
Microenvironmental Features Driving Immune Evasion in Myelodysplastic Syndromes and Acute Myeloid Leukemia



Abstract
1. Introduction
1.1. BM in Immunity and Inflammation
1.2. Lymphopoiesis
1.3. Interplay between Lymphoid Lineage Cells and the BM
1.4. Myelopoiesis
1.5. HSCs and Allo-HSCT
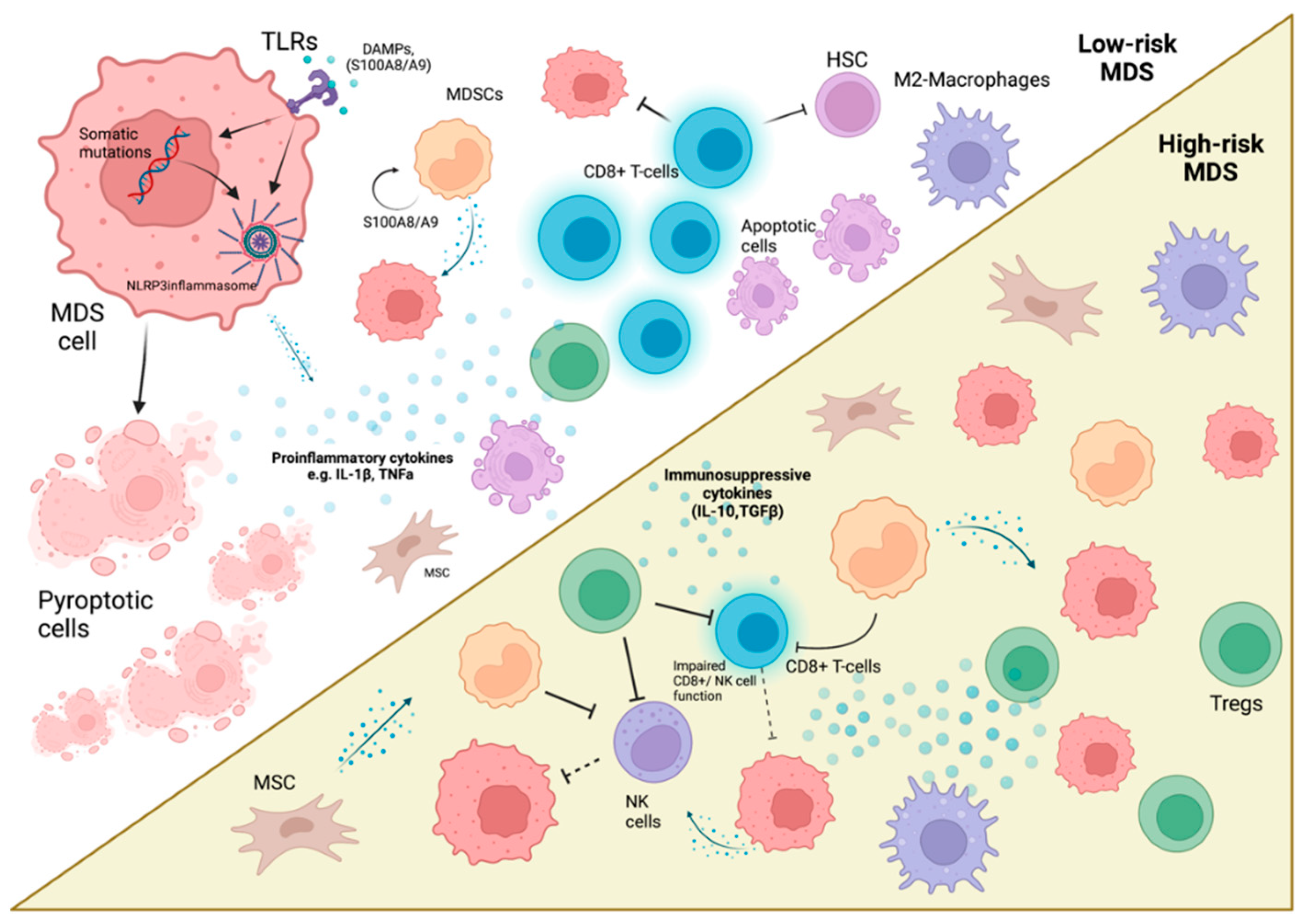
2. The Immune Landscape in MDS
2.1. The Inflammatory Microenvironment of MDS
2.1.1. Predisposing Factors Driving Inflammation (Mutations, Aging/Chronic Immune Stimulation)
2.1.2. Significant Signalling Pathways
2.1.3. Cytokines
2.2. Defective Cellular Immune Responses Result in MDS Progression
2.2.1. T-Lymphocytes in MDS
2.2.2. Dendritic Cells in MDS
2.2.3. Natural Killer Cells in MDS
2.2.4. Myeloid-Derived Suppressor Cells in MDS
2.2.5. Macrophages in MDS
2.2.6. Mesenchymal Stem Cells in MDS
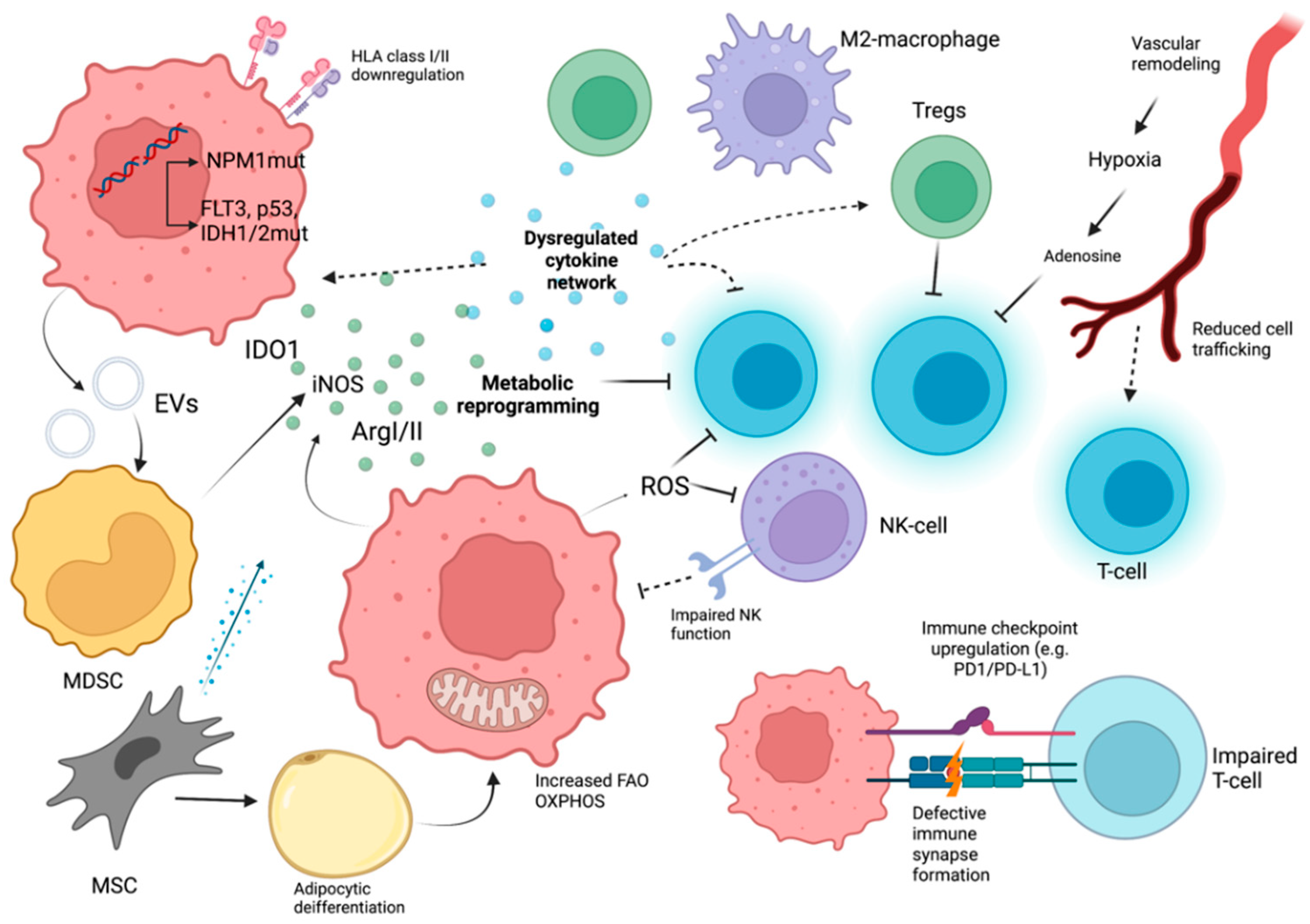
3. Immune Evasion in Acute Myeloid Leukemia
3.1. Cell Intrinsic Factors in AML
3.2. Cytokines in AML
3.3. Metabolic, Soluble, and Vascular Factors with Immunological Significance in AML
3.4. Cellular Immune Response Dysregulation
3.4.1. T-Cells in AML
3.4.2. Dendritic Cells in AML
3.4.3. NK Cells in AML
3.4.4. MDSCs in AML
3.4.5. Macrophages in AML
3.4.6. MSCs in AML
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sezaki, M.; Hayashi, Y.; Wang, Y.; Johansson, A.; Umemoto, T.; Takizawa, H. Immuno-Modulation of Hematopoietic Stem and Progenitor Cells in Inflammation. Front. Immunol. 2020, 11, 585367. [Google Scholar] [CrossRef]
- Mercier, F.E.; Ragu, C.; Scadden, D.T. The bone marrow at the crossroads of blood and immunity. Nat. Rev. Immunol. 2011, 12, 49–60. [Google Scholar] [CrossRef] [PubMed]
- Man, Y.; Yao, X.; Yang, T.; Wang, Y. Hematopoietic Stem Cell Niche During Homeostasis, Malignancy, and Bone Marrow Transplantation. Front. Cell Dev. Biol. 2021, 9, 621214. [Google Scholar] [CrossRef] [PubMed]
- Cazzola, M. Myelodysplastic Syndromes. N. Engl. J. Med. 2020, 383, 1358–1374. [Google Scholar] [CrossRef] [PubMed]
- Barreyro, L.; Chlon, T.M.; Starczynowski, D.T. Chronic immune response dysregulation in MDS pathogenesis. Blood 2018, 132, 1553–1560. [Google Scholar] [CrossRef]
- Sallman, D.A.; List, A. The central role of inflammatory signaling in the pathogenesis of myelodysplastic syndromes. Blood 2019, 133, 1039–1048. [Google Scholar] [CrossRef]
- Winter, S.; Shoaie, S.; Kordasti, S.; Platzbecker, U. Integrating the “Immunome“ in the Stratification of Myelodysplastic Syndromes and Future Clinical Trial Design. J. Clin. Oncol. 2020, 38, 1723–1735. [Google Scholar] [CrossRef]
- Wang, C.; Yang, Y.; Gao, S.; Chen, J.; Yu, J.; Zhang, H.; Li, M.; Zhan, X.; Li, W. Immune dysregulation in myelodysplastic syndrome: Clinical features, pathogenesis and therapeutic strategies. Crit. Rev. Oncol. Hematol. 2018, 122, 123–132. [Google Scholar] [CrossRef]
- Dohner, H.; Weisdorf, D.J.; Bloomfield, C.D. Acute Myeloid Leukemia. N. Engl. J. Med. 2015, 373, 1136–1152. [Google Scholar] [CrossRef]
- Vago, L.; Gojo, I. Immune escape and immunotherapy of acute myeloid leukemia. J. Clin. Investig. 2020, 130, 1552–1564. [Google Scholar] [CrossRef]
- Khaldoyanidi, S.; Nagorsen, D.; Stein, A.; Ossenkoppele, G.; Subklewe, M. Immune Biology of Acute Myeloid Leukemia: Implications for Immunotherapy. J. Clin. Oncol. 2021, 39, 419–432. [Google Scholar] [CrossRef] [PubMed]
- Slamanig, S.A.; Nolte, M.A. The Bone Marrow as Sanctuary for Plasma Cells and Memory T-Cells: Implications for Adaptive Immunity and Vaccinology. Cells 2021, 10, 1508. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.D.; Tokoyoda, K.; Radbruch, A. Immunological memories of the bone marrow. Immunol. Rev. 2018, 283, 86–98. [Google Scholar] [CrossRef]
- Boyer, S.W.; Schroeder, A.V.; Smith-Berdan, S.; Forsberg, E.C. All hematopoietic cells develop from hematopoietic stem cells through Flk2/Flt3-positive progenitor cells. Cell Stem Cell 2011, 9, 64–73. [Google Scholar] [CrossRef] [PubMed]
- Galan-Diez, M.; Cuesta-Dominguez, A.; Kousteni, S. The Bone Marrow Microenvironment in Health and Myeloid Malignancy. Cold Spring Harb. Perspect. Med. 2018, 8, a031328. [Google Scholar] [CrossRef] [PubMed]
- Beerman, I.; Bhattacharya, D.; Zandi, S.; Sigvardsson, M.; Weissman, I.L.; Bryder, D.; Rossi, D.J. Functionally distinct hematopoietic stem cells modulate hematopoietic lineage potential during aging by a mechanism of clonal expansion. Proc. Natl. Acad. Sci. USA 2010, 107, 5465–5470. [Google Scholar] [CrossRef]
- Wei, Q.; Frenette, P.S. Niches for Hematopoietic Stem Cells and Their Progeny. Immunity 2018, 48, 632–648. [Google Scholar] [CrossRef]
- Sapoznikov, A.; Pewzner-Jung, Y.; Kalchenko, V.; Krauthgamer, R.; Shachar, I.; Jung, S. Perivascular clusters of dendritic cells provide critical survival signals to B cells in bone marrow niches. Nat. Immunol. 2008, 9, 388–395. [Google Scholar] [CrossRef]
- Feuerer, M.; Beckhove, P.; Garbi, N.; Mahnke, Y.; Limmer, A.; Hommel, M.; Hammerling, G.J.; Kyewski, B.; Hamann, A.; Umansky, V.; et al. Bone marrow as a priming site for T-cell responses to blood-borne antigen. Nat. Med. 2003, 9, 1151–1157. [Google Scholar] [CrossRef]
- Di Rosa, F.; Gebhardt, T. Bone Marrow T Cells and the Integrated Functions of Recirculating and Tissue-Resident Memory T Cells. Front. Immunol. 2016, 7, 51. [Google Scholar] [CrossRef]
- Pierini, A.; Nishikii, H.; Baker, J.; Kimura, T.; Kwon, H.S.; Pan, Y.; Chen, Y.; Alvarez, M.; Strober, W.; Velardi, A.; et al. Foxp3(+) regulatory T cells maintain the bone marrow microenvironment for B cell lymphopoiesis. Nat. Commun. 2017, 8, 15068. [Google Scholar] [CrossRef] [PubMed]
- Baldridge, M.T.; King, K.Y.; Boles, N.C.; Weksberg, D.C.; Goodell, M.A. Quiescent haematopoietic stem cells are activated by IFN-gamma in response to chronic infection. Nature 2010, 465, 793–797. [Google Scholar] [CrossRef] [PubMed]
- de Bruin, A.M.; Demirel, O.; Hooibrink, B.; Brandts, C.H.; Nolte, M.A. Interferon-gamma impairs proliferation of hematopoietic stem cells in mice. Blood 2013, 121, 3578–3585. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, M.; Passegue, E. TNF-alpha Coordinates Hematopoietic Stem Cell Survival and Myeloid Regeneration. Cell Stem Cell 2019, 25, 357–372.e7. [Google Scholar] [CrossRef]
- Benson, M.J.; Dillon, S.R.; Castigli, E.; Geha, R.S.; Xu, S.; Lam, K.P.; Noelle, R.J. Cutting edge: The dependence of plasma cells and independence of memory B cells on BAFF and APRIL. J. Immunol. 2008, 180, 3655–3659. [Google Scholar] [CrossRef]
- O’Connor, B.P.; Raman, V.S.; Erickson, L.D.; Cook, W.J.; Weaver, L.K.; Ahonen, C.; Lin, L.L.; Mantchev, G.T.; Bram, R.J.; Noelle, R.J. BCMA is essential for the survival of long-lived bone marrow plasma cells. J. Exp. Med. 2004, 199, 91–98. [Google Scholar] [CrossRef]
- Nguyen, D.C.; Joyner, C.J.; Sanz, I.; Lee, F.E. Factors Affecting Early Antibody Secreting Cell Maturation Into Long-Lived Plasma Cells. Front. Immunol. 2019, 10, 2138. [Google Scholar] [CrossRef]
- Tripp, R.A.; Topham, D.J.; Watson, S.R.; Doherty, P.C. Bone marrow can function as a lymphoid organ during a primary immune response under conditions of disrupted lymphocyte trafficking. J. Immunol. 1997, 158, 3716–3720. [Google Scholar]
