
When a Multidisciplinary Approach Is Life-Saving: A Case Report of Cardiogenic Shock Induced by a Large Pheochromocytoma

 , ,
, ,
Abstract
1. Introduction
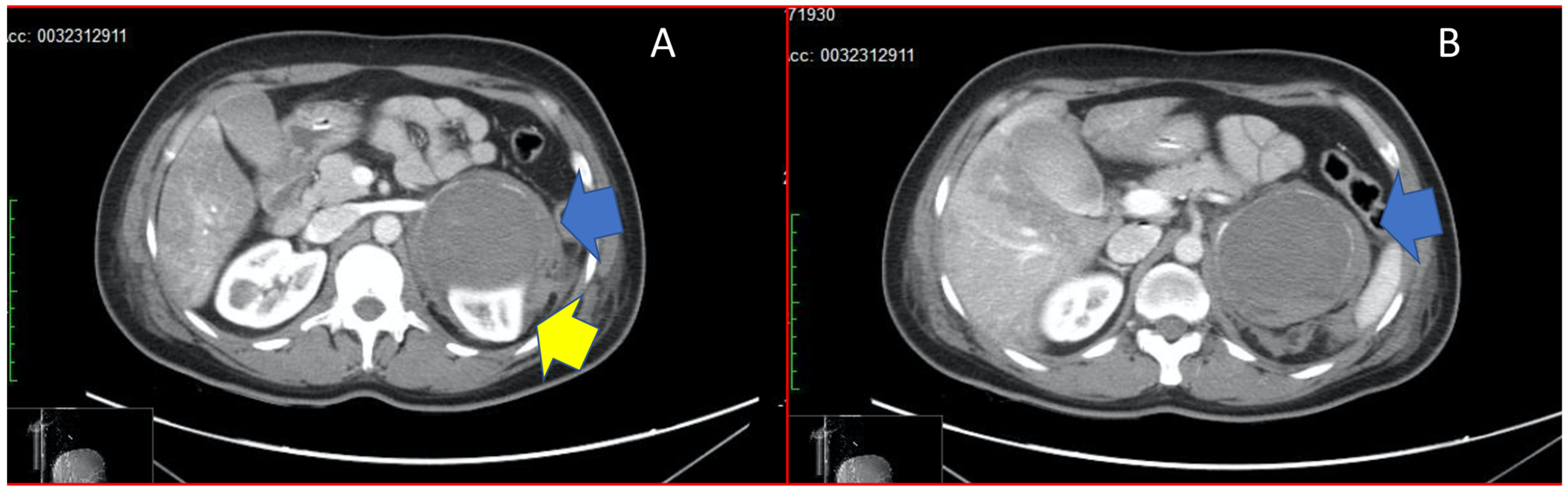
2. Case Report
- -
- Kinetic alterations (anterolateral akinesia and hypokinesia of the apex);
- -
- A moderate depression of the ejection fraction (about 40%);
- -
- Ascending aorta of normal size;
- -
- Absence of significant valvulopathies;
- -
- No evidence of pericardial effusion.
