eaax1159. [Google Scholar] [CrossRef]
- Mehta, N.; Maddineni, S.; Kelly, R.L.; Lee, R.B.; Hunter, S.A.; Silberstein, J.L.; Sperberg, R.A.P.; Miller, C.L.; Rabe, A.; Labanieh, L.; et al. An engineered antibody binds a distinct epitope and is a potent inhibitor of murine and human VISTA. Sci. Rep. 2020, 10, 15171. [Google Scholar] [CrossRef]
- Lee, D.H. Update of early phase clinical trials in cancer immunotherapy. BMB Rep. 2021, 54, 70–88. [Google Scholar] [CrossRef]
- Weinberg, A.D.; Rivera, M.-M.; Prell, R.; Morris, A.; Ramstad, T.; Vetto, J.T.; Urba, W.J.; Alvord, G.; Bunce, C.; Shields, J. Engagement of the OX-40 Receptor In Vivo Enhances Antitumor Immunity. J. Immunol. 2000, 164, 2160–2169. [Google Scholar] [CrossRef]
- Imura, A.; Hori, T.; Imada, K.; Ishikawa, T.; Tanaka, Y.; Maeda, M.; Imamura, S.; Uchiyama, T. The human OX40/gp34 system directly mediates adhesion of activated T cells to vascular endothelial cells. J. Exp. Med. 1996, 183, 2185–2195. [Google Scholar] [CrossRef]
- Gramaglia, I.; Weinberg, A.D.; Lemon, M.; Croft, M. Ox-40 ligand: A potent costimulatory molecule for sustaining primary CD4 T cell responses. J. Immunol. 1998, 161, 6510–6517. [Google Scholar]
- Alves Costa Silva, C.; Facchinetti, F.; Routy, B.; Derosa, L. New pathways in immune stimulation: Targeting OX40. ESMO Open 2020, 5, e000573. [Google Scholar] [CrossRef] [Green Version]
- Trimble, E.L.; Ungerleider, R.S.; Abrams, J.A.; Kaplan, R.S.; Feigal, E.G.; Smith, M.A.; Carter, C.L.; Friedman, M.A. Neoadjuvant therapy in cancer treatment. Cancer 1993, 72, 3515–3524. [Google Scholar] [CrossRef]
- Fu, Y.; Lin, Q.; Zhang, Z.; Zhang, L. Therapeutic strategies for the costimulatory molecule OX40 in T-cell-mediated immunity. Acta Pharm. Sin. B 2020, 10, 414–433. [Google Scholar] [CrossRef]
- Linch, S.N.; McNamara, M.J.; Redmond, W.L. OX40 agonists and combination immunotherapy: Putting the pedal to the metal. Front. Oncol. 2015, 5, 34. [Google Scholar] [CrossRef]
- Croft, M.; So, T.; Duan, W.; Soroosh, P. The significance of OX40 and OX40L to T-cell biology and immune disease. Immunol. Rev. 2009, 229, 173–191. [Google Scholar] [CrossRef]
- Duhen, R.; Ballesteros-Merino, C.; Frye, A.K.; Tran, E.; Rajamanickam, V.; Chang, S.-C.; Koguchi, Y.; Bifulco, C.B.; Bernard, B.; Leidner, R.S.; et al. Neoadjuvant anti-OX40 (MEDI6469) therapy in patients with head and neck squamous cell carcinoma activates and expands antigen-specific tumor-infiltrating T cells. Nat. Commun. 2021, 12, 1047. [Google Scholar] [CrossRef]
- Borg, N.; Alter, C.; Görldt, N.; Jacoby, C.; Ding, Z.; Steckel, B.; Quast, C.; Bönner, F.; Friebe, D.; Temme, S.; et al. CD73 on T Cells Orchestrates Cardiac Wound Healing After Myocardial Infarction by Purinergic Metabolic Reprogramming. Circulation 2017, 136, 297–313. [Google Scholar] [CrossRef]
- Jiang, T.; Xu, X.; Qiao, M.; Li, X.; Zhao, C.; Zhou, F.; Gao, G.; Wu, F.; Chen, X.; Su, C.; et al. Comprehensive evaluation of NT5E/CD73 expression and its prognostic significance in distinct types of cancers. BMC Cancer 2018, 18, 267. [Google Scholar] [CrossRef] [PubMed]
- Vijayan, D.; Young, A.; Teng, M.W.L.; Smyth, M.J. Targeting immunosuppressive adenosine in cancer. Nat. Rev. Cancer 2017, 17, 709–724. [Google Scholar] [CrossRef] [PubMed]
- Buisseret, L.; Rottey, S.; De Bono, J.; Mossakowski, M.; Delafontaine, B.; Manickavasagar, T.; Kotecki, N.; Martinoli, C.; Schneider, M.; De Henau, O.; et al. Abstract CT152: First in human study with EOS100850, a novel potent A2A antagonist, shows excellent tolerance and clinical benefit in immune resistant advanced cancers. Cancer Res. 2020, 80, CT152. [Google Scholar] [CrossRef]
- Luke, J.J.; Powderly, J.D.; Merchan, J.R.; Barve, M.A.; Hotson, A.N.; Mobasher, M.; Kwei, L.; Luciano, G.; Buggy, J.J.; Piccione, E.; et al. Immunobiology, preliminary safety, and efficacy of CPI-006, an anti-CD73 antibody with immune modulating activity, in a phase 1 trial in advanced cancers. J. Clin. Oncol. 2019, 37, 2505. [Google Scholar] [CrossRef]
- Creelan, B.C.; Antonia, S.J. The NKG2A immune checkpoint—a new direction in cancer immunotherapy. Nat. Rev. Clin. Oncol. 2019, 16, 277–278. [Google Scholar] [CrossRef]
- Ducoin, K.; Oger, R.; Mutala, L.B.; Deleine, C.; Jouand, N.; Desfrançois, J.; Podevin, J.; Duchalais, E.; Cruard, J.; Benlalam, H.; et al. Targeting NKG2A to boost anti-tumor CD8 T-cell responses in human colorectal cancer. OncoImmunology 2022, 11, 2046931. [Google Scholar] [CrossRef]
- Kamiya, T.; Seow, S.V.; Wong, D.; Robinson, M.; Campana, D. Blocking expression of inhibitory receptor NKG2A overcomes tumor resistance to NK cells. J. Clin. Investig. 2019, 129, 2094–2106. [Google Scholar] [CrossRef]
- Van Hall, T.; André, P.; Horowitz, A.; Ruan, D.F.; Borst, L.; Zerbib, R.; Narni-Mancinelli, E.; van der Burg, S.H.; Vivier, E. Monalizumab: Inhibiting the novel immune checkpoint NKG2A. J. Immunother. Cancer 2019, 7, 263. [Google Scholar] [CrossRef]
- Matlung, H.L.; Szilagyi, K.; Barclay, N.A.; van den Berg, T.K. The CD47-SIRPα signaling axis as an innate immune checkpoint in cancer. Immunol. Rev. 2017, 276, 145–164. [Google Scholar] [CrossRef] [PubMed]
- Huang, B.; Bai, Z.; Ye, X.; Zhou, C.; Xie, X.; Zhong, Y.; Lin, K.; Ma, L. Structural analysis and binding sites of inhibitors targeting the CD47/SIRPα interaction in anticancer therapy. Comput. Struct. Biotechnol. J. 2021, 19, 5494–5503. [Google Scholar] [CrossRef]
- Veillette, A.; Chen, J. SIRPα–CD47 Immune Checkpoint Blockade in Anticancer Therapy. Trends Immunol. 2018, 39, 173–184. [Google Scholar] [CrossRef]
- Feng, R.; Zhao, H.; Xu, J.; Shen, C. CD47: The next checkpoint target for cancer immunotherapy. Crit. Rev. Oncol. Hematol. 2020, 152, 103014. [Google Scholar] [CrossRef]
- Zhang, W.; Huang, Q.; Xiao, W.; Zhao, Y.; Pi, J.; Xu, H.; Zhao, H.; Xu, J.; Evans, C.E.; Jin, H. Advances in Anti-Tumor Treatments Targeting the CD47/SIRPα Axis. Front. Immunol. 2020, 11, 18. [Google Scholar] [CrossRef]
- Qu, T.; Li, B.; Wang, Y. Targeting CD47/SIRPα as a therapeutic strategy, where we are and where we are headed. Biomark. Res. 2022, 10, 20. [Google Scholar] [CrossRef]
- Lin, F.; Xiong, M.; Hao, W.; Song, Y.; Liu, R.; Yang, Y.; Yuan, X.; Fan, D.; Zhang, Y.; Hao, M.; et al. A Novel Blockade CD47 Antibody with Therapeutic Potential for Cancer. Front. Oncol. 2021, 10, 615534. [Google Scholar] [CrossRef]
- Feng, M.; Jiang, W.; Kim, B.Y.S.; Zhang, C.C.; Fu, Y.-X.; Weissman, I.L. Phagocytosis checkpoints as new targets for cancer immunotherapy. Nat. Rev. Cancer 2019, 19, 568–586. [Google Scholar] [CrossRef]
- Zhao, J.; Zhong, S.; Niu, X.; Jiang, J.; Zhang, R.; Li, Q. The MHC class I-LILRB1 signalling axis as a promising target in cancer therapy. Scand. J. Immunol. 2019, 90, e12804. [Google Scholar] [CrossRef]