- Di Rosa, F.; Pabst, R. The bone marrow: A nest for migratory memory T cells. Trends Immunol. 2005, 26, 360–366. [Google Scholar] [CrossRef]
- Siracusa, F.; Durek, P.; McGrath, M.A.; Sercan-Alp, O.; Rao, A.; Du, W.; Cendon, C.; Chang, H.D.; Heinz, G.A.; Mashreghi, M.F.; et al. CD69(+) memory T lymphocytes of the bone marrow and spleen express the signature transcripts of tissue-resident memory T lymphocytes. Eur. J. Immunol. 2019, 49, 966–968. [Google Scholar] [CrossRef]
- Zhao, E.; Xu, H.; Wang, L.; Kryczek, I.; Wu, K.; Hu, Y.; Wang, G.; Zou, W. Bone marrow and the control of immunity. Cell Mol. Immunol. 2012, 9, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Lucas, D. Structural organization of the bone marrow and its role in hematopoiesis. Curr. Opin. Hematol. 2021, 28, 36–42. [Google Scholar] [CrossRef] [PubMed]
- Herault, A.; Binnewies, M.; Leong, S.; Calero-Nieto, F.J.; Zhang, S.Y.; Kang, Y.A.; Wang, X.; Pietras, E.M.; Chu, S.H.; Barry-Holson, K.; et al. Myeloid progenitor cluster formation drives emergency and leukaemic myelopoiesis. Nature 2017, 544, 53–58. [Google Scholar] [CrossRef] [PubMed]
- Takizawa, H.; Boettcher, S.; Manz, M.G. Demand-adapted regulation of early hematopoiesis in infection and inflammation. Blood 2012, 119, 2991–3002. [Google Scholar] [CrossRef] [PubMed]
- Pietras, E.M. Inflammation: A key regulator of hematopoietic stem cell fate in health and disease. Blood 2017, 130, 1693–1698. [Google Scholar] [CrossRef]
- Clapes, T.; Lefkopoulos, S.; Trompouki, E. Stress and Non-Stress Roles of Inflammatory Signals during HSC Emergence and Maintenance. Front. Immunol. 2016, 7, 487. [Google Scholar] [CrossRef]
- Walter, D.; Lier, A.; Geiselhart, A.; Thalheimer, F.B.; Huntscha, S.; Sobotta, M.C.; Moehrle, B.; Brocks, D.; Bayindir, I.; Kaschutnig, P.; et al. Exit from dormancy provokes DNA-damage-induced attrition in haematopoietic stem cells. Nature 2015, 520, 549–552. [Google Scholar] [CrossRef]
- Nagai, Y.; Garrett, K.P.; Ohta, S.; Bahrun, U.; Kouro, T.; Akira, S.; Takatsu, K.; Kincade, P.W. Toll-like receptors on hematopoietic progenitor cells stimulate innate immune system replenishment. Immunity 2006, 24, 801–812. [Google Scholar] [CrossRef]
- Boettcher, S.; Ziegler, P.; Schmid, M.A.; Takizawa, H.; van Rooijen, N.; Kopf, M.; Heikenwalder, M.; Manz, M.G. Cutting edge: LPS-induced emergency myelopoiesis depends on TLR4-expressing nonhematopoietic cells. J. Immunol. 2012, 188, 5824–5828. [Google Scholar] [CrossRef]
- Burberry, A.; Zeng, M.Y.; Ding, L.; Wicks, I.; Inohara, N.; Morrison, S.J.; Nunez, G. Infection mobilizes hematopoietic stem cells through cooperative NOD-like receptor and Toll-like receptor signaling. Cell Host Microbe 2014, 15, 779–791. [Google Scholar] [CrossRef]
- Netea, M.G.; Joosten, L.A.; Latz, E.; Mills, K.H.; Natoli, G.; Stunnenberg, H.G.; O’Neill, L.A.; Xavier, R.J. Trained immunity: A program of innate immune memory in health and disease. Science 2016, 352, aaf1098. [Google Scholar] [CrossRef] [PubMed]
- Mitroulis, I.; Ruppova, K.; Wang, B.; Chen, L.S.; Grzybek, M.; Grinenko, T.; Eugster, A.; Troullinaki, M.; Palladini, A.; Kourtzelis, I.; et al. Modulation of Myelopoiesis Progenitors Is an Integral Component of Trained Immunity. Cell 2018, 172, 147–161.e12. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.; Yu, H.; Liang, P.H.; Cheng, H.; XuFeng, R.; Yuan, Y.; Zhang, P.; Smith, C.A.; Cheng, T. An acute negative bystander effect of gamma-irradiated recipients on transplanted hematopoietic stem cells. Blood 2012, 119, 3629–3637. [Google Scholar] [CrossRef]
- Ganuza, M.; McKinney-Freeman, S. Hematopoietic stem cells under pressure. Curr. Opin. Hematol. 2017, 24, 314–321. [Google Scholar] [CrossRef] [PubMed]
- Sasine, J.P.; Yeo, K.T.; Chute, J.P. Concise Review: Paracrine Functions of Vascular Niche Cells in Regulating Hematopoietic Stem Cell Fate. Stem Cells Transl. Med. 2017, 6, 482–489. [Google Scholar] [CrossRef] [PubMed]
- Lapidot, T.; Dar, A.; Kollet, O. How do stem cells find their way home? Blood 2005, 106, 1901–1910. [Google Scholar] [CrossRef]
- Spencer, J.A.; Ferraro, F.; Roussakis, E.; Klein, A.; Wu, J.; Runnels, J.M.; Zaher, W.; Mortensen, L.J.; Alt, C.; Turcotte, R.; et al. Direct measurement of local oxygen concentration in the bone marrow of live animals. Nature 2014, 508, 269–273. [Google Scholar] [CrossRef]
- Simsek, T.; Kocabas, F.; Zheng, J.; Deberardinis, R.J.; Mahmoud, A.I.; Olson, E.N.; Schneider, J.W.; Zhang, C.C.; Sadek, H.A. The distinct metabolic profile of hematopoietic stem cells reflects their location in a hypoxic niche. Cell Stem Cell 2010, 7, 380–390. [Google Scholar] [CrossRef]
- Takubo, K.; Nagamatsu, G.; Kobayashi, C.I.; Nakamura-Ishizu, A.; Kobayashi, H.; Ikeda, E.; Goda, N.; Rahimi, Y.; Johnson, R.S.; Soga, T.; et al. Regulation of glycolysis by Pdk functions as a metabolic checkpoint for cell cycle quiescence in hematopoietic stem cells. Cell Stem Cell 2013, 12, 49–61. [Google Scholar] [CrossRef]
- Seggewiss, R.; Einsele, H. Immune reconstitution after allogeneic transplantation and expanding options for immunomodulation: An update. Blood 2010, 115, 3861–3868. [Google Scholar] [CrossRef]
- Danby, R.; Rocha, V. Improving engraftment and immune reconstitution in umbilical cord blood transplantation. Front. Immunol. 2014, 5, 68. [Google Scholar] [CrossRef] [PubMed]
- Ogonek, J.; Kralj Juric, M.; Ghimire, S.; Varanasi, P.R.; Holler, E.; Greinix, H.; Weissinger, E. Immune Reconstitution after Allogeneic Hematopoietic Stem Cell Transplantation. Front. Immunol. 2016, 7, 507. [Google Scholar] [CrossRef] [PubMed]
- Dominietto, A.; Raiola, A.M.; Bruno, B.; van Lint, M.T.; Frassoni, F.; Grazia, C.D.; Gualandi, F.; Bregante, S.; Varaldo, R.; Ghiso, A.; et al. Rapid Immune Reconstitution Following Unmanipulated Haploidentical BMT with Post-Transplant High Dose Cyclophosphamide. Blood 2011, 118, 3050. [Google Scholar] [CrossRef]
- Chang, Y.J.; Zhao, X.Y.; Huang, X.J. Immune reconstitution after haploidentical hematopoietic stem cell transplantation. Biol. Blood Marrow Transplant. 2014, 20, 440–449. [Google Scholar] [CrossRef]
- Pfeilstocker, M.; Tuechler, H.; Sanz, G.; Schanz, J.; Garcia-Manero, G.; Sole, F.; Bennett, J.M.; Bowen, D.; Fenaux, P.; Dreyfus, F.; et al. Time-dependent changes in mortality and transformation risk in MDS. Blood 2016, 128, 902–910. [Google Scholar] [CrossRef]
- Matos, A.; Magalhaes, S.M.M.; Rauh, M.J. Immune Dysregulation and Recurring Mutations in Myelodysplastic Syndromes Pathogenesis. Adv. Exp. Med. Biol. 2021, 1326, 1–10. [Google Scholar] [CrossRef]
- Basiorka, A.A.; McGraw, K.L.; Eksioglu, E.A.; Chen, X.; Johnson, J.; Zhang, L.; Zhang, Q.; Irvine, B.A.; Cluzeau, T.; Sallman, D.A.; et al. The NLRP3 inflammasome functions as a driver of the myelodysplastic syndrome phenotype. Blood 2016, 128, 2960–2975. [Google Scholar] [CrossRef]
- Starczynowski, D.T.; Kuchenbauer, F.; Argiropoulos, B.; Sung, S.; Morin, R.; Muranyi, A.; Hirst, M.; Hogge, D.; Marra, M.; Wells, R.A.; et al. Identification of miR-145 and miR-146a as mediators of the 5q- syndrome phenotype. Nat. Med. 2010, 16, 49–58. [Google Scholar] [CrossRef]
- Varney, M.E.; Niederkorn, M.; Konno, H.; Matsumura, T.; Gohda, J.; Yoshida, N.; Akiyama, T.; Christie, S.; Fang, J.; Miller, D.; et al. Loss of Tifab, a del(5q) MDS gene, alters hematopoiesis through derepression of Toll-like receptor-TRAF6 signaling. J. Exp. Med. 2015, 212, 1967–1985. [Google Scholar] [CrossRef]