3. Discussion and Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Baguet, J.P.; Hammer, L.; Mazzuco, T.L.; Chabre, O.; Mallion, J.M.; Sturm, N.; Chaffanjon, P. Circumstances of discovery of phaeochromocytoma: A retrospective study of 41 consecutive patients. Eur. J. Endocrinol. 2004, 150, 681. [Google Scholar] [CrossRef] [PubMed]
- Kakoki, K.; Miyata, Y.; Shida, Y.; Hakariya, T.; Takehara, K.; Izumida, S.; Sekino, M.; Kinoshita, N.; Igawa, T.; Fukuoka, J.; et al. Pheochromocytoma multisystem crisis treated with emergency surgery: A case report and literature review. BMC Res. Notes 2015, 8, 758. [Google Scholar] [CrossRef] [PubMed]
- Carter, Y.M.; Mazeh, H.; Sippel, R.S.; Chen, H. Safety and feasibility of laparoscopic resection for large (≥ 6 CM) pheochromocytomas without suspected malignancy. Endocr. Pract. 2012, 18, 720–726. [Google Scholar] [CrossRef]
- Natkaniec, M.; Pędziwiatr, M.; Wierdakm, M.; Białasm, M.; Major, P.; Matłok, M.; Budzyński, P.; Dworak, J.; Buziak-Bereza, M.; Budzyński, A. Laparoscopic adrenalectomy for pheochromocytoma is more difficult compared to other adrenal tumors. Wideochir. Inne Tech. Maloinwazyjne 2015, 10, 466–471. [Google Scholar] [CrossRef] [PubMed]
- Gagner, M.; Lacroix, A.; Bolte, E. Laparoscopic adrenalectomy in Cushing’s syndrome and pheochromocytoma. N. Engl. J. Med. 1992, 327, 1033–1036. [Google Scholar] [PubMed]
- Gill, I.S. The case for laparoscopic adrenalectomy. J. Urol. 2001, 166, 429–436. [Google Scholar] [CrossRef]
- Winfield, H.N.; Hamilton, B.D.; Bravo, E.L.; Novick, A.C. Laparoscopic adrenalectomy: The preferred choice? A comparison to open adrenalectomy. J. Urol. 1998, 160, 325–329. [Google Scholar] [CrossRef]
- Gill, I.S.; Hobart, M.G.; Schweizer, D.; Bravo, E.L. Outpatient adrenalectomy. J. Urol. 2000, 163, 717–719. [Google Scholar] [CrossRef]
- Gagner, M.; Pomp, A.; Heniford, B.T.; Pharand, D.; Lacroix, A. Laparoscopic adrenalectomy: Lessons learned from 100 consecutive procedures. Ann. Surg. 1997, 226, 238–247. [Google Scholar] [CrossRef]
- Hazzan, D.; Shiloni, E.; Goliljanin, D. Laparoscopic vs open adrenalectomy for benign adrenal neoplasm: A comparative study. Surg. Endosc. 2001, 15, 1356–1358. [Google Scholar] [CrossRef]
- Brunt, L.M.; Doherty, G.M.; Norton, J.A. Laparoscopic adrenalectomy compared to open adrenalectomy for benign adrenal neoplasms. J. Am. Coll. Surg. 1996, 183, 1–10. [Google Scholar] [PubMed]
- Korman, J.E.; Ho, T.; Hiatt, J.R. Comparison of laparoscopic and open adrenalectomy. Am. Surg. 1997, 63, 908–912. [Google Scholar] [PubMed]
- Barresi, R.V.; Prinz, R.A. Laparoscopic adrenalectomy. Arch. Surg. 1999, 143, 212–217. [Google Scholar] [CrossRef]
- Hobart, M.G.; Gill, I.S.; Schweizer, D.; Sung, G.T.; Bravo, E.L. Laparoscopic adrenalectomy for large-volume (>/= 5 cm) adrenal masses. J. Endourol. 2000, 14, 149–154. [Google Scholar] [CrossRef] [PubMed]
- Novitsky, Y.W.; Czerniach, D.R.; Kercher, K.W.; Perugini, R.A.; Kelly, J.J.; Litwin, D.E.M. Feasibility of laparoscopic adrenalectomy for large adrenal masses. Surg. Laparosc. Endosc. Percutaneous Tech. 2003, 13, 106–110. [Google Scholar] [CrossRef]
- Bausch, B.; Boedeker, C.C.; Berlis, A.; Brink, I.; Cybulla, M.; Walz, M.K.; Januszewicz, A.; Letizia, C.; Opocher, G.; Eng, C.; et al. Genetic and clinical investigation of pheochromocytoma: A 22-year experience, from Freiburg, Germany to international effort. Ann. N. Y. Acad. Sci. 2006, 1073, 122–137. [Google Scholar] [CrossRef]