- Zhang, C.; Zhang, J.; Liang, F.; Guo, H.; Gao, S.; Yang, F.; Guo, H.; Wang, G.; Wang, W.; Zhou, G. Innate immune checkpoint Siglec10 in cancers: Mining of comprehensive omics data and validation in patient samples. Front. Med. 2022, 16, 596–609. [Google Scholar] [CrossRef] [PubMed]
- Yin, S.S.; Gao, F.H. Molecular Mechanism of Tumor Cell Immune Escape Mediated by CD24/Siglec-10. Front. Immunol. 2020, 11, 1324. [Google Scholar] [CrossRef]
- Wang, J.; Manni, M.; Bärenwaldt, A.; Wieboldt, R.; Kirchhammer, N.; Ivanek, R.; Stanczak, M.; Zippelius, A.; König, D.; Manutano, N.R.; et al. Siglec Receptors Modulate Dendritic Cell Activation and Antigen Presentation to T Cells in Cancer. Front. Cell Dev. Biol. 2022, 10, 828916. [Google Scholar] [CrossRef]
- Zhang, P.; Lu, X.; Tao, K.; Shi, L.; Li, W.; Wang, G.; Wu, K. Siglec-10 is associated with survival and natural killer cell dysfunction in hepatocellular carcinoma. J. Surg. Res. 2014, 194, 107–113. [Google Scholar] [CrossRef]
- Barkal, A.A.; Brewer, R.E.; Markovic, M.; Kowarsky, M.; Barkal, S.A.; Zaro, B.; Krishnan, V.; Hatakeyama, J.; Dorigo, O.; Barkal, L.J.; et al. CD24 signalling through macrophage Siglec-10 is a target for cancer immunotherapy. Nature 2019, 572, 392–396. [Google Scholar] [CrossRef]
- Kamber, R.A.; Nishiga, Y.; Morton, B.; Banuelos, A.M.; Barkal, A.A.; Vences-Catalán, F.; Gu, M.; Fernandez, D.; Seoane, J.A.; Yao, D.; et al. Inter-cellular CRISPR screens reveal regulators of cancer cell phagocytosis. Nature 2021, 597, 549–554. [Google Scholar] [CrossRef] [PubMed]
- Waldman, A.D.; Fritz, J.M.; Lenardo, M.J. A guide to cancer immunotherapy: From T cell basic science to clinical practice. Nat. Rev. Immunol. 2020, 20, 651–668. [Google Scholar] [CrossRef]
- Michielin, O.; Lalani, A.K.; Robert, C.; Sharma, P.; Peters, S. Defining unique clinical hallmarks for immune checkpoint inhibitor-based therapies. J. Immunother. Cancer 2022, 10, e003024. [Google Scholar] [CrossRef]
- Borcoman, E.; Kanjanapan, Y.; Champiat, S.; Kato, S.; Servois, V.; Kurzrock, R.; Goel, S.; Bedard, P.; le Tourneau, C. Novel patterns of response under immunotherapy. Ann. Oncol. 2019, 30, 385–396. [Google Scholar] [CrossRef] [PubMed]
- Pons-Tostivint, E.; Latouche, A.; Vaflard, P.; Ricci, F.; Loirat, D.; Hescot, S.; Sablin, M.-P.; Rouzier, R.; Kamal, M.; Morel, C.; et al. Comparative Analysis of Durable Responses on Immune Checkpoint Inhibitors Versus Other Systemic Therapies: A Pooled Analysis of Phase III Trials. JCO Precis. Oncol. 2019, 3, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Park, H.J.; Kim, K.W.; Pyo, J.; Suh, C.H.; Yoon, S.; Hatabu, H.; Nishino, M. Incidence of pseudoprogression during immune checkpoint inhibitor therapy for solid tumors: A systematic review and meta-Analysis. Radiology 2020, 297, 87–96. [Google Scholar] [CrossRef] [PubMed]
- Jia, W.; Gao, Q.; Han, A.; Zhu, H.; Yu, J. The potential mechanism, recognition and clinical significance of tumor pseudoprogression after immunotherapy. Cancer Biol. Med. 2019, 16, 655–670. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Wang, Q.; Dong, Q.; Zhan, L.; Zhang, J. How to differentiate pseudoprogression from true progression in cancer patients treated with immunotherapy. Am. J. Cancer Res. 2019, 9, 1546–1553. [Google Scholar]
- Johnson, D.B.; Nebhan, C.A.; Moslehi, J.J.; Balko, J.M. Immune-checkpoint inhibitors: Long-term implications of toxicity. Nat. Rev. Clin. Oncol. 2022, 19, 254–267. [Google Scholar] [CrossRef]
- Kennedy, L.B.; Salama, A. A review of cancer immunotherapy toxicity. CA Cancer J. Clin. 2020, 70, 86–104. [Google Scholar] [CrossRef]
- Conroy, M.; Naidoo, J. Immune-related adverse events and the balancing act of immunotherapy. Nat. Commun. 2022, 13, 392. [Google Scholar] [CrossRef]
- Sharma, P.; Hu-Lieskovan, S.; Wargo, J.A.; Ribas, A. Primary, Adaptive, and Acquired Resistance to Cancer Immunotherapy. Cell 2017, 168, 707–723. [Google Scholar] [CrossRef]
- Jenkins, R.W.; Barbie, D.A.; Flaherty, K.T. Mechanisms of resistance to immune checkpoint inhibitors. Br. J. Cancer 2018, 118, 9–16. [Google Scholar] [CrossRef] [Green Version]
- Zaretsky, J.M.; Garcia-Diaz, A.; Shin, D.S.; Escuin-Ordinas, H.; Hugo, W.; Hu-Lieskovan, S.; Torrejon, D.Y.; Abril-Rodriguez, G.; Sandoval, S.; Barthly, L.; et al. Mutations Associated with Acquired Resistance to PD-1 Blockade in Melanoma. N. Engl. J. Med. 2016, 375, 819–829. [Google Scholar] [CrossRef]
- Łuksza, M.; Riaz, N.; Makarov, V.; Balachandran, V.P.; Hellmann, M.D.; Solovyov, A.; Rizvi, N.A.; Merghoub, T.; Levine, A.J.; Chan, T.A.; et al. A neoantigen fitness model predicts tumour response to checkpoint blockade immunotherapy. Nature 2017, 551, 517–520. [Google Scholar] [CrossRef]
- Saez-Ibañez, A.R.; Upadhaya, S.; Partridge, T.; Shah, M.; Correa, D.; Campbell, J. Landscape of cancer cell therapies: Trends and real-world data. Nat. Rev. Drug Discov. 2022. [Google Scholar] [CrossRef]
- Bora, C.R.; Mahmood, A.; Rajasekar, S.; Andrali, S.S. Trends in Adoptive Tumor Infiltrating Lymphocytes (TILs) Therapy. BAOJ Cancer Res. Ther. 2015, 1, 1–6. [Google Scholar] [CrossRef]
- Kumar, A.; Watkins, R.; Vilgelm, A.E. Cell Therapy With TILs: Training and Taming T Cells to Fight Cancer. Front. Immunol. 2021, 12, 690499. [Google Scholar] [CrossRef]
- Yin, H.; Guo, W.; Sun, X.; Li, R.; Feng, C.; Tan, Y. TILs and Anti-PD1 Therapy: An Alternative Combination Therapy for PDL1 Negative Metastatic Cervical Cancer. J. Immunol. Res. 2020, 2020, 8345235. [Google Scholar] [CrossRef] [PubMed]
- Wu, R.; Forget, M.A.; Chacon, J.; Bernatchez, C.; Haymaker, C.; Chen, J.Q.; Hwu, P.; Radvanyi, L.G. Adoptive T-cell therapy using autologous tumor-infiltrating lymphocytes for metastatic melanoma: Current status and future outlook. Cancer J. 2012, 18, 160–175. [Google Scholar] [CrossRef] [PubMed]
- Merhavi-Shoham, E.; Itzhaki, O.; Markel, G.; Schachter, J.; Besser, M.J. Adoptive Cell Therapy for Metastatic Melanoma. Cancer J. 2017, 23, 48–53. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Sun, J.; Chen, K.; Ma, P.; Lei, Q.; Xing, S.; Cao, Z.; Sun, S.; Yu, Z.; Liu, Y.; et al. Perspectives of tumor-infiltrating lymphocyte treatment in solid tumors. BMC Med. 2021, 19, 140. [Google Scholar] [CrossRef]
- Stevanović, S.; Helman, S.R.; Wunderlich, J.R.; Langhan, M.M.; Doran, S.L.; Kwong, M.L.M.; Somerville, R.P.; Klebanoff, C.A.; Kammula, U.S.; Sherry, R.M.; et al. A Phase II Study of Tumor-infiltrating Lymphocyte Therapy for Human Papillomavirus–associated Epithelial Cancers. Clin. Cancer Res. 2019, 25, 1486–1493. [Google Scholar] [CrossRef] [PubMed]
- Creelan, B.C.; Wang, C.; Teer, J.K.; Toloza, E.M.; Yao, J.; Kim, S.; Landin, A.M.; Mullinax, J.E.; Saller, J.J.; Saltos, A.N.; et al. Tumor-infiltrating lymphocyte treatment for anti-PD-1-resistant metastatic lung cancer: A phase 1 trial. Nat. Med. 2021, 27, 1410–1418. [Google Scholar] [CrossRef]
- Wang, C.; Li, M.; Wei, R.; Wu, J. Adoptive transfer of TILs plus anti-PD1 therapy: An alternative combination therapy for treating metastatic osteosarcoma. J. Bone Oncol. 2020, 25, 100332. [Google Scholar] [CrossRef]