- Franceschi, C.; Garagnani, P.; Vitale, G.; Capri, M.; Salvioli, S. Inflammaging and ‘Garb-aging’. Trends Endocrinol. Metab. 2017, 28, 199–212. [Google Scholar] [CrossRef]
- Boraschi, D.; Aguado, M.T.; Dutel, C.; Goronzy, J.; Louis, J.; Grubeck-Loebenstein, B.; Rappuoli, R.; Del Giudice, G. The gracefully aging immune system. Sci. Transl. Med. 2013, 5, 185ps8. [Google Scholar] [CrossRef] [PubMed]
- Naismith, E.; Pangrazzi, L. The impact of oxidative stress, inflammation, and senescence on the maintenance of immunological memory in the bone marrow in old age. Biosci. Rep. 2019, 39, BSR20190371. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, W.K.; de Vos, S.; Komor, M.; Hoelzer, D.; Wachsman, W.; Koeffler, H.P. Characterization of gene expression of CD34+ cells from normal and myelodysplastic bone marrow. Blood 2002, 100, 3553–3560. [Google Scholar] [CrossRef] [PubMed]
- Pellagatti, A.; Cazzola, M.; Giagounidis, A.; Perry, J.; Malcovati, L.; Della Porta, M.G.; Jadersten, M.; Killick, S.; Verma, A.; Norbury, C.J.; et al. Deregulated gene expression pathways in myelodysplastic syndrome hematopoietic stem cells. Leukemia 2010, 24, 756–764. [Google Scholar] [CrossRef]
- Esplin, B.L.; Shimazu, T.; Welner, R.S.; Garrett, K.P.; Nie, L.; Zhang, Q.; Humphrey, M.B.; Yang, Q.; Borghesi, L.A.; Kincade, P.W. Chronic exposure to a TLR ligand injures hematopoietic stem cells. J. Immunol. 2011, 186, 5367–5375. [Google Scholar] [CrossRef]
- Maratheftis, C.I.; Andreakos, E.; Moutsopoulos, H.M.; Voulgarelis, M. Toll-like receptor-4 is up-regulated in hematopoietic progenitor cells and contributes to increased apoptosis in myelodysplastic syndromes. Clin. Cancer Res. 2007, 13, 1154–1160. [Google Scholar] [CrossRef]
- Zeng, Q.; Shu, J.; Hu, Q.; Zhou, S.H.; Qian, Y.M.; Hu, M.H.; Hu, L.Y.; Wang, Y.G.; Zhou, Y.M.; Lu, J.H. Apoptosis in human myelodysplastic syndrome CD34+ cells is modulated by the upregulation of TLRs and histone H4 acetylation via a beta-arrestin 1 dependent mechanism. Exp. Cell Res. 2016, 340, 22–31. [Google Scholar] [CrossRef]
- Kornblau, S.M.; McCue, D.; Singh, N.; Chen, W.; Estrov, Z.; Coombes, K.R. Recurrent expression signatures of cytokines and chemokines are present and are independently prognostic in acute myelogenous leukemia and myelodysplasia. Blood 2010, 116, 4251–4261. [Google Scholar] [CrossRef]
- Shi, X.; Zheng, Y.; Xu, L.; Cao, C.; Dong, B.; Chen, X. The inflammatory cytokine profile of myelodysplastic syndromes: A meta-analysis. Medicine 2019, 98, e15844. [Google Scholar] [CrossRef]
- Shetty, V.; Mundle, S.; Alvi, S.; Showel, M.; Broady-Robinson, L.; Dar, S.; Borok, R.; Showel, J.; Gregory, S.; Rifkin, S.; et al. Measurement of apoptosis, proliferation and three cytokines in 46 patients with myelodysplastic syndromes. Leuk. Res. 1996, 20, 891–900. [Google Scholar] [CrossRef]
- Kordasti, S.Y.; Ingram, W.; Hayden, J.; Darling, D.; Barber, L.; Afzali, B.; Lombardi, G.; Wlodarski, M.W.; Maciejewski, J.P.; Farzaneh, F.; et al. CD4+CD25high Foxp3+ regulatory T cells in myelodysplastic syndrome (MDS). Blood 2007, 110, 847–850. [Google Scholar] [CrossRef] [PubMed]
- Fozza, C.; Contini, S.; Galleu, A.; Simula, M.P.; Virdis, P.; Bonfigli, S.; Longinotti, M. Patients with myelodysplastic syndromes display several T-cell expansions, which are mostly polyclonal in the CD4(+) subset and oligoclonal in the CD8(+) subset. Exp. Hematol. 2009, 37, 947–955. [Google Scholar] [CrossRef] [PubMed]
- Lopes, M.R.; Traina, F.; Campos Pde, M.; Pereira, J.K.; Machado-Neto, J.A.; Machado Hda, C.; Gilli, S.C.; Saad, S.T.; Favaro, P. IL10 inversely correlates with the percentage of CD8(+) cells in MDS patients. Leuk. Res. 2013, 37, 541–546. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Z.; Qianqiao, Z.; Qi, H.; Feng, X.; Chunkang, C.; Xiao, L. In vitro deprivation of CD8(+)CD57(+)T cells promotes the malignant growth of bone marrow colony cells in patients with lower-risk myelodysplastic syndrome. Exp. Hematol. 2010, 38, 677–684. [Google Scholar] [CrossRef]
- Zou, J.X.; Rollison, D.E.; Boulware, D.; Chen, D.T.; Sloand, E.M.; Pfannes, L.V.; Goronzy, J.J.; Bai, F.; Painter, J.S.; Wei, S.; et al. Altered naive and memory CD4+ T-cell homeostasis and immunosenescence characterize younger patients with myelodysplastic syndrome. Leukemia 2009, 23, 1288–1296. [Google Scholar] [CrossRef] [PubMed]
- Coats, T.; Smith, A.E.; Mourikis, A.; Shahin, T.; Kulasekararaj, A.G.; Best, S.; Chitre, S.; Ellis, R.; Petrov, N.; Heck, S.; et al. Neoantigens in MDS Are Associated with Two Novel CD4+ T Cell Subsets and Improved Overall Survival. Blood 2017, 130, 2958. [Google Scholar] [CrossRef]
- Ok, C.Y.; Young, K.H. Checkpoint inhibitors in hematological malignancies. J. Hematol. Oncol. 2017, 10, 103. [Google Scholar] [CrossRef]
- Haroun, F.; Solola, S.A.; Nassereddine, S.; Tabbara, I. PD-1 signaling and inhibition in AML and MDS. Ann. Hematol. 2017, 96, 1441–1448. [Google Scholar] [CrossRef]
- Yang, H.; Bueso-Ramos, C.; DiNardo, C.; Estecio, M.R.; Davanlou, M.; Geng, Q.R.; Fang, Z.; Nguyen, M.; Pierce, S.; Wei, Y.; et al. Expression of PD-L1, PD-L2, PD-1 and CTLA4 in myelodysplastic syndromes is enhanced by treatment with hypomethylating agents. Leukemia 2014, 28, 1280–1288. [Google Scholar] [CrossRef]
- Sand, K.; Theorell, J.; Bruserud, O.; Bryceson, Y.T.; Kittang, A.O. Reduced potency of cytotoxic T lymphocytes from patients with high-risk myelodysplastic syndromes. Cancer Immunol. Immunother. 2016, 65, 1135–1147. [Google Scholar] [CrossRef]
- Hamdi, W.; Ogawara, H.; Handa, H.; Tsukamoto, N.; Murakami, H. Clinical significance of Th1/Th2 ratio in patients with myelodysplastic syndrome. Int. J. Lab. Hematol. 2009, 31, 630–638. [Google Scholar] [CrossRef] [PubMed]
- Shao, L.L.; Zhang, L.; Hou, Y.; Yu, S.; Liu, X.G.; Huang, X.Y.; Sun, Y.X.; Tian, T.; He, N.; Ma, D.X.; et al. Th22 cells as well as Th17 cells expand differentially in patients with early-stage and late-stage myelodysplastic syndrome. PLoS ONE 2012, 7, e51339. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Yue, L.; Wang, H.; Liu, C.; Liu, H.; Tao, J.; Qi, W.; Wang, Y.; Zhang, W.; Fu, R.; et al. Th17 Cells Exhibit Antitumor Effects in MDS Possibly through Augmenting Functions of CD8+ T Cells. J. Immunol. Res. 2016, 2016, 9404705. [Google Scholar] [CrossRef] [PubMed]
- Mailloux, A.W.; Young, M.R. Regulatory T-cell trafficking: From thymic development to tumor-induced immune suppression. Crit. Rev. Immunol. 2010, 30, 435–447. [Google Scholar] [CrossRef] [PubMed]
- Kotsianidis, I.; Bouchliou, I.; Nakou, E.; Spanoudakis, E.; Margaritis, D.; Christophoridou, A.V.; Anastasiades, A.; Tsigalou, C.; Bourikas, G.; Karadimitris, A.; et al. Kinetics, function and bone marrow trafficking of CD4+CD25+FOXP3+ regulatory T cells in myelodysplastic syndromes (MDS). Leukemia 2009, 23, 510–518. [Google Scholar] [CrossRef] [PubMed]
- Mailloux, A.W.; Sugimori, C.; Komrokji, R.S.; Yang, L.; Maciejewski, J.P.; Sekeres, M.A.; Paquette, R.; Loughran, T.P., Jr.; List, A.F.; Epling-Burnette, P.K. Expansion of effector memory regulatory T cells represents a novel prognostic factor in lower risk myelodysplastic syndrome. J. Immunol. 2012, 189, 3198–3208. [Google Scholar] [CrossRef]