- Mantero, F.; Terzolo, M.; Arnaldi, G.; Osella, G.; Masini, A.M.; Alì, A.; Giovagnetti, M.; Opocher, G.; Angeli, A. A survey on adrenal incidentaloma in Italy. Study Group on Adrenal Tumors of the Italian Society of Endocrinology. J. Clin. Endocrinol. Metab. 2000, 85, 637–644. [Google Scholar]
- Sheps, S.G.; Jiang, N.S.; Klee, G.G.; Van Heerden, J.A. Mayo: Recent developments in the diagnosis and treatment of pheochromocytoma. Clin. Proc. 1990, 65, 88–95. [Google Scholar] [CrossRef]
- Neumann, H.P.; Bausch, B.; McWhinney, S.R.; Bender, B.U.; Gimm, O.; Franke, G.; Franke, G.; Schipper, J.; Klisch, J.; Altehoefer, C.; et al. Germ–line mutations in nonsyndromic pheochromocytoma. N. Engl. J. Med. 2002, 346, 1459–1466. [Google Scholar] [CrossRef]
- Toledo, S.P.; dos Santos, M.A.; Toledo Rde, A.; Delmar, M.L., Jr. Impact of RET protooncogene analysis on the clinical management of multiple endocrine neoplasia type 2. Clinics 2006, 61, 59–70. [Google Scholar] [CrossRef]
- Maher, E.R.; Yates, J.R.; Harries, R.; Benjamin, C.; Harris, R.; Moore, A.T.; Ferguson-Smith, M.A. Clinical features and natural history of von Hippel-Lindau disease. Q. J. Med. 1990, 77, 1151–1163. [Google Scholar] [CrossRef] [PubMed]
- Walther, M.M.; Herring, J.; Enquist, E.; Keiser, H.R.; Linehan, W.M. von Recklinghausen’s disease and pheochromocytomas. J. Urol. 1999, 162, 1582–1586. [Google Scholar] [CrossRef]
- Eng, C.; Kiuru, M.; Fernandez, M.J.; Aaltonen, L.A. A role for mitochondrial enzymes in inherited neoplasia and beyond. Nat. Rev. Cancer 2003, 3, 193–202. [Google Scholar] [CrossRef] [PubMed]
- Gimenez-Roqueplo, A.P.; Favier, J.; Rustin, P.; Kerlan, V.; Plouin, P.; Rötig, A.; Jeunemaitre, X. Functional consequences of a SDHB gene mutation in an apparently sporadic pheochromocytoma. J. Clin. Endocrinol. Metab. 2002, 87, 4771–4774. [Google Scholar] [CrossRef]
- Burnichon, N.; Rohmer, V.; Amar, L.; Herman, P.; Leboulleux, S.; Darrouzet, V.; Niccoli, P.; Gaillard, D.; Chabrier, G.; PGL.NET network; et al. The succinate dehydrogenase genetic testing in a large prospective series of patients with paragangliomas. J. Clin. Endocrinol. Metab. 2009, 94, 2817–2827. [Google Scholar] [CrossRef] [PubMed]
- Motta–Ramirez, G.A.; Remer, E.M.; Herts, B.R.; Gill, I.S.; Hamrahian, A.H. Comparison of CT findings in symptomatic and incidentally discovered pheochromocytomas. Am. J. Roentgenol. 2005, 185, 684. [Google Scholar] [CrossRef] [PubMed]
- Lenders, J.; Eisenhofer, G.; Mannelli, M.; Pacak, K. Phaeochromocytoma. Lancet 2005, 366, 665–675. [Google Scholar] [CrossRef]
- Kobayashi, T.; Iwai, A.; Takahashi, R.; Ide, Y.; Nishizawa, K.; Mitsumori, K. Spontaneous rupture of adrenal pheochromocytoma: Review and analysis of prognostic factors. J. Surg. Oncol. 2005, 90, 31–35. [Google Scholar] [CrossRef]
- Hasson, J.; Monges, G.; Giraud, P.; Henry, J.F.; Charpin, C.; Payan, H.; Toga, M. Immunohistochemical study of pheochromocytomas. An investigation of methionine- enkephalin vasoactive intestinal peptide, somatostatin, corticotropin, b- endorphin and calcitonin in 16 cases. Am. J. Pathol. 1914, 14, 56–63. [Google Scholar]