- Song, H.; Wang, H.; Gong, M.; Wu, L.; Liu, X.; Cao, W.; Gao, X.; Dou, R.; Chen, Q.; Hu, H. Augmentation of antitumor function of tumor-infiltrating lymphocytes against triple-negative breast cancer by PD-1 blockade. Cell Biol. Int. 2021, 46, 278–287. [Google Scholar] [CrossRef]
- Robbins, P.F.; Dudley, M.E.; Wunderlich, J.; El-Gamil, M.; Li, Y.F.; Zhou, J.; Huang, J.; Powell, D.J.; Rosenberg, S.A. Cutting Edge: Persistence of Transferred Lymphocyte Clonotypes Correlates with Cancer Regression in Patients Receiving Cell Transfer Therapy. J. Immunol. 2004, 173, 7125–7130. [Google Scholar] [CrossRef]
- Guo, J.; Luo, N.; Ai, G.; Yang, W.; Zhu, J.; Li, C.; Chen, R.; Zhang, C.; Liu, S.; Jin, H.; et al. Eradicating tumor in a recurrent cervical cancer patient with autologous tumor-infiltrating lymphocytes and a modified lymphodepleting regimen. J. Immunother. Cancer 2022, 10, e003887. [Google Scholar] [CrossRef]
- Heemskerk, B.; Liu, K.; Dudley, M.E.; Johnson, L.A.; Kaiser, A.; Downey, S.; Zheng, Z.; Shelton, T.E.; Matsuda, K.; Robbins, P.F.; et al. Adoptive Cell Therapy for Patients with Melanoma, Using Tumor-Infiltrating Lymphocytes Genetically Engineered to Secrete Interleukin-2. Hum. Gene Ther. 2008, 19, 496–510. [Google Scholar] [CrossRef]
- Ritthipichai, K.; Machin, M.; Juillerat, A.; Poirot, L.; Fardis, M.; Chartier, C. 1052P Genetic modification of Iovance’s TIL through TALEN-mediated knockout of PD-1 as a strategy to empower TIL therapy for cancer. Ann. Oncol. 2020, 31, S720. [Google Scholar] [CrossRef]
- Palmer, D.C.; Webber, B.R.; Patel, Y.; Johnson, M.J.; Kariya, C.M.; Lahr, W.S.; Parkhurst, M.R.; Gartner, J.J.; Prickett, T.D.; Lowery, F.J.; et al. Internal Checkpoint Regulates T Cell Neoantigen Reactivity and Susceptibility to PD1 Blockade. SSRN Electron. J. 2020. [Google Scholar] [CrossRef]
- Chamberlain, C.A.; Bennett, E.P.; Kverneland, A.H.; Svane, I.M.; Donia, M.; Met, Ö. Highly efficient PD-1-targeted CRISPR-Cas9 for tumor-infiltrating lymphocyte-based adoptive T cell therapy. Mol. Ther. Oncolytics 2022, 24, 417–428. [Google Scholar] [CrossRef]
- Ou, X.; Ma, Q.; Yin, W.; Ma, X.; He, Z. CRISPR/Cas9 Gene-Editing in Cancer Immunotherapy: Promoting the Present Revolution in Cancer Therapy and Exploring More. Front. Cell Dev. Biol. 2021, 9, 674467. [Google Scholar] [CrossRef]
- Shifrut, E.; Carnevale, J.; Tobin, V.; Roth, T.L.; Woo, J.M.; Bui, C.T.; Li, P.J.; Diolaiti, M.E.; Ashworth, A.; Marson, A. Genome-wide CRISPR Screens in Primary Human T Cells Reveal Key Regulators of Immune Function. Cell 2018, 175, 1958–1971.e15. [Google Scholar] [CrossRef]
- Yin, T. Improving T cell therapy: In vivo CRISPR-Cas9 screens tell us how to do. Precis. Clin. Med. 2021, 4, 176–178. [Google Scholar] [CrossRef]
- Bradley, C.A. In vivo T cell CRISPR screens reveal immunotherapeutic targets. Nat. Cancer 2019, 19, 606. [Google Scholar] [CrossRef]
- Tsimberidou, A.M.; van Morris, K.; Vo, H.H.; Eck, S.; Lin, Y.F.; Rivas, J.M.; Andersson, B.S. T-cell receptor-based therapy: An innovative therapeutic approach for solid tumors. J. Hematol. Oncol. 2021, 14, 102. [Google Scholar] [CrossRef]
- Gaissmaier, L.; Elshiaty, M.; Christopoulos, P. Breaking Bottlenecks for the TCR Therapy of Cancer. Cells 2020, 9, 2095. [Google Scholar] [CrossRef] [PubMed]
- Shafer, P.; Kelly, L.M.; Hoyos, V. Cancer Therapy With TCR-Engineered T Cells: Current Strategies, Challenges, and Prospects. Front. Immunol. 2022, 13, 835762. [Google Scholar] [CrossRef] [PubMed]
- Scanlan, M.J.; Gure, A.O.; Jungbluth, A.A.; Old, L.J.; Chen, Y.-T. Cancer/testis antigens: An expanding family of targets for cancer immunotherapy. Immunol. Rev. 2002, 188, 22–32. [Google Scholar] [CrossRef] [PubMed]
- Häffner, A.C.; Tassis, A.; Zepter, K.; Storz, M.; Tureci, O.; Burg, G.; Nestle, F.O. Expression of cancer/testis antigens in cutaneous T cell lymphomas. Int. J. Cancer 2001, 97, 668–670. [Google Scholar] [CrossRef] [PubMed]
- Rapoport, A.P.; Stadtmauer, E.A.; Binder-Scholl, G.K.; Goloubeva, O.; Vogl, D.T.; Lacey, S.F.; Badros, A.Z.; Garfall, A.; Weiss, B.M.; Finklestein, J.; et al. NY-ESO-1–specific TCR–engineered T cells mediate sustained antigen-specific antitumor effects in myeloma. Nat. Med. 2015, 21, 914–921. [Google Scholar] [CrossRef]
- Morgan, R.A.; Dudley, M.E.; Wunderlich, J.R.; Hughes, M.S.; Yang, J.C.; Sherry, R.M.; Royal, R.E.; Topalian, S.L.; Kammula, U.S.; Restifo, N.P.; et al. Cancer Regression in Patients After Transfer of Genetically Engineered Lymphocytes. Science 2006, 314, 126–129. [Google Scholar] [CrossRef] [Green Version]
- Parkhurst, M.R.; Yang, J.C.; Langan, R.C.; Dudley, M.E.; Nathan, D.-A.N.; A. Feldman, S.; Davis, J.L.; A. Morgan, R.; Merino, M.J.; Sherry, R.M.; et al. T Cells Targeting Carcinoembryonic Antigen Can Mediate Regression of Metastatic Colorectal Cancer but Induce Severe Transient Colitis. Mol. Ther. 2011, 19, 620–626. [Google Scholar] [CrossRef]
- Lu, Y.-C.; Parker, L.L.; Lu, T.; Zheng, Z.; Yao, X.; Robbins, P.F.; Van Der Bruggen, P.; Klebanoff, C.A.; Hinrichs, C.S.; Goff, S.L.; et al. A Phase I study of an HLA-DPB1*0401-restricted T cell receptor targeting MAGE-A3 for patients with metastatic cancers. J. Immunother. Cancer 2015, 3, P158. [Google Scholar] [CrossRef]
- Johnson, L.A.; Morgan, R.A.; Dudley, M.E.; Cassard, L.; Yang, J.C.; Hughes, M.S.; Kammula, U.S.; Royal, R.E.; Sherry, R.M.; Wunderlich, J.R.; et al. Gene therapy with human and mouse T-cell receptors mediates cancer regression and targets normal tissues expressing cognate antigen. Blood 2009, 114, 535–546. [Google Scholar] [CrossRef]
- Mullard, A. FDA approval of Immunocore’s first-in-class TCR therapeutic broadens depth of the T cell engager platform. Nat. Rev. Drug Discov. 2022, 21, 170. [Google Scholar] [CrossRef]
- Nathan, P.; Hassel, J.C.; Rutkowski, P.; Baurain, J.-F.; Butler, M.O.; Schlaak, M.; Sullivan, R.J.; Ochsenreither, S.; Dummer, R.; Kirkwood, J.M.; et al. Overall Survival Benefit with Tebentafusp in Metastatic Uveal Melanoma. N. Engl. J. Med. 2021, 385, 1196–1206. [Google Scholar] [CrossRef]
- Ping, Y.; Liu, C.; Zhang, Y. T-cell receptor-engineered T cells for cancer treatment: Current status and future directions. Protein Cell 2018, 9, 254–266. [Google Scholar] [CrossRef]
- Abramson, J.S. Anti-CD19 CAR T-Cell Therapy for B-Cell Non-Hodgkin Lymphoma. Transfus. Med. Rev. 2020, 34, 29–33. [Google Scholar] [CrossRef]
- Shah, N.N.; Maatman, T.; Hari, P.; Johnson, B. Multi targeted CAR-T cell therapies for B-cell malignancies. Front. Oncol. 2019, 9, 146. [Google Scholar] [CrossRef]
- Frigault, M.J.; Maus, M.V. State of the art in CAR T cell therapy for CD19+ B cell malignancies. J. Clin. Investig. 2020, 130, 1586–1594. [Google Scholar] [CrossRef]
- Albinger, N.; Hartmann, J.; Ullrich, E. Current status and perspective of CAR-T and CAR-NK cell therapy trials in Germany. Gene Ther. 2021, 28, 513–527. [Google Scholar] [CrossRef]