- Sallman, D.A.; McLemore, A.F.; Aldrich, A.L.; Komrokji, R.S.; McGraw, K.L.; Dhawan, A.; Geyer, S.; Hou, H.A.; Eksioglu, E.A.; Sullivan, A.; et al. TP53 mutations in myelodysplastic syndromes and secondary AML confer an immunosuppressive phenotype. Blood 2020, 136, 2812–2823. [Google Scholar] [CrossRef]
- Banchereau, J.; Briere, F.; Caux, C.; Davoust, J.; Lebecque, S.; Liu, Y.J.; Pulendran, B.; Palucka, K. Immunobiology of dendritic cells. Annu. Rev. Immunol. 2000, 18, 767–811. [Google Scholar] [CrossRef]
- Kufner, S.; Fleischer, R.P.; Kroell, T.; Schmid, C.; Zitzelsberger, H.; Salih, H.; de Valle, F.; Treder, W.; Schmetzer, H.M. Serum-free generation and quantification of functionally active Leukemia-derived DC is possible from malignant blasts in acute myeloid leukemia and myelodysplastic syndromes. Cancer Immunol. Immunother. 2005, 54, 953–970. [Google Scholar] [CrossRef]
- Ma, L.; Delforge, M.; van Duppen, V.; Verhoef, G.; Emanuel, B.; Boogaerts, M.; Hagemeijer, A.; Vandenberghe, P. Circulating myeloid and lymphoid precursor dendritic cells are clonally involved in myelodysplastic syndromes. Leukemia 2004, 18, 1451–1456. [Google Scholar] [CrossRef]
- Ma, L.; Ceuppens, J.; Kasran, A.; Delforge, M.; Boogaerts, M.; Vandenberghe, P. Immature and mature monocyte-derived dendritic cells in myelodysplastic syndromes of subtypes refractory anemia or refractory anemia with ringed sideroblasts display an altered cytokine profile. Leuk. Res. 2007, 31, 1373–1382. [Google Scholar] [CrossRef] [PubMed]
- Saft, L.; Bjorklund, E.; Berg, E.; Hellstrom-Lindberg, E.; Porwit, A. Bone marrow dendritic cells are reduced in patients with high-risk myelodysplastic syndromes. Leuk. Res. 2013, 37, 266–273. [Google Scholar] [CrossRef] [PubMed]
- Hejazi, M.; Manser, A.R.; Frobel, J.; Kundgen, A.; Zhao, X.; Schonberg, K.; Germing, U.; Haas, R.; Gattermann, N.; Uhrberg, M. Impaired cytotoxicity associated with defective natural killer cell differentiation in myelodysplastic syndromes. Haematologica 2015, 100, 643–652. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, N.; Swerdlow, S.H.; TenEyck, S.P.; Boyiadzis, M.; Felgar, R.E. Natural killer cell (NK) subsets and NK-like T-cell populations in acute myeloid leukemias and myelodysplastic syndromes. Cytom. B Clin. Cytom. 2016, 90, 349–357. [Google Scholar] [CrossRef]
- Epling-Burnette, P.K.; Bai, F.; Painter, J.S.; Rollison, D.E.; Salih, H.R.; Krusch, M.; Zou, J.; Ku, E.; Zhong, B.; Boulware, D.; et al. Reduced natural killer (NK) function associated with high-risk myelodysplastic syndrome (MDS) and reduced expression of activating NK receptors. Blood 2007, 109, 4816–4824. [Google Scholar] [CrossRef]
- Zhang, W.; Xie, X.; Mi, H.; Sun, J.; Ding, S.; Li, L.; Liu, H.; Wang, H.; Fu, R.; Shao, Z. Abnormal populations and functions of natural killer cells in patients with myelodysplastic syndromes. Oncol. Lett. 2018, 15, 5497–5504. [Google Scholar] [CrossRef]
- Carlsten, M.; Jaras, M. Natural Killer Cells in Myeloid Malignancies: Immune Surveillance, NK Cell Dysfunction, and Pharmacological Opportunities to Bolster the Endogenous NK Cells. Front. Immunol. 2019, 10, 2357. [Google Scholar] [CrossRef]
- Marvel, D.; Gabrilovich, D.I. Myeloid-derived suppressor cells in the tumor microenvironment: Expect the unexpected. J. Clin. Investig. 2015, 125, 3356–3364. [Google Scholar] [CrossRef]
- Filipazzi, P.; Huber, V.; Rivoltini, L. Phenotype, function and clinical implications of myeloid-derived suppressor cells in cancer patients. Cancer Immunol. Immunother. 2012, 61, 255–263. [Google Scholar] [CrossRef]
- Salminen, A.; Kaarniranta, K.; Kauppinen, A. Immunosenescence: The potential role of myeloid-derived suppressor cells (MDSC) in age-related immune deficiency. Cell Mol. Life Sci. 2019, 76, 1901–1918. [Google Scholar] [CrossRef]
- Raza, A.; Galili, N. The genetic basis of phenotypic heterogeneity in myelodysplastic syndromes. Nat. Rev. Cancer 2012, 12, 849–859. [Google Scholar] [CrossRef] [PubMed]
- Gabrilovich, D.I.; Nagaraj, S. Myeloid-derived suppressor cells as regulators of the immune system. Nat. Rev. Immunol. 2009, 9, 162–174. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Eksioglu, E.A.; Zhou, J.; Zhang, L.; Djeu, J.; Fortenbery, N.; Epling-Burnette, P.; Van Bijnen, S.; Dolstra, H.; Cannon, J.; et al. Induction of myelodysplasia by myeloid-derived suppressor cells. J. Clin. Investig. 2013, 123, 4595–4611. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.J.; Fu, R.; Wang, H.Q.; Li, L.J.; Qu, W.; Liang, Y.; Wang, G.J.; Wang, X.M.; Wu, Y.H.; Liu, H.; et al. Increased circulating of myeloid-derived suppressor cells in myelodysplastic syndrome. Chin. Med. J. 2013, 126, 2582–2584. [Google Scholar] [PubMed]
- Kirkwood, K.L.; Zhang, L.; Thiyagarajan, R.; Seldeen, K.L.; Troen, B.R. Myeloid-Derived Suppressor Cells at the Intersection of Inflammaging and Bone Fragility. Immunol. Invest. 2018, 47, 844–854. [Google Scholar] [CrossRef]
- Kittang, A.O.; Kordasti, S.; Sand, K.E.; Costantini, B.; Kramer, A.M.; Perezabellan, P.; Seidl, T.; Rye, K.P.; Hagen, K.M.; Kulasekararaj, A.; et al. Expansion of myeloid derived suppressor cells correlates with number of T regulatory cells and disease progression in myelodysplastic syndrome. Oncoimmunology 2016, 5, e1062208. [Google Scholar] [CrossRef]
- Wynn, T.A.; Chawla, A.; Pollard, J.W. Macrophage biology in development, homeostasis and disease. Nature 2013, 496, 445–455. [Google Scholar] [CrossRef]
- Han, Y.; Wang, H.; Shao, Z. Monocyte-Derived Macrophages Are Impaired in Myelodysplastic Syndrome. J. Immunol. Res. 2016, 2016, 5479013. [Google Scholar] [CrossRef]
- Yang, Y.; Wu, Y. The Clinical Significance of Tumor Associated Macrophages in Myelodysplastic Syndromes. Blood 2018, 132, 5505. [Google Scholar] [CrossRef]
- Sica, A.; Mantovani, A. Macrophage plasticity and polarization: In vivo veritas. J. Clin. Investig. 2012, 122, 787–795. [Google Scholar] [CrossRef]
- Zhang, G.; Yang, L.; Han, Y.; Niu, H.; Yan, L.; Shao, Z.; Xing, L.; Wang, H. Abnormal Macrophage Polarization in Patients with Myelodysplastic Syndrome. Mediat. Inflamm. 2021, 2021, 9913382. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.; Greenberger, J.S.; Epperly, M.W.; Goff, J.P.; Adler, C.; Leboff, M.S.; Glowacki, J. Age-related intrinsic changes in human bone-marrow-derived mesenchymal stem cells and their differentiation to osteoblasts. Aging Cell 2008, 7, 335–343. [Google Scholar] [CrossRef] [PubMed]
- Medyouf, H.; Mossner, M.; Jann, J.C.; Nolte, F.; Raffel, S.; Herrmann, C.; Lier, A.; Eisen, C.; Nowak, V.; Zens, B.; et al. Myelodysplastic cells in patients reprogram mesenchymal stromal cells to establish a transplantable stem cell niche disease unit. Cell Stem Cell 2014, 14, 824–837. [Google Scholar] [CrossRef] [PubMed]
- Zambetti, N.A.; Ping, Z.; Chen, S.; Kenswil, K.J.G.; Mylona, M.A.; Sanders, M.A.; Hoogenboezem, R.M.; Bindels, E.M.J.; Adisty, M.N.; Van Strien, P.M.H.; et al. Mesenchymal Inflammation Drives Genotoxic Stress in Hematopoietic Stem Cells and Predicts Disease Evolution in Human Pre-leukemia. Cell Stem Cell 2016, 19, 613–627. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Zambetti, N.A.; Bindels, E.M.; Kenswill, K.; Mylona, A.M.; Adisty, N.M.; Hoogenboezem, R.M.; Sanders, M.A.; Cremers, E.M.; Westers, T.M.; et al. Massive parallel RNA sequencing of highly purified mesenchymal elements in low-risk MDS reveals tissue-context-dependent activation of inflammatory programs. Leukemia 2016, 30, 1938–1942. [Google Scholar] [CrossRef]