- Heath, H.; Edis, A.J. Pheochromocytoma associated with hypercalcaemia and ectopic secretion of calcitonin. Ann. Int. Med. 1979, 91, 208–210. [Google Scholar] [CrossRef]
- Wood, R.; Commerford, P.J.; Rose, A.G.; Tooke, A. Reversible catecholamine induced cardiomyopathy. Am. Heart J. 1991, 121, 610–613. [Google Scholar] [CrossRef]
- Mohamed, H.A.; Aldakar, M.O.; Habib, N. Cardiogenic shock due to acute hemorrhagic necrosis of a pheochromocytoma: A case report and review of the literature. Can. J. Cardiol. 2003, 19, 573–576. [Google Scholar] [PubMed]
- Haas, G.J.; Tzagournis, M.; Boudoulas, H. Pheochromocytoma: Catecholamine mediated electrocardiographic changes mimicking ischaemia. Am. Heart J. 1988, 116, 1363–1365. [Google Scholar] [CrossRef]
- Connolly, D.L.; Mariathas, D.A. Phaeochromocytoma presenting acutely as severe cardiac failure. J. Accid. Emerg. Med. 1994, 11, 125–126. [Google Scholar] [CrossRef] [PubMed]
- Schurmeyer, T.H.; Engeroff, B.; Dralle, E.; von zur Muhlen, A. Cardiological effects of catecholamine-secreting tumors. Eur. J. Clin. Investig. 2005, 27, 189–195. [Google Scholar] [CrossRef]
- Liao, W.B.; Liu, C.F.; Chiang, C.W.; Kung, C.T.; Lee, C.W. Cardiovascular manifestations of pheochromocytoma. Am. J. Emerg. Med. 2000, 18, 622–625. [Google Scholar] [CrossRef]
- Brouwers, F.M.; Lenders, J.W.; Eisenhofer, G.; Pacak, K. Pheochromocytoma as an endocrine emergency. Rev. Endocr. Metab. Disord. 2003, 4, 121–128. [Google Scholar] [CrossRef]
- Lenders, J.W.; Keiser, H.R.; Goldstein, D.S.; Willemsen, J.J.; Friberg, P.; Jacobs, M.C.; Kloppenborg, P.W.; Thien, T.; Eisenhofer, G. Plasma metanephrines in the diagnosis of pheochromocytoma. Ann. Intern. Med. 1995, 123, 101–109. [Google Scholar] [CrossRef]
- Heron, E.; Chatellier, G.; Billaud, E.; Foos, E.; Plouin, P.F. The urinary metanephrine-to-creatinine ratio for the diagnosis of pheochromocytoma. Ann. Intern. Med. 1996, 125, 300–303. [Google Scholar] [CrossRef]
- Lenders, J.W.; Pacak, K.; Walther, M.M.; Linehan, W.M.; Mannelli, M.; Friberg, P.; Kaiser, H.R.; Goldstein, D.S.; Eisenhofer, G. Biochemical diagnosis of pheochromocytoma: Which test is best? JAMA 2002, 287, 1427–1434. [Google Scholar] [CrossRef]
- Eisenhofer, G.; Keiser, H.; Friberg, P.; Mezey, E.; Huynh, T.T.; Hiremagalur, B.; Ellingson, T.; Duddempudi, S.; Eijsbouts, A.; Lenders, J.W. Plasma metanephrines are markers of pheochromocytoma produced by catechol-O-methyltransferase within tumors. J. Clin. Endocrinol. Metab. 1998, 83, 2175–2185. [Google Scholar] [CrossRef]
- Lawrance, A.M. Glucagon provocative test for pheochromocytoma. Ann. Int. Med. 1967, 66, 1091–1096. [Google Scholar] [CrossRef] [PubMed]
- Bravo, E.L.; Tarazi, R.C.; Fouad, F.M.; Vidt, D.G.; Gifford, R.W., Jr. Clonidine suppression test: A useful aid in the diagnosis of pheochromocytoma. N. Engl. J. Med. 1981, 305, 623–626. [Google Scholar] [CrossRef] [PubMed]
- Niemann, U.; Hiller, W.; Behrend, M. 25 years experience of the surgical treatment of phaeochromocytoma. Eur. J. Surg. 2002, 168, 716–719. [Google Scholar] [CrossRef] [PubMed]
- Castillo, O.A.; Vitagliano, G.; Secin, F.P.; Kerkebe, M.; Arellano, L. Laparoscopic adrenalectomy for adrenal masses: Does size matter? Urology 2008, 71, 1138–1141. [Google Scholar] [CrossRef]