- Fesnak, A.; Lin, C.; Siegel, D.L.; Maus, M.V. CAR-T Cell Therapies From the Transfusion Medicine Perspective. Transfus. Med. Rev. 2016, 30, 139–145. [Google Scholar] [CrossRef]
- D’Ippolito, E.; Schober, K.; Nauerth, M.; Busch, D.H. T cell engineering for adoptive T cell therapy: Safety and receptor avidity. Cancer Immunol. Immunother. 2019, 68, 1701–1712. [Google Scholar] [CrossRef]
- Zhang, H.; Zhao, P.; Huang, H. Engineering better chimeric antigen receptor T cells. Exp. Hematol. Oncol. 2020, 9, 34. [Google Scholar] [CrossRef]
- Gross, G.; Gorochov, G.; Waks, T.; Eshhar, Z. Generation of effector T cells expressing chimeric T cell receptor with antibody type-specificity. Transplant. Proc. 1989, 21, 127–130. [Google Scholar]
- Gross, G.; Waks, T.; Eshhar, Z. Expression of immunoglobulin-T-cell receptor chimeric molecules as functional receptors with antibody-type specificity. Proc. Natl. Acad. Sci. USA 1989, 86, 10024–10028. [Google Scholar] [CrossRef]
- Chimeric Antigen Receptor–Modified T Cells in Chronic Lymphoid Leukemia; Chimeric Antigen Receptor–Modified T Cells for Acute Lymphoid Leukemia; Chimeric Antigen Receptor T Cells for Sustained Remissions in Leukemia. N. Engl. J. Med. 2016, 374, 998. [CrossRef] [PubMed]
- Porter, D.L.; Levine, B.L.; Kalos, M.; Bagg, A.; June, C.H. Chimeric Antigen Receptor–Modified T Cells in Chronic Lymphoid Leukemia. N. Engl. J. Med. 2011, 365, 725–733. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, K.; Kitaura, M.; Tsunei, A.; Kusabuka, H.; Ogaki, E.; Okada, N. Structure of the Signal Transduction Domain in Second-Generation CAR Regulates the Input Efficiency of CAR Signals. Int. J. Mol. Sci. 2021, 22, 2476. [Google Scholar] [CrossRef] [PubMed]
- Ramos, C.A.; Rouce, R.; Robertson, C.S.; Reyna, A.; Narala, N.; Vyas, G.; Mehta, B.; Zhang, H.; Dakhova, O.; Carrum, G.; et al. In Vivo Fate and Activity of Second- versus Third-Generation CD19-Specific CAR-T Cells in B Cell Non-Hodgkin’s Lymphomas. Mol. Ther. 2018, 26, 2727–2737. [Google Scholar] [CrossRef]
- Ruella, M.; Kenderian, S.S. Next-Generation Chimeric Antigen Receptor T-Cell Therapy: Going off the Shelf. BioDrugs 2017, 31, 473–481. [Google Scholar] [CrossRef]
- Castellarin, M.; Watanabe, K.; June, C.H.; Kloss, C.C.; Posey, A.D., Jr. Driving cars to the clinic for solid tumors. Gene Ther. 2018, 25, 165–175. [Google Scholar] [CrossRef] [Green Version]
- Skorka, K.; Ostapinska, K.; Malesa, A.; Giannopoulos, K. The Application of CAR-T Cells in Haematological Malignancies. Arch. Immunol. Ther. Exp. 2020, 68, 34. [Google Scholar] [CrossRef]
- Subklewe, M.; von Bergwelt-Baildon, M.; Humpe, A. Chimeric Antigen Receptor T Cells: A Race to Revolutionize Cancer Therapy. Transfus. Med. Hemother. 2019, 46, 15–24. [Google Scholar] [CrossRef]
- Scarfò, I.; Maus, M.V. Current approaches to increase CAR T cell potency in solid tumors: Targeting the tumor microenvironment. J. Immunother. Cancer 2017, 5, 28. [Google Scholar] [CrossRef]
- Sachdeva, M.; Busser, B.W.; Temburni, S.; Jahangiri, B.; Gautron, A.-S.; Maréchal, A.; Juillerat, A.; Williams, A.; Depil, S.; Duchateau, P.; et al. Repurposing endogenous immune pathways to tailor and control chimeric antigen receptor T cell functionality. Nat. Commun. 2019, 10, 5100. [Google Scholar] [CrossRef]
- FDA Approval Letter 30 August 2017, KYMRIAH. Available online: https://www.fda.gov/media/106989/ (accessed on 1 July 2022).
- FDA Approval Letter 27 May 2022, KYMRIAH. Available online: https://www.fda.gov/media/158835/ (accessed on 1 July 2022).
- Xie, G.; Dong, H.; Liang, Y.; Ham, J.D.; Rizwan, R.; Chen, J. CAR-NK cells: A promising cellular immunotherapy for cancer. EBioMedicine 2020, 59, 102975. [Google Scholar] [CrossRef]
- Gong, Y.; Klein Wolterink, R.G.J.; Wang, J.; Bos, G.M.J.; Germeraad, W.T.V. Chimeric antigen receptor natural killer (CAR-NK) cell design and engineering for cancer therapy. J. Hematol. Oncol. 2021, 14, 73. [Google Scholar] [CrossRef]
- Fowler, M. Celularity receives fast track designation from U.S. FDA for its NK cell therapy CYNK-001 in development for the treatment of AML. News Release 2022. [Google Scholar]
- Marofi, F.; Abdul-Rasheed, O.F.; Rahman, H.S.; Budi, H.S.; Jalil, A.T.; Yumashev, A.V.; Hassanzadeh, A.; Yazdanifar, M.; Motavalli, R.; Chartrand, M.S.; et al. CAR-NK cell in cancer immunotherapy; A promising frontier. Cancer Sci. 2021, 112, 3427–3436. [Google Scholar] [CrossRef]
- Wrona, E.; Borowiec, M.; Potemski, P. CAR-NK Cells in the Treatment of Solid Tumors. Int. J. Mol. Sci. 2021, 22, 5899. [Google Scholar] [CrossRef]
- Gurney, M.; O’dwyer, M. Realizing innate potential: Car-nk cell therapies for acute myeloid leukemia. Cancers 2021, 13, 1568. [Google Scholar] [CrossRef]
- Demaria, O.; Gauthier, L.; Debroas, G.; Vivier, E. Natural killer cell engagers in cancer immunotherapy: Next generation of immuno-oncology treatments. Eur. J. Immunol. 2021, 51, 1934–1942. [Google Scholar] [CrossRef]
- Schmohl, J.U.; Felices, M.; Oh, F.; Lenvik, A.J.; Lebeau, A.M.; Panyam, J.; Miller, J.S.; Vallera, D.A. Engineering of Anti-CD133 Tri-Specific Molecule Capable of Inducing NK Expansion and Driving Antibody-Dependent Cell-Mediated Cytotoxicity. Cancer Res. Treat. 2017, 49, 1140–1152. [Google Scholar] [CrossRef]
- Vallera, D.A.; Oh, F.; Kodal, B.; Hinderlie, P.; Geller, M.A.; Miller, J.S.; Felices, M. A HER2 Tri-Specific NK Cell Engager Mediates Efficient Targeting of Human Ovarian Cancer. Cancers 2021, 13, 3994. [Google Scholar] [CrossRef]
- Gauthier, L.; Morel, A.; Anceriz, N.; Rossi, B.; Blanchard-Alvarez, A.; Grondin, G.; Trichard, S.; Cesari, C.; Sapet, M.; Bosco, F.; et al. Multifunctional Natural Killer Cell Engagers Targeting NKp46 Trigger Protective Tumor Immunity. Cell 2019, 177, 1701–1713.e16. [Google Scholar] [CrossRef]
- Marofi, F.; Saleh, M.M.; Rahman, H.S.; Suksatan, W.; Al-Gazally, M.E.; Abdelbasset, W.K.; Thangavelu, L.; Yumashev, A.V.; Hassanzadeh, A.; Yazdanifar, M.; et al. CAR-engineered NK cells; a promising therapeutic option for treatment of hematological malignancies. Stem Cell Res. Ther. 2021, 12, 374. [Google Scholar] [CrossRef]
- Sterner, R.C.; Sterner, R.M. CAR-T cell therapy: Current limitations and potential strategies. Blood Cancer J. 2021, 11, 69. [Google Scholar] [CrossRef] [PubMed]
- Majzner, R.G.; Mackall, C.L. Tumor antigen escape from car t-cell therapy. Cancer Discov. 2018, 8, 1219–1226. [Google Scholar] [CrossRef] [PubMed]
- Maude, S.L.; Teachey, D.; Porter, D.L.; Grupp, S.A. CD19-targeted chimeric antigen receptor T-cell therapy for acute lymphoblastic leukemia. Blood 2015, 125, 4017–4023. [Google Scholar] [CrossRef] [PubMed]
- Brudno, J.N.; Maric, I.; Hartman, S.D.; Rose, J.J.; Wang, M.; Lam, N.; Stetler-Stevenson, M.; Salem, D.; Yuan, C.; Pavletic, S.; et al. T Cells Genetically Modified to Express an Anti–B-Cell Maturation Antigen Chimeric Antigen Receptor Cause Remissions of Poor-Prognosis Relapsed Multiple Myeloma. J. Clin. Oncol. 2018, 36, 2267–2280. [Google Scholar] [CrossRef]