- Lopes, M.R.; Pereira, J.K.; de Melo Campos, P.; Machado-Neto, J.A.; Traina, F.; Saad, S.T.; Favaro, P. De novo AML exhibits greater microenvironment dysregulation compared to AML with myelodysplasia-related changes. Sci. Rep. 2017, 7, 40707. [Google Scholar] [CrossRef]
- Zhao, Z.; Wang, Z.; Li, Q.; Li, W.; You, Y.; Zou, P. The different immunoregulatory functions of mesenchymal stem cells in patients with low-risk or high-risk myelodysplastic syndromes. PLoS ONE 2012, 7, e45675. [Google Scholar] [CrossRef]
- Wang, Z.; Tang, X.; Xu, W.; Cao, Z.; Sun, L.; Li, W.; Li, Q.; Zou, P.; Zhao, Z. The different immunoregulatory functions on dendritic cells between mesenchymal stem cells derived from bone marrow of patients with low-risk or high-risk myelodysplastic syndromes. PLoS ONE 2013, 8, e57470. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research Network; Ley, T.J.; Miller, C.; Ding, L.; Raphael, B.J.; Mungall, A.J.; Robertson, A.; Hoadley, K.; Triche, T.J., Jr.; Laird, P.W.; et al. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N. Engl. J. Med. 2013, 368, 2059–2074. [Google Scholar] [CrossRef]
- Dohner, H.; Estey, E.; Grimwade, D.; Amadori, S.; Appelbaum, F.R.; Buchner, T.; Dombret, H.; Ebert, B.L.; Fenaux, P.; Larson, R.A.; et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood 2017, 129, 424–447. [Google Scholar] [CrossRef]
- Mendez, L.M.; Posey, R.R.; Pandolfi, P.P. The Interplay Between the Genetic and Immune Landscapes of AML: Mechanisms and Implications for Risk Stratification and Therapy. Front. Oncol. 2019, 9, 1162. [Google Scholar] [CrossRef] [PubMed]
- Rickmann, M.; Krauter, J.; Stamer, K.; Heuser, M.; Salguero, G.; Mischak-Weissinger, E.; Ganser, A.; Stripecke, R. Elevated frequencies of leukemic myeloid and plasmacytoid dendritic cells in acute myeloid leukemia with the FLT3 internal tandem duplication. Ann. Hematol. 2011, 90, 1047–1058. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez, L.; Jang, M.; Zhang, T.; Akhtari, M.; Alachkar, H. Midostaurin reduces Regulatory T cells markers in Acute Myeloid Leukemia. Sci. Rep. 2018, 8, 17544. [Google Scholar] [CrossRef] [PubMed]
- Vago, L.; Perna, S.K.; Zanussi, M.; Mazzi, B.; Barlassina, C.; Stanghellini, M.T.; Perrelli, N.F.; Cosentino, C.; Torri, F.; Angius, A.; et al. Loss of mismatched HLA in leukemia after stem-cell transplantation. N. Engl. J. Med. 2009, 361, 478–488. [Google Scholar] [CrossRef] [PubMed]
- Crucitti, L.; Crocchiolo, R.; Toffalori, C.; Mazzi, B.; Greco, R.; Signori, A.; Sizzano, F.; Chiesa, L.; Zino, E.; Lupo Stanghellini, M.T.; et al. Incidence, risk factors and clinical outcome of leukemia relapses with loss of the mismatched HLA after partially incompatible hematopoietic stem cell transplantation. Leukemia 2015, 29, 1143–1152. [Google Scholar] [CrossRef]
- McCurdy, S.R.; Iglehart, B.S.; Batista, D.A.; Gocke, C.D.; Ning, Y.; Knaus, H.A.; Jackson, A.M.; Leffell, M.S.; Luznik, L.; Gojo, I. Loss of the mismatched human leukocyte antigen haplotype in two acute myelogenous leukemia relapses after haploidentical bone marrow transplantation with post-transplantation cyclophosphamide. Leukemia 2016, 30, 2102–2106. [Google Scholar] [CrossRef]
- Christopher, M.J.; Petti, A.A.; Rettig, M.P.; Miller, C.A.; Chendamarai, E.; Duncavage, E.J.; Klco, J.M.; Helton, N.M.; O’Laughlin, M.; Fronick, C.C.; et al. Immune Escape of Relapsed AML Cells after Allogeneic Transplantation. N. Engl. J. Med. 2018, 379, 2330–2341. [Google Scholar] [CrossRef]
- Toffalori, C.; Zito, L.; Gambacorta, V.; Riba, M.; Oliveira, G.; Bucci, G.; Barcella, M.; Spinelli, O.; Greco, R.; Crucitti, L.; et al. Immune signature drives leukemia escape and relapse after hematopoietic cell transplantation. Nat. Med. 2019, 25, 603–611. [Google Scholar] [CrossRef]
- Sendker, S.; Reinhardt, D.; Niktoreh, N. Redirecting the Immune Microenvironment in Acute Myeloid Leukemia. Cancers 2021, 13, 1423. [Google Scholar] [CrossRef]
- van Luijn, M.M.; van den Ancker, W.; Chamuleau, M.E.; Ossenkoppele, G.J.; van Ham, S.M.; van de Loosdrecht, A.A. Impaired antigen presentation in neoplasia: Basic mechanisms and implications for acute myeloid leukemia. Immunotherapy 2010, 2, 85–97. [Google Scholar] [CrossRef]
- Guo, Q.Y.; Chen, B.G.; Ruan, Y.Y.; Lin, A.; Yan, W.H. HLA-G expression is irrelevant to prognosis in patients with acute myeloid leukemia. Leuk. Res. 2011, 35, 1350–1354. [Google Scholar] [CrossRef] [PubMed]
- Zhong, C.; Wang, R.; Hua, M.; Zhang, C.; Han, F.; Xu, M.; Yang, X.; Li, G.; Hu, X.; Sun, T.; et al. NLRP3 Inflammasome Promotes the Progression of Acute Myeloid Leukemia via IL-1beta Pathway. Front. Immunol. 2021, 12, 661939. [Google Scholar] [CrossRef]
- Jia, Y.; Zhang, C.; Hua, M.; Wang, M.; Chen, P.; Ma, D. Aberrant NLRP3 inflammasome associated with aryl hydrocarbon receptor potentially contributes to the imbalance of T-helper cells in patients with acute myeloid leukemia. Oncol. Lett. 2017, 14, 7031–7044. [Google Scholar] [CrossRef] [PubMed]
- Binder, S.; Luciano, M.; Horejs-Hoeck, J. The cytokine network in acute myeloid leukemia (AML): A focus on pro- and anti-inflammatory mediators. Cytokine Growth Factor Rev. 2018, 43, 8–15. [Google Scholar] [CrossRef] [PubMed]
- Ersvaer, E.; Skavland, J.; Ulvestad, E.; Gjertsen, B.T.; Bruserud, O. Effects of interferon gamma on native human acute myelogenous leukaemia cells. Cancer Immunol. Immunother. 2007, 56, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Carey, A.; Edwards, D.K.t.; Eide, C.A.; Newell, L.; Traer, E.; Medeiros, B.C.; Pollyea, D.A.; Deininger, M.W.; Collins, R.H.; Tyner, J.W.; et al. Identification of Interleukin-1 by Functional Screening as a Key Mediator of Cellular Expansion and Disease Progression in Acute Myeloid Leukemia. Cell Rep. 2017, 18, 3204–3218. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Chen, P.; Huang, H.F.; Huang, M.J.; Chen, Y.Z. Reduction of transforming growth factor-beta1 expression in leukemia and its possible role in leukemia development. Leuk. Lymphoma 2012, 53, 145–151. [Google Scholar] [CrossRef]
- Sanchez-Correa, B.; Bergua, J.M.; Campos, C.; Gayoso, I.; Arcos, M.J.; Banas, H.; Morgado, S.; Casado, J.G.; Solana, R.; Tarazona, R. Cytokine profiles in acute myeloid leukemia patients at diagnosis: Survival is inversely correlated with IL-6 and directly correlated with IL-10 levels. Cytokine 2013, 61, 885–891. [Google Scholar] [CrossRef]
- Diaz de la Guardia, R.; Lopez-Millan, B.; Lavoie, J.R.; Bueno, C.; Castano, J.; Gomez-Casares, M.; Vives, S.; Palomo, L.; Juan, M.; Delgado, J.; et al. Detailed Characterization of Mesenchymal Stem/Stromal Cells from a Large Cohort of AML Patients Demonstrates a Definitive Link to Treatment Outcomes. Stem Cell Rep. 2017, 8, 1573–1586. [Google Scholar] [CrossRef]
- Tettamanti, S.; Pievani, A.; Biondi, A.; Dotti, G.; Serafini, M. Catch me if you can: How AML and its niche escape immunotherapy. Leukemia 2022, 36, 13–22. [Google Scholar] [CrossRef]