- Feo, C.V.; Portinari, M.; Maestroni, U.; Del Rio, P.; Severi, S.; Viani, L.; Pravisani, R.; Soliani, G.; Zatelli, M.C.; Ambrosio, M.R.; et al. Applicability of laparoscopic approach to the resection of large adrenal tumours: A retrospective cohort study on 200 patients. Surg. Endosc. 2016, 30, 3532–3540. [Google Scholar] [CrossRef]
- Maestroni, U.; Vicente, D.; Del Rio, P.; Ziglioli, F.; Dinale, F.; Campobasso, D.; Ferretti, S.; Stojadinovic, A.; Avital, I. Laparoscopic adrenalectomy for large adrenal masses: A challenge or a routine? Minerva Chir. 2014, 69, 59–64. [Google Scholar]
- Walz, M.K.; Peitgen, K.; Neumann, H.P.; Janssen, O.E.; Philipp, T.; Mann, K. Endoscopic treatment of solitary, bilateral, multiple, and recurrent pheochromocytomas and paragangliomas. World J. Surg. 2002, 26, 1005–1012. [Google Scholar] [CrossRef]
- Jaroszewski, D.E.; Tessier, D.J.; Schlinkert, R.T. Laparoscopic adrenalectomy for pheochromocytoma. Mayo Clin. Proc. 2003, 78, 1501–1504. [Google Scholar] [CrossRef]
- Cheah, W.K.; Clark, O.H.; Horn, J.K.; Siperstein, A.E.; Duh, Q.Y. Laparoscopic adrenalectomy for pheochromocytoma. World J. Surg. 2002, 26, 1048–1051. [Google Scholar] [CrossRef]
- Lenders, J.W.; Duh, Q.Y.; Eisenhofer, G.; Gimenez–Roqueplo, A.P.; Grebe, S.K.; Murad, M.H.; Naruse, M.; Pacak, K.; Young, W.F., Jr.; Endocrine Society. Pheochromocytoma and paraganglioma: An endocrine society clinical practice guideline. J. Clin. Endocrinol. Metab. 2014, 99, 1915–1942. [Google Scholar] [CrossRef]
- de Fourmestraux, A.; Salomon, L.; Abbou, C.C.; Grise, P. Ten year experience of retroperitoneal laparoscopic resection for pheochromocytomas: A dual–Centre study of 72 cases. World J. Urol. 2015, 33, 1103–1107. [Google Scholar] [CrossRef]
- Naya, Y.; Suzuki, H.; Komiya, A.; Nagata, M.; Tobe, T.; Ueda, T. Laparoscopic adrenalectomy in patients with large adrenal tumors. Int. J. Urol. 2005, 12, 134–139. [Google Scholar] [CrossRef]
- Shen, W.T.; Sturgeon, C.; Clark, O.H.; Duh, Q.Y.; Kebebew, E. Should pheochromocytoma size influence surgical approach? A comparison of 90 malignant and 60 benign pheochromocytomas. Surgery 2004, 136, 1129–1137. [Google Scholar] [CrossRef] [PubMed]
- Conzo, G.; Musella, M.; Corcione, F.; De Palma, M.; Avenia, N.; Milone, M.; della Pietra, C.; Palazzo, A.; Parmeggiani, D.; Pasquali, D.; et al. Laparoscopic treatment of pheochromocytomas smaller or larger than 6 cm. A clinical retrospective study on 44 patients. Laparoscopic adrenalectomy for pheochromocytoma. Ann. Ital. Chir. 2013, 84, 417–422. [Google Scholar] [PubMed]
- Kasahara, T.; Nishiyama, T.; Takahashi, K. Laparoscopic adrenalectomy for pheochromocytoma: Evaluation of experience and strategy at a single institute. BJU Int. 2009, 103, 218–222. [Google Scholar] [CrossRef] [PubMed]
- Kercher, K.W.; Park, A.; Matthews, B.D.; Rolband, G.; Sing, R.F.; Heniford, B.T. Laparoscopic adrenalectomy for pheochromocytoma. Surg. Endosc. 2002, 16, 100–102. [Google Scholar] [CrossRef]
- Newell, K.A. Pheochromocytoma multisystem crisis: A surgical emergency. Arch. Surg. 1988, 123, 956–959. [Google Scholar] [CrossRef]
- Solorzano, C.C. Pheocromocytoma presenting with multiple organ failure. Am. Surg. 2008, 74, 1119–1121. [Google Scholar] [CrossRef]
- Quezado, Z.N.; Keiser, H.R.; Parker, M.M. Reversible myocardial depression after massive catecholamine release from a pheochromocytoma. Crit. Care Med. 1992, 20, 549–551. [Google Scholar] [CrossRef]