- Green, D.J.; Pont, M.; Sather, B.D.; Cowan, A.J.; Turtle, C.J.; Till, B.G.; Nagengast, A.M.; Libby, E.N., III; Becker, P.S.; Coffey, D.G.; et al. Fully Human Bcma Targeted Chimeric Antigen Receptor T Cells Administered in a Defined Composition Demonstrate Potency at Low Doses in Advanced Stage High Risk Multiple Myeloma. Blood 2018, 132, 1011. [Google Scholar] [CrossRef]
- Majzner, R.G.; Theruvath, J.L.; Nellan, A.; Heitzeneder, S.; Cui, Y.; Mount, C.W.; Rietberg, S.P.; Linde, M.H.; Xu, P.; Rota, C.; et al. CAR T Cells Targeting B7-H3, a Pan-Cancer Antigen, Demonstrate Potent Preclinical Activity Against Pediatric Solid Tumors and Brain Tumors. Clin. Cancer Res. 2019, 25, 2560–2574. [Google Scholar] [CrossRef]
- Koneru, M.; O’Cearbhaill, R.; Pendharkar, S.; Spriggs, D.R.; Brentjens, R.J. A phase I clinical trial of adoptive T cell therapy using IL-12 secreting MUC-16ecto directed chimeric antigen receptors for recurrent ovarian cancer. J. Transl. Med. 2015, 13, 102. [Google Scholar] [CrossRef]
- Steentoft, C.; Migliorini, D.; King, T.R.; Mandel, U.; June, C.H.; Posey, A.D., Jr. Glycan-directed CAR-T cells. Glycobiology 2018, 28, 656–669. [Google Scholar] [CrossRef]
- Whilding, L.; Halim, L.; Draper, B.; Parente-Pereira, A.; Zabinski, T.; Davies, D.; Maher, J. CAR T-Cells Targeting the Integrin αvβ6 and Co-Expressing the Chemokine Receptor CXCR2 Demonstrate Enhanced Homing and Efficacy against Several Solid Malignancies. Cancers 2019, 11, 674. [Google Scholar] [CrossRef]
- Yin, Y.; Boesteanu, A.C.; Binder, Z.A.; Xu, C.; Reid, R.A.; Rodriguez, J.L.; Cook, D.R.; Thokala, R.; Blouch, K.; McGettigan-Croce, B.; et al. Checkpoint Blockade Reverses Anergy in IL-13Rα2 Humanized scFv-Based CAR T Cells to Treat Murine and Canine Gliomas. Mol. Ther. Oncolytics 2018, 11, 20–38. [Google Scholar] [CrossRef] [PubMed]
- Chong, E.A.; Melenhorst, J.J.; Lacey, S.F.; Ambrose, D.E.; Gonzalez, V.; Levine, B.L.; June, C.H.; Schuster, S.J. PD-1 blockade modulates chimeric antigen receptor (CAR)-modified T cells: Refueling the CAR. Blood 2017, 129, 1039–1041. [Google Scholar] [CrossRef]
- Milone, M.C.; Bhoj, V.G. The Pharmacology of T Cell Therapies. Mol. Ther. Methods Clin. Dev. 2018, 8, 210–221. [Google Scholar] [CrossRef]
- Mehrabadi, A.Z.; Ranjbar, R.; Farzanehpour, M.; Shahriary, A.; Dorostkar, R.; Hamidinejad, M.A.; Ghaleh, H.E.G. Therapeutic potential of CAR T cell in malignancies: A scoping review. Biomed. Pharmacother. 2022, 146, 112512. [Google Scholar] [CrossRef] [PubMed]
- Milone, M.C.; Xu, J.; Chen, S.J.; Collins, M.A.; Zhou, J.; Powell, D.J., Jr.; Melenhorst, J.J. Engineering-enhanced CAR T cells for improved cancer therapy. Nat. Cancer 2021, 2, 780–793. [Google Scholar] [CrossRef]
- Zhang, H.; Gao, L.; Liu, L.; Wang, J.; Wang, S.; Zhang, C.; Liu, Y.; Kong, P.; Liu, J.; He, J.; et al. A Bcma and CD19 Bispecific CAR-T for Relapsed and Refractory Multiple Myeloma. Blood 2019, 134, 3147. [Google Scholar] [CrossRef]
- Kimiz-Gebologlu, I.; Gulce-Iz, S.; Biray-Avci, C. Monoclonal antibodies in cancer immunotherapy. Mol. Biol. Rep. 2018, 45, 2935–2940. [Google Scholar] [CrossRef]
- Simpson, A.; Caballero, O. Monoclonal antibodies for the therapy of cancer. BMC Proc. 2014, 8, O6. [Google Scholar] [CrossRef]
- Zahavi, D.; AlDeghaither, D.; O’Connell, A.; Weiner, L.M. Enhancing antibody-dependent cell-mediated cytotoxicity: A strategy for improving antibody-based immunotherapy. Antib. Ther. 2018, 1, 7–12. [Google Scholar] [CrossRef]
- Bayer, V. An Overview of Monoclonal Antibodies. Semin. Oncol. Nurs. 2019, 35, 150927. [Google Scholar] [CrossRef]
- Kaplon, H.; Reichert, J.M. Antibodies to watch in 2021. mAbs 2021, 13, 1860476. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.; Sun, Y.; Liang, X.; Gu, X.; Ning, J.; Xu, Y.; Chen, S.; Pan, L. Emerging new therapeutic antibody derivatives for cancer treatment. Signal Transduct. Target. Ther. 2022, 7, 39. [Google Scholar] [CrossRef]
- Taylor, R.P.; Lindorfer, M.A. Cytotoxic mechanisms of immunotherapy: Harnessing complement in the action of anti-tumor monoclonal antibodies. Semin Immunol. 2016, 28, 309–316. [Google Scholar] [CrossRef]
- Golay, J.; Taylor, R. The Role of Complement in the Mechanism of Action of Therapeutic Anti-Cancer mAbs. Antibodies 2020, 9, 58. [Google Scholar] [CrossRef]
- Trivedi, S.; Srivastava, R.M.; Concha-Benavente, F.; Ferrone, S.; Garcia-Bates, T.M.; Li, J.; Ferris, R.L. Anti-EGFR Targeted Monoclonal Antibody Isotype Influences Antitumor Cellular Immunity in Head and Neck Cancer Patients. Clin. Cancer Res. 2016, 22, 5229–5237. [Google Scholar] [CrossRef] [PubMed]
- Boross, P.; Leusen, J.H.W. Mechanisms of action of CD20 antibodies. Am. J. Cancer Res. 2012, 2, 676–690. [Google Scholar]
- Tarantino, P.; Uliano, J.; Morganti, S.; Giugliano, F.; Crimini, E.; Curigliano, G. Clinical development and current role of margetuximab for the treatment of breast cancer. Drugs Today 2021, 57, 551. [Google Scholar] [CrossRef]
- Rajabi, M.; Mousa, S.A. The role of angiogenesis in cancer treatment. Biomedicines 2017, 5, 34. [Google Scholar] [CrossRef] [Green Version]
- Yetkin-Arik, B.; Kastelein, A.W.; Klaassen, I.; Jansen, C.H.J.R.; Latul, Y.P.; Vittori, M.; Biri, A.; Kahraman, K.; Griffioen, A.W.; Amant, F.; et al. Angiogenesis in gynecological cancers and the options for anti-angiogenesis therapy. Biochim. Biophys. Acta Rev. Cancer 2021, 1875, 188446. [Google Scholar] [CrossRef]
- Carmeliet, P. VEGF as a key mediator of angiogenesis in cancer. Oncology 2005, 69, 4–10. [Google Scholar] [CrossRef]
- Salvatore, L.; Bria, E.; Sperduti, I.; Hinke, A.; Hegewisch-Becker, S.; Aparicio, T.; le Malicot, K.; Boige, V.; Koeberle, D.; Baertschi, D.; et al. Bevacizumab as maintenance therapy in patients with metastatic colorectal cancer: A meta-analysis of individual patients’ data from 3 phase III studies. Cancer Treat. Rev. 2021, 97, 102202. [Google Scholar] [CrossRef] [PubMed]
- Oza, A.M.; Dubois, F.; Hegg, R.; Hernández, C.A.; Finocchiaro, G.; Ghiringhelli, F.; Zamagni, C.; Nick, S.; Irahara, N.; Perretti, T.; et al. A Long-Term Extension Study of Bevacizumab in Patients with Solid Tumors. Oncologist 2021, 26, e2254–e2264. [Google Scholar] [CrossRef]
- Reardon, D.A.; Brandes, A.A.; Omuro, A.; Mulholland, P.; Lim, M.; Wick, A.; Baehring, J.; Ahluwalia, M.S.; Roth, P.; Bähr, O.; et al. Effect of Nivolumab vs Bevacizumab in Patients with Recurrent Glioblastoma: The CheckMate 143 Phase 3 Randomized Clinical Trial. JAMA Oncol. 2020, 6, 1003–1010. [Google Scholar] [CrossRef] [PubMed]