- Curti, A.; Pandolfi, S.; Valzasina, B.; Aluigi, M.; Isidori, A.; Ferri, E.; Salvestrini, V.; Bonanno, G.; Rutella, S.; Durelli, I.; et al. Modulation of tryptophan catabolism by human leukemic cells results in the conversion of CD25- into CD25+ T regulatory cells. Blood 2007, 109, 2871–2877. [Google Scholar] [CrossRef] [PubMed]
- Folgiero, V.; Goffredo, B.M.; Filippini, P.; Masetti, R.; Bonanno, G.; Caruso, R.; Bertaina, V.; Mastronuzzi, A.; Gaspari, S.; Zecca, M.; et al. Indoleamine 2,3-dioxygenase 1 (IDO1) activity in leukemia blasts correlates with poor outcome in childhood acute myeloid leukemia. Oncotarget 2014, 5, 2052–2064. [Google Scholar] [CrossRef] [PubMed]
- Mussai, F.; De Santo, C.; Abu-Dayyeh, I.; Booth, S.; Quek, L.; McEwen-Smith, R.M.; Qureshi, A.; Dazzi, F.; Vyas, P.; Cerundolo, V. Acute myeloid leukemia creates an arginase-dependent immunosuppressive microenvironment. Blood 2013, 122, 749–758. [Google Scholar] [CrossRef] [PubMed]
- Jacamo, R.; Hoang, N.-M.; Al Rawi, A.; Ly, C.; Parihar, R.; McQueen, T.; Ruvolo, P.P.; Williams, P.; Alatrash, G.; Konopleva, M.; et al. Up-Regulation of iNOS in AML Blasts Creates an Immunosuppressive Microenvironment, Inhibits T-Cell Proliferation and Transforms T-Cells Towards a Tumor-Tolerating Phenotype. Blood 2017, 130, 2443. [Google Scholar] [CrossRef]
- Mougiakakos, D. The Induction of a Permissive Environment to Promote T Cell Immune Evasion in Acute Myeloid Leukemia: The Metabolic Perspective. Front. Oncol. 2019, 9, 1166. [Google Scholar] [CrossRef] [PubMed]
- Azadniv, M.; Myers, J.R.; McMurray, H.R.; Guo, N.; Rock, P.; Coppage, M.L.; Ashton, J.; Becker, M.W.; Calvi, L.M.; Liesveld, J.L. Bone marrow mesenchymal stromal cells from acute myelogenous leukemia patients demonstrate adipogenic differentiation propensity with implications for leukemia cell support. Leukemia 2020, 34, 391–403. [Google Scholar] [CrossRef]
- Shafat, M.S.; Oellerich, T.; Mohr, S.; Robinson, S.D.; Edwards, D.R.; Marlein, C.R.; Piddock, R.E.; Fenech, M.; Zaitseva, L.; Abdul-Aziz, A.; et al. Leukemic blasts program bone marrow adipocytes to generate a protumoral microenvironment. Blood 2017, 129, 1320–1332. [Google Scholar] [CrossRef]
- Tabe, Y.; Yamamoto, S.; Saitoh, K.; Sekihara, K.; Monma, N.; Ikeo, K.; Mogushi, K.; Shikami, M.; Ruvolo, V.; Ishizawa, J.; et al. Bone Marrow Adipocytes Facilitate Fatty Acid Oxidation Activating AMPK and a Transcriptional Network Supporting Survival of Acute Monocytic Leukemia Cells. Cancer Res. 2017, 77, 1453–1464. [Google Scholar] [CrossRef]
- O’Neill, L.A.; Kishton, R.J.; Rathmell, J. A guide to immunometabolism for immunologists. Nat. Rev. Immunol. 2016, 16, 553–565. [Google Scholar] [CrossRef]
- Moschoi, R.; Imbert, V.; Nebout, M.; Chiche, J.; Mary, D.; Prebet, T.; Saland, E.; Castellano, R.; Pouyet, L.; Collette, Y.; et al. Protective mitochondrial transfer from bone marrow stromal cells to acute myeloid leukemic cells during chemotherapy. Blood 2016, 128, 253–264. [Google Scholar] [CrossRef]
- Hole, P.S.; Zabkiewicz, J.; Munje, C.; Newton, Z.; Pearn, L.; White, P.; Marquez, N.; Hills, R.K.; Burnett, A.K.; Tonks, A.; et al. Overproduction of NOX-derived ROS in AML promotes proliferation and is associated with defective oxidative stress signaling. Blood 2013, 122, 3322–3330. [Google Scholar] [CrossRef] [PubMed]
- Aurelius, J.; Thoren, F.B.; Akhiani, A.A.; Brune, M.; Palmqvist, L.; Hansson, M.; Hellstrand, K.; Martner, A. Monocytic AML cells inactivate antileukemic lymphocytes: Role of NADPH oxidase/gp91(phox) expression and the PARP-1/PAR pathway of apoptosis. Blood 2012, 119, 5832–5837. [Google Scholar] [CrossRef]
- Vijayan, D.; Young, A.; Teng, M.W.L.; Smyth, M.J. Targeting immunosuppressive adenosine in cancer. Nat. Rev. Cancer 2017, 17, 709–724. [Google Scholar] [CrossRef] [PubMed]
- Beavis, P.A.; Stagg, J.; Darcy, P.K.; Smyth, M.J. CD73: A potent suppressor of antitumor immune responses. Trends Immunol. 2012, 33, 231–237. [Google Scholar] [CrossRef] [PubMed]
- Passaro, D.; Di Tullio, A.; Abarrategi, A.; Rouault-Pierre, K.; Foster, K.; Ariza-McNaughton, L.; Montaner, B.; Chakravarty, P.; Bhaw, L.; Diana, G.; et al. Increased Vascular Permeability in the Bone Marrow Microenvironment Contributes to Disease Progression and Drug Response in Acute Myeloid Leukemia. Cancer Cell 2017, 32, 324–341.e6. [Google Scholar] [CrossRef]
- Barbier, V.; Erbani, J.; Fiveash, C.; Davies, J.M.; Tay, J.; Tallack, M.R.; Lowe, J.; Magnani, J.L.; Pattabiraman, D.R.; Perkins, A.C.; et al. Endothelial E-selectin inhibition improves acute myeloid leukaemia therapy by disrupting vascular niche-mediated chemoresistance. Nat. Commun. 2020, 11, 2042. [Google Scholar] [CrossRef]
- Lamble, A.J.; Lind, E.F. Targeting the Immune Microenvironment in Acute Myeloid Leukemia: A Focus on T Cell Immunity. Front. Oncol. 2018, 8, 213. [Google Scholar] [CrossRef]
- Le Dieu, R.; Taussig, D.C.; Ramsay, A.G.; Mitter, R.; Miraki-Moud, F.; Fatah, R.; Lee, A.M.; Lister, T.A.; Gribben, J.G. Peripheral blood T cells in acute myeloid leukemia (AML) patients at diagnosis have abnormal phenotype and genotype and form defective immune synapses with AML blasts. Blood 2009, 114, 3909–3916. [Google Scholar] [CrossRef]
- Rey, J.; Fauriat, C.; Kochbati, E.; Orlanducci, F.; Charbonnier, A.; D’Incan, E.; Andre, P.; Romagne, F.; Barbarat, B.; Vey, N.; et al. Kinetics of Cytotoxic Lymphocytes Reconstitution after Induction Chemotherapy in Elderly AML Patients Reveals Progressive Recovery of Normal Phenotypic and Functional Features in NK Cells. Front. Immunol. 2017, 8, 64. [Google Scholar] [CrossRef]
- Panoskaltsis, N.; Reid, C.D.; Knight, S.C. Quantification and cytokine production of circulating lymphoid and myeloid cells in acute myelogenous leukaemia. Leukemia 2003, 17, 716–730. [Google Scholar] [CrossRef]
- Vidriales, M.B.; Orfao, A.; Lopez-Berges, M.C.; Gonzalez, M.; Hernandez, J.M.; Ciudad, J.; Lopez, A.; Moro, M.J.; Martinez, M.; San Miguel, J.F. Lymphoid subsets in acute myeloid leukemias: Increased number of cells with NK phenotype and normal T-cell distribution. Ann. Hematol. 1993, 67, 217–222. [Google Scholar] [CrossRef] [PubMed]
- Ismail, M.M.; Abdulateef, N.A.B. Bone marrow T-cell percentage: A novel prognostic indicator in acute myeloid leukemia. Int J. Hematol. 2017, 105, 453–464. [Google Scholar] [CrossRef] [PubMed]
- Behl, D.; Porrata, L.F.; Markovic, S.N.; Letendre, L.; Pruthi, R.K.; Hook, C.C.; Tefferi, A.; Elliot, M.A.; Kaufmann, S.H.; Mesa, R.A.; et al. Absolute lymphocyte count recovery after induction chemotherapy predicts superior survival in acute myelogenous leukemia. Leukemia 2006, 20, 29–34. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Munger, M.E.; Veenstra, R.G.; Weigel, B.J.; Hirashima, M.; Munn, D.H.; Murphy, W.J.; Azuma, M.; Anderson, A.C.; Kuchroo, V.K.; et al. Coexpression of Tim-3 and PD-1 identifies a CD8+ T-cell exhaustion phenotype in mice with disseminated acute myelogenous leukemia. Blood 2011, 117, 4501–4510. [Google Scholar] [CrossRef]
- Knaus, H.A.; Berglund, S.; Hackl, H.; Blackford, A.L.; Zeidner, J.F.; Montiel-Esparza, R.; Mukhopadhyay, R.; Vanura, K.; Blazar, B.R.; Karp, J.E.; et al. Signatures of CD8+ T cell dysfunction in AML patients and their reversibility with response to chemotherapy. JCI Insight 2018, 3, e120974. [Google Scholar] [CrossRef]
