1. Introduction
As a result of the advances in DNA/RNA sequencing technology in recent decades, a huge amount of data is generated daily, creating the immediate need for efficient storage. The standard solution for the distribution and processing of raw (non-assembled) sequencing data are the FASTQ files [
1], the uncompressed text files that store the fragments of sequenced nucleotide chains, together with the accompanying header information and associated quality scores. The need for compressing FASTQ files is obvious; various approaches to this task are listed in [
2], while [
3,
4,
5] describe important newer results. There exists no single “best” compression method as there are different usage aspects to consider: the compression ratio, speed of construction, speed of decompression, and memory requirements for compression and decompression. While FQSqueezer [
5] reaches the highest compression factor for DNA streams, when considering the decompression time we regard PgRC [
4] as the most useful compressor for DNA streams only, and SPRING [
3] as, currently, the best overall solution for whole FASTQ files. Some applications benefit from the capability to retrieve the specific record, implying random access to records, combined with an index [
6]. For some purposes one can use the k-mer index to access only the relevant reads and avoid mapping/aligning the whole dataset. There exists a large body of literature on k-mer indexing, mainly for the collections of datasets [
7]. It is straightforward to modify the index to include record IDs. Currently, the random access functionality is implemented only in the BEETL/BEETL-fastq [
6,
8], and to some extent in the SPRING software that supports decompression of a specific block of records.
All of the existing methods require the decompression stage. In order to use the HTS data, the original FASTQ file must be reconstructed, either as a separate operation on the disk or online in RAM. As for the random access, in the existing software it involves a standard decompression, only on a smaller block of the compressed data. This imposes overhead in retrieval time and in run-time RAM.
We propose a new paradigm for storing and accessing the vast amount of HTS data: a succinct data structure sFASTQ that is a compressed representation of a FASTQ file, which allows for fully random access to individual records, and can replace a flat (interleaved) FASTQ file. This means that the sFASTQ format can be listed and searched on a disk without decompression or loading into RAM. sFASTQ is stored as a directory with multiple subdirectories. In order to use sFASTQ as a source, the downstream application must accept STDIN stream as the input. Considering the lossless compression of full FASTQ data, the sizes of sFASTQ directories are comparable to those of Gzip archives. The reconstruction of the flat file is considerably slower than with the fastest of state-of-the-art methods. However, we do not need to reconstruct the flat file since the records are decompressed online and can be forwarded on to the downstream applications. While the slower reading of the FASTQ records can still be acceptable in certain set-ups, the major benefit of sFASTQ lies in the fast random access to specific records, with no memory overhead.
2. Methods—Implementation of sFASTQ Format
2.1. Overview
Each record in a FASTQ file consists of four lines: header, DNA sequence read, a single plus sign, and quality values. The plus sign separates sequence and quality entries; it is omitted from the compression process and simply added at the decompression stage. The rest of the entries can be of variable length throughout the FASTQ file, with the condition that the length of the sequence and quality entries in each record must be equal. With paired end methods of sequencing, where the same DNA segment is sequenced from two directions, the result is stored in two corresponding FASTQ files—forward and reverse. The matching of the corresponding records in both files is achieved either via ordering of records or through the headers. We assume the case where corresponding records are listed in both files in the same order. Alternatively, forward and reverse records can be stored in a single file in an interleaved manner.
The core of our implementation of a succinct FASTQ format is the
LZ trie, the state-of-the-art method for automata compression [
9] that is incorporated into our
SFQ (
https://github.com/lisp-rbi/sfq, accessed on 10 May 2022) software for the construction and querying of sFASTQ. The drawbacks of employing automata are considerable memory consumption and the time needed for construction. However, the construction of sFASTQ format is an once-only process and does not require computing resources beyond those that can be found in a professional bioinformatics facility. While the LZ trie engine is implemented in C++, the interface and the functionalities of SFQ software are implemented in Rust. The SFQ usage instructions are given in the
Supplementary Material.
SFQ works both with single- and paired-end FASTQ files, and supports long reads and records of variable lengths. The records in the sFASTQ are accessed through their ID; with paired end sequencing two paired records are retrieved with a single ID. As an option, sFASTQ can be loaded into RAM for (approximately) three times faster reading than on the disk. SFQ implements four different methods of lossy compression that include binning of qualities, removal of the headers, removal of duplicate sequences, and averaging of the associated qualities.