- Arora, S.; Balasubramaniam, S.; Zhang, H.; Berman, T.; Narayan, P.; Suzman, D.; Bloomquist, E.; Tang, S.; Gong, Y.; Sridhara, R.; et al. FDA Approval Summary: Olaparib Monotherapy or in Combination with Bevacizumab for the Maintenance Treatment of Patients with Advanced Ovarian Cancer. Oncologist 2020, 26, e164–e172. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.S.; Qin, S.; Ikeda, M.; Galle, P.R.; Ducreux, M.; Kim, T.-Y.; Kudo, M.; Breder, V.; Merle, P.; Kaseb, A.O.; et al. Atezolizumab plus Bevacizumab in Unresectable Hepatocellular Carcinoma. N. Engl. J. Med. 2020, 382, 1894–1905. [Google Scholar] [CrossRef] [PubMed]
- Garcia, J.; Hurwitz, H.I.; Sandler, A.B.; Miles, D.; Coleman, R.L.; Deurloo, R.; Chinot, O.L. Bevacizumab (Avastin®) in cancer treatment: A review of 15 years of clinical experience and future outlook. Cancer Treat. Rev. 2020, 86, 102017. [Google Scholar] [CrossRef] [PubMed]
- De Luca, E.; Marino, D.; di Maio, M. Ramucirumab, a second-line option for patients with hepatocellular carcinoma: A review of the evidence. Cancer Manag. Res. 2020, 12, 3721–3729. [Google Scholar] [CrossRef]
- De Vita, F.; Borg, C.; Farina, G.; Geva, R.; Carton, I.; Cuku, H.; Wei, R.; Muro, K. Ramucirumab and paclitaxel in patients with gastric cancer and prior trastuzumab: Subgroup analysis from RAINBOW study. Futur. Oncol. 2019, 15, 2723–2731. [Google Scholar] [CrossRef]
- Uprety, D. Clinical utility of ramucirumab in non-small-cell lung cancer. Biologics 2019, 13, 133–137. [Google Scholar] [CrossRef]
- Bornstein, G.G. Antibody Drug Conjugates: Preclinical Considerations. AAPS J. 2015, 17, 525–534. [Google Scholar] [CrossRef]
- Lucas, A.T.; Moody, A.; Schorzman, A.N.; Zamboni, W.C. Importance and considerations of antibody engineering in antibody-drug conjugates development from a clinical pharmacologist’s perspective. Antibodies 2021, 10, 30. [Google Scholar] [CrossRef]
- Esnault, C.; Schrama, D.; Houben, R.; Guyétant, S.; Desgranges, A.; Martin, C.; Berthon, P.; Viaud-Massuard, M.-C.; Touzé, A.; Kervarrec, T.; et al. Antibody–Drug Conjugates as an Emerging Therapy in Oncodermatology. Cancers 2022, 14, 778. [Google Scholar] [CrossRef]
- Hasan, M.M.; Laws, M.; Jin, P.; Rahman, K.M. Factors influencing the choice of monoclonal antibodies for antibody–drug conjugates. Drug Discov. Today 2022, 27, 354–361. [Google Scholar] [CrossRef]
- Jin, Y.; Schladetsch, M.A.; Huang, X.; Balunas, M.J.; Wiemer, A.J. Stepping forward in antibody-drug conjugate development. Pharmacol. Ther. 2022, 229, 107917. [Google Scholar] [CrossRef]
- Sheyi, R.; de la Torre, B.G.; Albericio, F. Linkers: An Assurance for Controlled Delivery of Antibody-Drug Conjugate. Pharmaceutics 2022, 14, 396. [Google Scholar] [CrossRef]
- Grzywa, A. Antibody-drug conjugates for cancer therapy. Farm. Polska 2021, 77, 581–587. [Google Scholar] [CrossRef]
- Su, Z.; Xiao, D.; Xie, F.; Liu, L.; Wang, Y.; Fan, S.; Zhou, X.; Li, S. Antibody–drug conjugates: Recent advances in linker chemistry. Acta Pharm. Sin. B 2021, 11, 3889–3907. [Google Scholar] [CrossRef]
- Khongorzul, P.; Ling, C.J.; Khan, F.U.; Ihsan, A.U.; Zhang, J. Antibody-drug conjugates: A comprehensive review. Mol. Cancer Res. 2020, 18, 3–19. [Google Scholar] [CrossRef] [Green Version]
- Baah, S.; Laws, M.; Rahman, K.M. Antibody–drug conjugates—A tutorial review. Molecules 2021, 26, 2943. [Google Scholar] [CrossRef]
- Mckertish, C.; Kayser, V. Advances and Limitations of Antibody Drug Conjugates for Cancer. Biomedicines 2021, 9, 872. [Google Scholar] [CrossRef]
- Gandullo-sánchez, L.; Ocaña, A.; Pandiella, A. Generation of antibody-drug conjugate resistant models. Cancers 2021, 13, 4631. [Google Scholar] [CrossRef] [PubMed]
- Fu, Z.; Li, S.; Han, S.; Shi, C.; Zhang, Y. Antibody drug conjugate: The “biological missile” for targeted cancer therapy. Signal Transduct. Target. Ther. 2022, 7, 93. [Google Scholar] [CrossRef] [PubMed]
- Ali, S.; Dunmore, H.-M.; Karres, D.; Hay, J.L.; Salmonsson, T.; Gisselbrecht, C.; Sarac, S.B.; Bjerrum, O.W.; Hovgaard, D.; Barbachano, Y.; et al. The EMA Review of Mylotarg (Gemtuzumab Ozogamicin) for the Treatment of Acute Myeloid Leukemia. Oncologist 2019, 24, e171–e179. [Google Scholar] [CrossRef] [PubMed]
- Barbet, J.; Bardiès, M.; Bourgeois, M.; Chatal, J.; Chérel, M.; Davodeau, F.; Faivre-Chauvet, A.; Gestin, J.; Kraeber-Bodéré, F. Radiolabeled antibodies for cancer imaging and therapy. Methods Mol. Biol. 2012, 907, 681–697. [Google Scholar]
- Parakh, S.; Lee, S.T.; Gan, H.K.; Scott, A.M. Radiolabeled Antibodies for Cancer Imaging and Therapy. Cancers 2022, 14, 1454. [Google Scholar] [CrossRef]
- D’Huyvetter, M.; Xavier, C.; Caveliers, V.; Lahoutte, T.; Muyldermans, S.; Devoogdt, N. Radiolabeled nanobodies as theranostic tools in targeted radionuclide therapy of cancer. Expert Opin. Drug Deliv. 2014, 11, 1939–1954. [Google Scholar] [CrossRef]
- Delaloye, A.B. The role of nuclear medicine in the treatment of non-Hodgkin’s lymphoma (NHL). Leuk. Lymphoma 2003, 44, S29–S36. [Google Scholar] [CrossRef]
- Etrych, T.; Braunova, A.; Zogala, D.; Lambert, L.; Renesova, N.; Klener, P. Targeted Drug Delivery and Theranostic Strategies in Malignant Lymphomas. Cancers 2022, 14, 626. [Google Scholar] [CrossRef]
- Liu, W.; Li, K.; Deng, H.; Wang, J.; Zhao, P.; Liao, W.; Zhuo, L.; Wei, H.; Yang, X.; Chen, Y. In vitro and in vivo evaluation of a novel anti-EGFR antibody labeled with 89Zr and 177Lu. J. Radioanal. Nucl. Chem. Artic. 2022, 331, 747–754. [Google Scholar] [CrossRef]
- Chen, S.; Li, J.; Li, Q.; Wang, Z. Bispecific antibodies in cancer immunotherapy. Hum. Vaccines Immunother. 2016, 12, 3–17. [Google Scholar] [CrossRef]
- Suurs, F.V.; Lub-de Hooge, M.N.; de Vries, E.G.E.; de Groot, D.J.A. A review of bispecific antibodies and antibody constructs in oncology and clinical challenges. Pharmacol. Ther. 2019, 201, 103–119. [Google Scholar] [CrossRef]
- Fan, G.; Wang, Z.; Hao, M.; Li, J. Bispecific antibodies and their applications. J. Hematol. Oncol. 2015, 8, 130. [Google Scholar] [CrossRef]
- Shim, H. Bispecific antibodies and antibody–drug conjugates for cancer therapy: Technological considerations. Biomolecules 2020, 10, 360. [Google Scholar] [CrossRef]
- Schuster, S.J. Bispecific antibodies for the treatment of lymphomas: Promises and challenges. Hematol. Oncol. 2021, 39, 113–116. [Google Scholar] [CrossRef]
- Dahlén, E.; Veitonmäki, N.; Norlén, P. Bispecific antibodies in cancer immunotherapy. Ther. Adv. Vaccines Immunother. 2018, 6, 3–17. [Google Scholar] [CrossRef]
- Spiess, C.; Zhai, Q.; Carter, P.J. Alternative molecular formats and therapeutic applications for bispecific antibodies. Mol. Immunol. 2015, 67, 95–106. [Google Scholar] [CrossRef]
- Brinkmann, U.; Kontermann, R.E. The making of bispecific antibodies. MAbs 2017, 9, 182–212. [Google Scholar] [CrossRef]
- Chulpanova, D.S.; Kitaeva, K.V.; Green, A.R.; Rizvanov, A.A.; Solovyeva, V.V. Molecular Aspects and Future Perspectives of Cytokine-Based Anti-cancer Immunotherapy. Front. Cell Dev. Biol. 2022, 8, 402. [Google Scholar] [CrossRef]
- Oppenheim, J.; Fujiwara, H. The role of cytokines in cancer. Cytokine Growth Factor Rev. 1996, 7, 279–288. [Google Scholar] [CrossRef]