- Kikushige, Y.; Shima, T.; Takayanagi, S.; Urata, S.; Miyamoto, T.; Iwasaki, H.; Takenaka, K.; Teshima, T.; Tanaka, T.; Inagaki, Y.; et al. TIM-3 is a promising target to selectively kill acute myeloid leukemia stem cells. Cell Stem Cell 2010, 7, 708–717. [Google Scholar] [CrossRef]
- Radwan, S.M.; Elleboudy, N.S.; Nabih, N.A.; Kamal, A.M. The immune checkpoints Cytotoxic T lymphocyte antigen-4 and Lymphocyte activation gene-3 expression is up-regulated in acute myeloid leukemia. Hla 2020, 96, 3–12. [Google Scholar] [CrossRef]
- Schnorfeil, F.M.; Lichtenegger, F.S.; Emmerig, K.; Schlueter, M.; Neitz, J.S.; Draenert, R.; Hiddemann, W.; Subklewe, M. T cells are functionally not impaired in AML: Increased PD-1 expression is only seen at time of relapse and correlates with a shift towards the memory T cell compartment. J. Hematol. Oncol. 2015, 8, 93. [Google Scholar] [CrossRef]
- Li, Z.; Philip, M.; Ferrell, P.B. Alterations of T-cell-mediated immunity in acute myeloid leukemia. Oncogene 2020, 39, 3611–3619. [Google Scholar] [CrossRef]
- Jia, B.; Wang, L.; Claxton, D.F.; Ehmann, W.C.; Rybka, W.B.; Mineishi, S.; Rizvi, S.; Shike, H.; Bayerl, M.; Schell, T.D.; et al. Bone marrow CD8 T cells express high frequency of PD-1 and exhibit reduced anti-leukemia response in newly diagnosed AML patients. Blood Cancer J. 2018, 8, 34. [Google Scholar] [CrossRef]
- Paley, M.A.; Kroy, D.C.; Odorizzi, P.M.; Johnnidis, J.B.; Dolfi, D.V.; Barnett, B.E.; Bikoff, E.K.; Robertson, E.J.; Lauer, G.M.; Reiner, S.L.; et al. Progenitor and terminal subsets of CD8+ T cells cooperate to contain chronic viral infection. Science 2012, 338, 1220–1225. [Google Scholar] [CrossRef] [PubMed]
- Jia, B.; Zhao, C.; Rakszawski, K.L.; Claxton, D.F.; Ehmann, W.C.; Rybka, W.B.; Mineishi, S.; Wang, M.; Shike, H.; Bayerl, M.G.; et al. Eomes(+)T-bet(low) CD8(+) T Cells Are Functionally Impaired and Are Associated with Poor Clinical Outcome in Patients with Acute Myeloid Leukemia. Cancer Res. 2019, 79, 1635–1645. [Google Scholar] [CrossRef] [PubMed]
- Radpour, R.; Riether, C.; Simillion, C.; Hopner, S.; Bruggmann, R.; Ochsenbein, A.F. CD8(+) T cells expand stem and progenitor cells in favorable but not adverse risk acute myeloid leukemia. Leukemia 2019, 33, 2379–2392. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Wang, S.; Wang, F.; Chen, Q.; Peng, S.; Zhang, Y.; Qian, J.; Jin, J.; Xu, H. Increased frequencies of T helper type 17 cells in the peripheral blood of patients with acute myeloid leukaemia. Clin. Exp. Immunol. 2009, 158, 199–204. [Google Scholar] [CrossRef]
- Shenghui, Z.; Yixiang, H.; Jianbo, W.; Kang, Y.; Laixi, B.; Yan, Z.; Xi, X. Elevated frequencies of CD4(+) CD25(+) CD127lo regulatory T cells is associated to poor prognosis in patients with acute myeloid leukemia. Int J. Cancer 2011, 129, 1373–1381. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Dong, Y.; Yang, Q.; Xu, W.; Jiang, S.; Yu, Z.; Yu, K.; Zhang, S. Acute Myeloid Leukemia Cells Express ICOS Ligand to Promote the Expansion of Regulatory T Cells. Front. Immunol. 2018, 9, 2227. [Google Scholar] [CrossRef]
- Zou, L.; Barnett, B.; Safah, H.; Larussa, V.F.; Evdemon-Hogan, M.; Mottram, P.; Wei, S.; David, O.; Curiel, T.J.; Zou, W. Bone marrow is a reservoir for CD4+CD25+ regulatory T cells that traffic through CXCL12/CXCR4 signals. Cancer Res. 2004, 64, 8451–8455. [Google Scholar] [CrossRef]
- Lichtenegger, F.S.; Lorenz, R.; Gellhaus, K.; Hiddemann, W.; Beck, B.; Subklewe, M. Impaired NK cells and increased T regulatory cell numbers during cytotoxic maintenance therapy in AML. Leuk Res. 2014, 38, 964–969. [Google Scholar] [CrossRef]
- Ersvaer, E.; Liseth, K.; Skavland, J.; Gjertsen, B.T.; Bruserud, O. Intensive chemotherapy for acute myeloid leukemia differentially affects circulating TC1, TH1, TH17 and TREG cells. BMC Immunol. 2010, 11, 38. [Google Scholar] [CrossRef]
- Menter, T.; Kuzmanic, B.; Bucher, C.; Medinger, M.; Halter, J.; Dirnhofer, S.; Tzankov, A. Beneficial role of increased FOXP3(+) regulatory T-cells in acute myeloid leukaemia therapy response. Br. J. Haematol. 2018, 182, 581–583. [Google Scholar] [CrossRef]
- Derolf, A.R.; Laane, E.; Bjorklund, E.; Saft, L.; Bjorkholm, M.; Porwit, A. Dendritic cells in bone marrow at diagnosis and after chemotherapy in adult patients with acute myeloid leukaemia. Scand. J. Immunol. 2014, 80, 424–431. [Google Scholar] [CrossRef] [PubMed]
- Xiao, W.; Goldberg, A.D.; Famulare, C.A.; Devlin, S.M.; Nguyen, N.T.; Sim, S.; Kabel, C.C.; Patel, M.A.; McGovern, E.M.; Patel, A.; et al. Loss of plasmacytoid dendritic cell differentiation is highly predictive for post-induction measurable residual disease and inferior outcomes in acute myeloid leukemia. Haematologica 2019, 104, 1378–1387. [Google Scholar] [CrossRef] [PubMed]
- Fatehchand, K.; Mehta, P.; Colvin, C.B.; Buteyn, N.J.; Santhanam, R.; Merchand-Reyes, G.; Inshaar, H.; Shen, B.; Mo, X.; Mundy-Bosse, B.; et al. Activation of plasmacytoid dendritic cells promotes AML-cell fratricide. Oncotarget 2021, 12, 878–890. [Google Scholar] [CrossRef] [PubMed]
- Xiao, W.; Chan, A.; Waarts, M.R.; Mishra, T.; Liu, Y.; Cai, S.F.; Yao, J.; Gao, Q.; Bowman, R.L.; Koche, R.P.; et al. Plasmacytoid dendritic cell expansion defines a distinct subset of RUNX1-mutated acute myeloid leukemia. Blood 2021, 137, 1377–1391. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Feng, Y.; Deng, X.; Liu, S.; Qiang, X.; Gou, Y.; Li, J.; Yang, W.; Peng, X.; Zhang, X. Tumor-forming plasmacytoid dendritic cells in acute myelocytic leukemia: A report of three cases and literature review. Int. J. Clin. Exp. Pathol. 2017, 10, 7285–7291. [Google Scholar] [PubMed]
- Szczepanski, M.J.; Szajnik, M.; Welsh, A.; Whiteside, T.L.; Boyiadzis, M. Blast-derived microvesicles in sera from patients with acute myeloid leukemia suppress natural killer cell function via membrane-associated transforming growth factor-beta1. Haematologica 2011, 96, 1302–1309. [Google Scholar] [CrossRef]
- Ghiringhelli, F.; Menard, C.; Terme, M.; Flament, C.; Taieb, J.; Chaput, N.; Puig, P.E.; Novault, S.; Escudier, B.; Vivier, E.; et al. CD4+CD25+ regulatory T cells inhibit natural killer cell functions in a transforming growth factor-beta-dependent manner. J. Exp. Med. 2005, 202, 1075–1085. [Google Scholar] [CrossRef]
- Costello, R.T.; Sivori, S.; Marcenaro, E.; Lafage-Pochitaloff, M.; Mozziconacci, M.J.; Reviron, D.; Gastaut, J.A.; Pende, D.; Olive, D.; Moretta, A. Defective expression and function of natural killer cell-triggering receptors in patients with acute myeloid leukemia. Blood 2002, 99, 3661–3667. [Google Scholar] [CrossRef]
- Stringaris, K.; Sekine, T.; Khoder, A.; Alsuliman, A.; Razzaghi, B.; Sargeant, R.; Pavlu, J.; Brisley, G.; de Lavallade, H.; Sarvaria, A.; et al. Leukemia-induced phenotypic and functional defects in natural killer cells predict failure to achieve remission in acute myeloid leukemia. Haematologica 2014, 99, 836–847. [Google Scholar] [CrossRef]
- Nowbakht, P.; Ionescu, M.C.; Rohner, A.; Kalberer, C.P.; Rossy, E.; Mori, L.; Cosman, D.; De Libero, G.; Wodnar-Filipowicz, A. Ligands for natural killer cell-activating receptors are expressed upon the maturation of normal myelomonocytic cells but at low levels in acute myeloid leukemias. Blood 2005, 105, 3615–3622. [Google Scholar] [CrossRef]