- Gil–Barrionuevo, E.; Balibrea, J.M.; Caubet, E.; Gonzalez, O.; Vilallonga, R.; Fort, J.M.; Ciudin, A.; Armengol, M. Adrenergic cardiomyopathy and cardiogenic shock as initial presentation of pheochromocytoma. A case report and review of the literature. Int. J. Surg. Case Rep. 2018, 49, 145–148. [Google Scholar] [CrossRef] [PubMed]
- Y-Hassan, S. Clinical features and outcome of Pheochromocytoma–induced Takotsubo syndrome: Analysis of 80 published cases. Am. J. Cardiol. 2016, 117, 1836–1844. [Google Scholar] [CrossRef] [PubMed]
- Templin, C.; Ghadri, J.R.; Diekmann, J.; Napp, L.C.; Bataiosu, D.R.; Jaguszewski, M.; Cammann, V.L.; Sarcon, A.; Geyer, V.; Neumann, C.A.; et al. Clinical features and outcomes of Takotsubo (stress) cardiomyopathy. N. Engl. J. Med. 2015, 373, 929–938. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Malignant Arrhythmia |
|---|
| Cardiomyopathy |
| Acute coronary syndrome |
| Acute heart failure |
| Pathological Conditions |
|---|
| Aldosteronoma |
| Pheochromocytoma |
| Cortisol producing adenoma |
| Nonfunctioning adenomas |
| Rare entities (cysts and myelolipomas) |
| Patient Value | Normal Range | |
|---|---|---|
| White blood cell count | 15.400 | 4.000–10.000/mm3 |
| Troponin I | 64.7 | ≤40 ng/L |
| Myoglobin | 162.7 | 14.3–65.8 ng/mL |
| Blood glucose | 360 | 60–100 mg/dL |
| Lactate | 8.9 | <4 mEq/L |
| CK-MB | 3.10 | 0.6–6.3 ng/mL |
| Patient Value | Normal Range | |
|---|---|---|
| Cortisolemia | 65 | 6.2–19.4 mcg/dL (in the morning) 2.3–11.9 mcg/dL (in the afternoon) |
| Adrenaline | 340 | 20–190 pg/mL |
| Metanephrine | 7.260 | 0–90 pg/mL |
| Normetanephrine | 7.860 | 0–180 pg/mL |
| Renin | 310 | 3–40 pg/mL |
| Multiple endocrine neoplasia type II |
| Von Hippel–Lindau syndrome |
| Neurofibromatosis type I |
| Carney syndrome |
| PGL 1 syndrome |
| PGL 4 syndrome |
| Intravascular volume depletion |
| Abrupt cessation of catecholamine secretion due to tumour necrosis |
| Desensitisation of adrenergic receptors |
| Hypocalcaemia |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Baio, R.; Pagano, T.; Molisso, G.; Di Mauro, U.; Intilla, O.; Albano, F.; Scarpato, F.; Giacometti, S.; Sanseverino, R. When a Multidisciplinary Approach Is Life-Saving: A Case Report of Cardiogenic Shock Induced by a Large Pheochromocytoma. Diseases 2022, 10, 29. https://doi.org/10.3390/diseases10020029
Baio R, Pagano T, Molisso G, Di Mauro U, Intilla O, Albano F, Scarpato F, Giacometti S, Sanseverino R. When a Multidisciplinary Approach Is Life-Saving: A Case Report of Cardiogenic Shock Induced by a Large Pheochromocytoma. Diseases. 2022; 10(2):29. https://doi.org/10.3390/diseases10020029
Chicago/Turabian StyleBaio, Raffaele, Tommaso Pagano, Giovanni Molisso, Umberto Di Mauro, Olivier Intilla, Francesco Albano, Fulvio Scarpato, Stefania Giacometti, and Roberto Sanseverino. 2022. "When a Multidisciplinary Approach Is Life-Saving: A Case Report of Cardiogenic Shock Induced by a Large Pheochromocytoma" Diseases 10, no. 2: 29. https://doi.org/10.3390/diseases10020029
APA StyleBaio, R., Pagano, T., Molisso, G., Di Mauro, U., Intilla, O., Albano, F., Scarpato, F., Giacometti, S., & Sanseverino, R. (2022). When a Multidisciplinary Approach Is Life-Saving: A Case Report of Cardiogenic Shock Induced by a Large Pheochromocytoma. Diseases, 10(2), 29. https://doi.org/10.3390/diseases10020029