- Qiu, Y.; Su, M.; Liu, L.; Tang, Y.; Pan, Y.; Sun, J. Clinical application of cytokines in cancer immunotherapy. Drug Des. Devel. Ther. 2021, 15, 2269–2287. [Google Scholar] [CrossRef]
- Rosenberg, S.A. IL-2: The First Effective Immunotherapy for Human Cancer. J. Immunol. 2014, 192, 5451–5458. [Google Scholar] [CrossRef]
- Jiang, T.; Zhou, C.; Ren, S. Role of IL-2 in cancer immunotherapy. Oncoimmunology 2016, 5, e1163462. [Google Scholar] [CrossRef]
- Di Trolio, R.; Simeone, E.; di Lorenzo, G.; Grimaldi, A.M.; Romano, A.; Ayala, F.; Caracò, C.; Mozzillo, N.; Ascierto, P.A. Update on PEG-interferon α-2b as adjuvant therapy in melanoma. Anticancer. Res. 2012, 32, 3901–3909. [Google Scholar]
- Lee, S.; Margolin, K. Cytokines in cancer immunotherapy. Cancers 2011, 3, 3856–3893. [Google Scholar] [CrossRef]
- Rallis, K.S.; Corrigan, A.E.; Dadah, H.; George, A.M.; Keshwara, S.M.; Sideris, M.; Szabados, B. Cytokine-based cancer immunotherapy: Challenges and opportunities for IL-10. Anticancer. Res. 2021, 41, 3247–3252. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, K.G.; Vrabel, M.R.; Mantooth, S.M.; Hopkins, J.J.; Wagner, E.S.; Gabaldon, T.A.; Zaharoff, D.A. Localized Interleukin-12 for Cancer Immunotherapy. Front. Immunol. 2020, 11, 575597. [Google Scholar] [CrossRef]
- Berraondo, P.; Sanmamed, M.F.; Ochoa, M.C.; Etxeberria, I.; Aznar, M.A.; Pérez-Gracia, J.L.; Rodríguez-Ruiz, M.E.; Ponz-Sarvise, M.; Melero, E.C.I. Cytokines in clinical cancer immunotherapy. Br. J. Cancer 2019, 120, 6–15. [Google Scholar] [CrossRef]
- Mao, C.; Ding, Y.; Xu, N. A Double-Edged Sword Role of Cytokines in Prostate Cancer Immunotherapy. Front. Oncol. 2021, 11, 688489. [Google Scholar] [CrossRef] [PubMed]
- Zheng, M.; Huang, J.; Tong, A.; Yang, H. Oncolytic Viruses for Cancer Therapy: Barriers and Recent Advances. Mol. Ther. Oncolytics 2019, 15, 234–247. [Google Scholar] [CrossRef] [PubMed]
- Kaufman, H.L.; Kohlhapp, F.J.; Zloza, A. Oncolytic viruses: A new class of immunotherapy drugs. Nat. Rev. Drug Discov. 2015, 14, 642–662. [Google Scholar] [CrossRef] [PubMed]
- Russell, S.J.; Barber, G.N. Oncolytic Viruses as Antigen-Agnostic Cancer Vaccines. Cancer Cell 2018, 33, 599–605. [Google Scholar] [CrossRef] [PubMed]
- Alberts, P.; Tilgase, A.; Rasa, A.; Bandere, K.; Venskus, D. The advent of oncolytic virotherapy in oncology: The Rigvir® story. Eur. J. Pharmacol. 2018, 837, 117–126. [Google Scholar] [CrossRef] [PubMed]
- Liang, M. Oncorine, the World First Oncolytic Virus Medicine and its Update in China. Curr. Cancer Drug Targets 2018, 18, 171–176. [Google Scholar] [CrossRef] [PubMed]
- Andtbacka, R.H.; Kaufman, H.L.; Collichio, F.; Amatruda, T.; Senzer, N.; Chesney, J.; Delman, K.A.; Spitler, L.E.; Puzanov, I.; Agarwala, S.S.; et al. Talimogene Laherparepvec Improves Durable Response Rate in Patients with Advanced Melanoma. J. Clin. Oncol. 2015, 33, 2780–2788. [Google Scholar] [CrossRef]
- Tian, Y.; Xie, D.; Yang, L. Engineering strategies to enhance oncolytic viruses in cancer immunotherapy. Signal Transduct. Target Ther. 2022, 7, 117. [Google Scholar] [CrossRef]
- Russell, S.J.; Bell, J.C.; Engeland, C.E.; McFadden, G. Advances in oncolytic virotherapy. Commun. Med. 2022, 2, 1–3. [Google Scholar] [CrossRef]
- Todo, T.; Ito, H.; Ino, Y.; Ohtsu, H.; Ota, Y.; Shibahara, J.; Tanaka, M. Intratumoral oncolytic herpes virus G47∆ for residual or recurrent glioblastoma: A phase 2 trial. Nat. Med. 2022, 28, 1630–1639. [Google Scholar] [CrossRef]
- Todo, T.; Martuza, R.L.; Rabkin, S.D.; Johnson, P.A. Oncolytic herpes simplex virus vector with enhanced MHC class I presentation and tumor cell killing. Proc. Natl. Acad. Sci. USA 2001, 98, 6396–6401. [Google Scholar] [CrossRef]
- Macedo, N.; Miller, D.M.; Haq, R.; Kaufman, H.L. Clinical landscape of oncolytic virus research in 2020. J. Immunother. Cancer 2020, 8, e001486. [Google Scholar] [CrossRef]
- Donninger, H.; Li, C.; Eaton, J.W.; Yaddanapudi, K. Cancer vaccines: Promising therapeutics or an unattainable dream. Vaccines 2021, 9, 668. [Google Scholar] [CrossRef]
- Finn, O.J. The dawn of vaccines for cancer prevention. Nat. Rev. Immunol. 2017, 18, 183–194. [Google Scholar] [CrossRef]
- Cutts, F.T.; Hall, A.J. Vaccines for neonatal viral infections: Hepatitis B vaccine. Expert Rev. Vaccines 2004, 3, 349–352. [Google Scholar] [CrossRef]
- Pattyn, J.; Hendrickx, G.; Vorsters, A.; Van Damme, P. Hepatitis B Vaccines. J. Infect. Dis. 2021, 224, S343–S351. [Google Scholar] [CrossRef]
- Zhou, X.; Sun, L.; Yao, X.; Li, G.; Wang, Y.; Lin, Y. Progress in Vaccination of Prophylactic Human Papillomavirus Vaccine. Front. Immunol. 2020, 11, 1434. [Google Scholar] [CrossRef]
- Lowy, D.R.; Schiller, J.T. Prophylactic human papillomavirus vaccines. J. Clin. Investig. 2006, 116, 1167–1173. [Google Scholar] [CrossRef]
- Morales, A.; Eidinger, D.; Bruce, A.W. Intracavitary Bacillus Calmette-guerin in the Treatment of Superficial Bladder Tumors. J. Urol. 1976, 116, 180–182. [Google Scholar] [CrossRef]
- Cheever, M.A.; Higano, C.S. PROVENGE (sipuleucel-T) in prostate cancer: The first FDA-approved therapeutic cancer vaccine. Clin. Cancer Res. 2011, 17, 3520–3526. [Google Scholar] [CrossRef]
- A Madan, R.; Gulley, J.L. Therapeutic cancer vaccine fulfills the promise of immunotherapy in prostate cancer. Immunotherapy 2011, 3, 27–31. [Google Scholar] [CrossRef]
- Kantoff, P.W.; Higano, C.S.; Shore, N.D.; Berger, E.R.; Small, E.J.; Penson, D.F.; Redfern, C.H.; Ferrari, A.C.; Dreicer, R.; Sims, R.B.; et al. Sipuleucel-T Immunotherapy for Castration-Resistant Prostate Cancer. N. Engl. J. Med. 2010, 363, 411–422. [Google Scholar] [CrossRef]
- Hollingsworth, R.E.; Jansen, K. Turning the corner on therapeutic cancer vaccines. NPJ Vaccines 2019, 4, 7. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Fu, M.; Wang, M.; Wan, D.; Wei, Y.; Wei, X. Cancer vaccines as promising immuno-therapeutics: Platforms and current progress. J. Hematol. Oncol. 2022, 15, 28. [Google Scholar] [CrossRef] [PubMed]
- Antonarelli, G.; Corti, C.; Tarantino, P.; Ascione, L.; Cortes, J.; Romero, P.; Mittendorf, E.A.; Disis, M.L.; Curigliano, G. Therapeutic cancer vaccines revamping: Technology advancements and pitfalls. Ann. Oncol. 2021, 32, 1537–1551. [Google Scholar] [CrossRef] [PubMed]
- Peng, M.; Mo, Y.; Wang, Y.; Wu, P.; Zhang, Y.; Xiong, F.; Guo, C.; Wu, X.; Li, Y.; Li, X.; et al. Neoantigen vaccine: An emerging tumor immunotherapy. Mol. Cancer 2019, 18, 128. [Google Scholar] [CrossRef] [PubMed]
- Gopalakrishnan, V.; Helmink, B.A.; Spencer, C.N.; Reuben, A.; Wargo, J.A. The Influence of the Gut Microbiome on Cancer, Immunity, and Cancer Immunotherapy. Cancer Cell 2018, 33, 570–580. [Google Scholar] [CrossRef] [PubMed]
- Gopalakrishnan, V.; Spencer, C.N.; Nezi, L.; Reuben, A.; Andrews, M.C.; Karpinets, T.V.; Prieto, P.A.; Vicente, D.; Hoffman, K.; Wei, S.C.; et al. Gut microbiome modulates response to anti–PD-1 immunotherapy in melanoma patients. Science 2018, 359, 97–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Disease Condition | Drug Combination | Phase | Participants | Status | NCT Number |