- Della Chiesa, M.; Carlomagno, S.; Frumento, G.; Balsamo, M.; Cantoni, C.; Conte, R.; Moretta, L.; Moretta, A.; Vitale, M. The tryptophan catabolite L-kynurenine inhibits the surface expression of NKp46- and NKG2D-activating receptors and regulates NK-cell function. Blood 2006, 108, 4118–4125. [Google Scholar] [CrossRef] [PubMed]
- Baragano Raneros, A.; Martin-Palanco, V.; Fernandez, A.F.; Rodriguez, R.M.; Fraga, M.F.; Lopez-Larrea, C.; Suarez-Alvarez, B. Methylation of NKG2D ligands contributes to immune system evasion in acute myeloid leukemia. Genes Immun. 2015, 16, 71–82. [Google Scholar] [CrossRef] [PubMed]
- Paczulla, A.M.; Rothfelder, K.; Raffel, S.; Konantz, M.; Steinbacher, J.; Wang, H.; Tandler, C.; Mbarga, M.; Schaefer, T.; Falcone, M.; et al. Absence of NKG2D ligands defines leukaemia stem cells and mediates their immune evasion. Nature 2019, 572, 254–259. [Google Scholar] [CrossRef] [PubMed]
- Elias, S.; Yamin, R.; Golomb, L.; Tsukerman, P.; Stanietsky-Kaynan, N.; Ben-Yehuda, D.; Mandelboim, O. Immune evasion by oncogenic proteins of acute myeloid leukemia. Blood 2014, 123, 1535–1543. [Google Scholar] [CrossRef]
- Lehmann, C.; Zeis, M.; Schmitz, N.; Uharek, L. Impaired binding of perforin on the surface of tumor cells is a cause of target cell resistance against cytotoxic effector cells. Blood 2000, 96, 594–600. [Google Scholar] [CrossRef] [PubMed]
- Pyzer, A.R.; Stroopinsky, D.; Rajabi, H.; Washington, A.; Tagde, A.; Coll, M.; Fung, J.; Bryant, M.P.; Cole, L.; Palmer, K.; et al. MUC1-mediated induction of myeloid-derived suppressor cells in patients with acute myeloid leukemia. Blood 2017, 129, 1791–1801. [Google Scholar] [CrossRef] [PubMed]
- Tohumeken, S.; Baur, R.; Bottcher, M.; Stoll, A.; Loschinski, R.; Panagiotidis, K.; Braun, M.; Saul, D.; Volkl, S.; Baur, A.S.; et al. Palmitoylated Proteins on AML-Derived Extracellular Vesicles Promote Myeloid-Derived Suppressor Cell Differentiation via TLR2/Akt/mTOR Signaling. Cancer Res. 2020, 80, 3663–3676. [Google Scholar] [CrossRef]
- Yang, Y.; Li, C.; Liu, T.; Dai, X.; Bazhin, A.V. Myeloid-Derived Suppressor Cells in Tumors: From Mechanisms to Antigen Specificity and Microenvironmental Regulation. Front. Immunol. 2020, 11, 1371. [Google Scholar] [CrossRef]
- Miari, K.E.; Guzman, M.L.; Wheadon, H.; Williams, M.T.S. Macrophages in Acute Myeloid Leukaemia: Significant Players in Therapy Resistance and Patient Outcomes. Front. Cell Dev. Biol. 2021, 9, 692800. [Google Scholar] [CrossRef]
- Jiang, M.; Zhang, J.; Qian, L.; Miao, Y.; Song, W.; Liu, H.; Li, R. MOZ Forms an Autoregulatory Feedback Loop with miR-223 in AML and Monocyte/Macrophage Development. iScience 2019, 11, 189–204. [Google Scholar] [CrossRef]
- Al-Matary, Y.S.; Botezatu, L.; Opalka, B.; Hones, J.M.; Lams, R.F.; Thivakaran, A.; Schutte, J.; Koster, R.; Lennartz, K.; Schroeder, T.; et al. Acute myeloid leukemia cells polarize macrophages towards a leukemia supporting state in a Growth factor independence 1 dependent manner. Haematologica 2016, 101, 1216–1227. [Google Scholar] [CrossRef] [PubMed]
- Mansour, I.; Zayed, R.A.; Said, F.; Latif, L.A. Indoleamine 2,3-dioxygenase and regulatory T cells in acute myeloid leukemia. Hematology 2016, 21, 447–453. [Google Scholar] [CrossRef] [PubMed]
- Vasold, J.; Wagner, M.; Drolle, H.; Deniffel, C.; Kutt, A.; Oostendorp, R.; Sironi, S.; Rieger, C.; Fiegl, M. The bone marrow microenvironment is a critical player in the NK cell response against acute myeloid leukaemia in vitro. Leuk. Res. 2015, 39, 257–262. [Google Scholar] [CrossRef]
- Lu, Y.; Liu, J.; Liu, Y.; Qin, Y.; Luo, Q.; Wang, Q.; Duan, H. TLR4 plays a crucial role in MSC-induced inhibition of NK cell function. Biochem. Biophys. Res. Commun. 2015, 464, 541–547. [Google Scholar] [CrossRef] [PubMed]
- Wang, A.; Zhong, H. Roles of the bone marrow niche in hematopoiesis, leukemogenesis, and chemotherapy resistance in acute myeloid leukemia. Hematology 2018, 23, 729–739. [Google Scholar] [CrossRef]
- Vijay, V.; Miller, R.; Vue, G.S.; Pezeshkian, M.B.; Maywood, M.; Ast, A.M.; Drusbosky, L.M.; Pompeu, Y.; Salgado, A.D.; Lipten, S.D.; et al. Interleukin-8 blockade prevents activated endothelial cell mediated proliferation and chemoresistance of acute myeloid leukemia. Leuk. Res. 2019, 84, 106180. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; de Haan, G. Inflammation and Aging of Hematopoietic Stem Cells in Their Niche. Cells 2021, 10, 1849. [Google Scholar] [CrossRef]
- Guo, R.; Lu, M.; Cao, F.; Wu, G.; Gao, F.; Pang, H.; Li, Y.; Zhang, Y.; Xing, H.; Liang, C.; et al. Single-cell map of diverse immune phenotypes in the acute myeloid leukemia microenvironment. Biomark. Res. 2021, 9, 15. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Low-Risk MDS | High-Risk MDS | AML | |
|---|---|---|---|
| Cell intrinsic factors | Increased NLRP3-inflammasome activation and Pyroptosis Apoptosis induction | NLRP3 inflammasome activation | Downregulation of mismatched HLAs, class II HLAs (in alloHSCT) Impaired antigen presentation |
| Cytokines/Metabolites | Increased proinflammatory cytokine production (TNFa, IL-1β, etc.) | Immunosuppressive cytokine production (IL-10) | Increased Il-1β/ΙL-6/TNFa/IL-35 Decreased TGFβ Increased immunosuppressive metabolic factors (IDO1, ArgII, iNOS, FAO, etc.) Vascular remodeling with immunosuppressive sequelae |
| T-lymphocytes | Increased CD8+ lymphocytes with autoreactive potential Increased Th17 helper cell numbers Decreased Treg number | Diminished and dysfunctional CD8+ lymphocytes Increased immune checkpoint molecule expression Increased Th22 helper cells Tregs with increased number and functionality | Reduced CD8+ lymphocyte function/Increased immune checkpoint molecule expression Increased Treg numbers with immunosuppressive properties |
| NK-lymphocytes | Decreased NK-cell numbers with reduced function | NK-cell impairement | |
| MDSCs | MDSC expansion | MDSC expansion correlating with Treg increase | MDSC expansion |
| Macrophages | M2 macrophage expansion INOS+ subtype increase | M2 macrophage expansion | M2 macrophage expansion |
| MSCs | Reduced differentiation potential | Increased with immunosuppressive properties | Increased immunosuppressive behavior |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barakos, G.P.; Hatzimichael, E. Microenvironmental Features Driving Immune Evasion in Myelodysplastic Syndromes and Acute Myeloid Leukemia. Diseases 2022, 10, 33. https://doi.org/10.3390/diseases10020033
Barakos GP, Hatzimichael E. Microenvironmental Features Driving Immune Evasion in Myelodysplastic Syndromes and Acute Myeloid Leukemia. Diseases. 2022; 10(2):33. https://doi.org/10.3390/diseases10020033
Chicago/Turabian StyleBarakos, Georgios Petros, and Eleftheria Hatzimichael. 2022. "Microenvironmental Features Driving Immune Evasion in Myelodysplastic Syndromes and Acute Myeloid Leukemia" Diseases 10, no. 2: 33. https://doi.org/10.3390/diseases10020033
APA StyleBarakos, G. P., & Hatzimichael, E. (2022). Microenvironmental Features Driving Immune Evasion in Myelodysplastic Syndromes and Acute Myeloid Leukemia. Diseases, 10(2), 33. https://doi.org/10.3390/diseases10020033