|---|---|---|---|---|---|
| Non-small cell lung cancer | Durvalumab (anti-PDL1), Oleclumab (anti-CD73), Monalizumab (anti-NKG2A) Durvalumab (anti-PDL1), Domvanalimab (anti-TIGIT) Atezolizumab (anti-PDL1), Pemetrexed, Carboplatin, Cisplatin, Gemcitabine, Paclitaxel | III III III | 999 860 114 | Recruiting Recruiting Recruiting | NCT05221840 NCT05211895 NCT05047250 |
| Extensive stage small cell lung cancer | Atezolizumab (anti-PDL1), Chemotherapy (Carboplatin-etoposide) | III | 200 | Not yet recruiting | NCT05468489 |
| Metastatic non-small cell lung cancer | Pembrolizumab (anti-PD1), Datopotamab deruxtecan (ADC with TROP2 Ab and Dato-DXd, DS-1062a) Ipilimumab (anti-CTLA-4), Nivolumab (anti-PD1) | III III | 740 265 | Recruiting Active, not recruiting | NCT05215340 NCT03469960 |
| Squamous cell non-small cell lung cancer | Sintilimab (anti-PD1), Carboplatin, Albumin-Bound Paclitaxel | III | 236 | Not yet recruiting | NCT05429463 |
| Head and Neck squamous cell carcinoma | Monalizumab (anti-NKG2A), Cetuximab (anti-EGFR) | III | 624 | Recruiting | NCT04590963 |
| Nasopharyngeal carcinoma | PD-1 antibody, Capecitabine | III | 556 | Recruiting | NCT05342792 |
| Gastric cancer | SHR-1701 (bifunctional antibody against PD-L1 and TGF-βRII) | III | 896 | Enrolling by invitation | NCT05149807 |
| Colorectal carcinoma | Sintilimab (anti-PD1), Oxaliplatin, Capecitabine | III | 323 | Recruiting | NCT05236972 |
| Anal cancer | Sintilimab (anti- PD-1), Chemoradiotherapy | III | 102 | Recruiting | NCT05374252 |
| Metastatic urothelial cancer | Avelumab (anti-PDL1), Cabozantinib S-malate | III | 654 | Recruiting | NCT05092958 |
| Renal cell carcinoma | Nivolumab (anti-PD1), Tivozanib | III | 326 | Recruiting | NCT04987203 |
| Acute myeloid leukemia | Magrolimab (anti-CD47), Venetoclax, Azacitidine | III | 432 | Recruiting | NCT05079230 |
| Relapsed or refractory myeloma | Talquetamab (bispecific Ab binding CD3 and GPRC5D), Daratumumab (anti-CD38), Pomalidomide, Dexamethasone | III | 810 | Not yet Recruiting | NCT05455320 |
| Recurrent myeloma | Satuximab (anti-CD38), Dexamethasone, Pomalidomide | III | 534 | Recruiting | NCT05405166 |
| Melanoma | Fianlimab (anti-LAG3), Cemiplimab (anti-PD1), Pembrolizumab (anti-PD1) Nivolumab (anti-PD1) (subcutaneously versus intravenous), rHuPH20 | III III | 1100 286 | Recruiting Recruiting | NCT05352672 NCT05297565 |
| Breast cancer | Inetetamab (anti-HER2), Toripalimab (anti-PD1), Albumin-Bound Paclitaxel | IV | 70 | Not yet Recruiting | NCT05291910 |
| Disease Condition | Drug Combinations | Phase | Participants | Status | NCT Number |
|---|---|---|---|---|---|
| Breast cancer | 4SCAR T cells (CAR-T cells targeting Her2, GD2, and CD44v6) | I/II | 100 | Recruiting | NCT04430595 |
| Acute myeloid leukemia | CAR-T CD19 CD7 CAR-T cells | II/III I/II | 10 108 | Recruiting Recruiting | NCT04257175 NCT04599556 |
| Multiple myeloma | CAR-T cell targeting B-cell maturation antigen (BCMA), Bortezomib, Dexamethasone, Lenalidomide, Cyclophosphamide, Fludarabine. JNJ-68284528 (cilta-cel), Pomalidomide, Bortezomib, Dexamethasone, Daratumumab JNJ-68284528 (ciltacabtagene autoleucel [cilta-cel], Bortezomib, Lenalidomide, Dexamethasone, Cyclophosphamide, Fludarabine, Daratumumab | III III III | 650 419 750 | Recruiting Active not yet recruiting Not yet recruiting | NCT04923893 NCT04181827 NCT05257083 |
| B cell lymphoma | CAR-T-CD19, BTK inhibitor, Fludarabine, Cyclophosphamide | III | 24 | Recruiting | NCT05020392 |
| B Cell malignancies | CD19/CD22-CAR-T cells, fludarabine, cyclophosphamide | I/II | 146 | Not yet Recruiting | NCT05442515 |
| Solid tumor | CLDN6 CAR-T, CLDN6 RNA-LPX | I/II | 96 | Recruiting | NCT04503278 |
| Pancreatic cancer | CD276 CAR-T cells | I/II | 10 | Recruiting | NCT05143151 |
| Gastric cancer, Pancreatic cancer | CT041 (CAR-T cells targeting claudin18.2) | I/II | 110 | Recruiting | NCT04404595 |
| Prostate cancer | 4SCAR-PSMA T cells [CAR-T cells targeting Prostate-specific membrane antigen (PSMA)] | I/II | 100 | Recruiting | NCT04429451 |
| CD44v6 positive cancers (squamous cell carcinomas, adenocarcinomas, melanoma, lymphoma) | 4SCAR-CD44v6 [CAR-T cells targeting CD44v6] | I/II | 100 | Recruiting | NCT04427449 |
| Disease Condition | Drug Combination | Phase | Participants | Status | NCT Number |
|---|---|---|---|---|---|
| Breast cancer | AST-301[pNGVL3-hICD (DNA vaccine against HER2)], rhuGM-CSF, Pembrolizumab Adagloxad simolenin, OBI-821 (Vaccine with tumor-associated antigen Globo H linked to KLH). | II III | 146 668 | Recruiting Recruiting | NCT05163223 NCT03562637 |
| Cervical cancer | Recombinant Human Papillomavirus Bivalent Vaccine, Recombinant Human Papillomavirus Nonavalent Vaccine, Diphtheria Toxoid/Tetanus Toxoid/Acellular Pertussis Vaccine Cecolin® (bivalent HPV vaccine) Gardasil® (HPV 9-valent Vaccine) Gardasil-9 (9-valent HPV Vaccination) | IV III III | 5000 1025 1220 | Enrolling by invitation Active, not recruiting Not yet recruiting | NCT05237947 NCT04508309 NCT03848039 |
| Colorectal cancer | GRT-C901, GRT-R902[Chimpanzee adenovirus vector (ChAdV)twenty tumor-specific neoantigens (TSNAs)], Atezolizumab, Ipilimumab, Fluoropyrimidine, Bevacizumab, Oxaliplatin | II/III | 665 | Recruiting | NCT05141721 |
| Bladder cancer | Bacillus Calmette-Guérin (BCG) Bacillus Calmette Guerin (BCG), PF-06801591(anti-PD1 mAb) | III III | 32 1160 | Active not yet recruiting Recruiting | NCT04806178 NCT04165317 |
| Liver cancer | GP96 (Heat Shock Protein-Peptide Complex Vaccine) | II/III | 80 | Not yet Recruiting | NCT04206254 |
| Acute myeloid leukemia | DSP-7888 [vaccine with two synthetic peptides derived from Wilms’ tumor 1 (WT1) | II | 100 | Recruiting | NCT04747002 |
| Non-small cell lung cancer | UCPVax [vaccine with two peptides from hTERT], Nivolumab | II | 111 | Recruiting | NCT04263051 |
| Glioblastoma multiforme | ADCTA-SSI-G1 (Autologous Dendritic Cell/Tumor Antigen) | III | 118 | Recruiting | NCT04277221 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mishra, A.K.; Ali, A.; Dutta, S.; Banday, S.; Malonia, S.K. Emerging Trends in Immunotherapy for Cancer. Diseases 2022, 10, 60. https://doi.org/10.3390/diseases10030060
Mishra AK, Ali A, Dutta S, Banday S, Malonia SK. Emerging Trends in Immunotherapy for Cancer. Diseases. 2022; 10(3):60. https://doi.org/10.3390/diseases10030060
Chicago/Turabian StyleMishra, Alok K., Amjad Ali, Shubham Dutta, Shahid Banday, and Sunil K. Malonia. 2022. "Emerging Trends in Immunotherapy for Cancer" Diseases 10, no. 3: 60. https://doi.org/10.3390/diseases10030060
APA StyleMishra, A. K., Ali, A., Dutta, S., Banday, S., & Malonia, S. K. (2022). Emerging Trends in Immunotherapy for Cancer. Diseases, 10(3), 60. https://doi.org/10.3390/diseases10030060