We regard three different streams—headers, reads, and qualities—as the collections of strings, and for each stream we build a compressed minimal acyclic deterministic finite automaton (MADFA) [
10] that stores (in the terminology of automata theory, the automaton
recognizes the language that is a collection of strings) the collection of all entries in the FASTQ file. The streams are compressed separately, as there is more redundancy in the same type of data than across the streams.
The MADFA has an important property that allows it to be queried with prefixes of strings that are stored. We explore this feature by enumerating the records in a FASTQ file and prefixing each data entry with the ordinal number of the record that entry belongs to. The purpose of this is twofold. Firstly, the enumeration enables fully random access, i.e., direct access to any given record in the FASTQ file. The second reason for the enumeration of the records is that the compression procedure requires sorting of the collection of input strings. Since the three streams are compressed separately, the enumeration is used to keep the original order of records across the compressed automata, while at the same time imposing a sorted order on the entries. For paired end datasets, the enumeration is complemented with forward/reverse annotation.
Compressed MADFA is a succinct data structure. Informally, a succinct data structure is a compressed structure that has a size close to its theoretical minimum, but that still supports various lookup operations. Our succinct data structure sFASTQ has a size comparable to that of a Gzip file, and supports sequential listing of the FASTQ records, as well as direct access to a specific record. A sFASTQ directory emulates a flat file in the sense that it can be searched on the disk without additional memory requirements. Alternatively, sFASTQ can be loaded into RAM for faster lookup. If the sFASTQ archive stores a paired end dataset, then its output is in the interleaved format.
The sFASTQ format supports arbitrarily long reads, as well as the reads of variable length, which is in line with the requirements of the emerging long-read sequencing technologies. Besides lossles compression of the FASTQ files, we have implemented four different methods of lossy compression.
2.2. sFASTQ Data Structures Implementation Details
2.2.1. LZ Trie Data Structure
The state-of-the-art in deterministic automata compression, regarding the compression factor and the construction speed, is the LZ trie algorithm. The details on LZ trie can be found in [
9]; here we give a brief description. A trie is a digital tree that represents a set of strings by storing the shared prefixes only once. LZ trie construction starts with a trie that stores a collection of strings, and finds the repeated parts in the trie and replaces them with pointers, which is a generic LZ compression method. Some further processing and the optimized bit assignment result in the most compact representation of an MADFA for most types of data. The advantage of this approach is based on an efficient discovery of within-string redundancies across the whole dataset, while most other MADFA compression methods focus only on the shared prefixes and suffixes, and the statistical coding.
The drawback of the LZ trie method is the large size of the initial trie and the correspondingly large auxiliary structures. Our implementation has the worst case RAM requirement of 55 bytes per input symbol. This roughly translates to the worst case memory footprint of 25 times the size of the processed FASTQ input. The time complexity of LZ trie construction is quasilinear. In this case, this means that large files can be processed within realistic time bounds.
Alternatively, in order to produce a succinct FASTQ representation, it would be possible to use some other approach to compress of a set of strings, with different trade-offs in construction parameters. Some of the competing methods are listed in [
11]. Still, we believe that LZ trie is the most useful compromise—especially so if the input strings are long, as in a FASTQ record.
2.2.2. On LZ Trie Implementation
The distinctive feature of the SFQ tool is its ability to list and query the compressed FASTQ data directly from the disk, without the overhead of either decompressing the data or loading it into RAM. This feature is implemented by persisting the LZ trie data structure in a format that mirrors its representation in RAM. The LZ trie structure is represented as an array that holds the nodes of the trie [
9]. This array is encapsulated as an abstract
TNodeArray type, which enabled us to transparently switch to disk storage by implementing the on-disk array with the same interface. Specifically, the trie array is composed of several sub-arrays containing distinct node-level information. These sub-arrays are optimized bit arrays that use a char array as a backing structure. The switch to on-disk storage is implemented by switching to an on-disk char array implemented using random file access. Crucially, the high-level trie array uses an in-memory node-level cache based on an FIFO structure (the oldest cached element is removed first). The caching enables the access time of the on-disk array to be of the same order of magnitude as that of the in-memory array. For more details, we refer the reader to the implementation, documentation, and usage of the
CompactArray,
BitSequenceArray, and the
DiskArrayChar classes (
https://github.com/lisp-rbi/sfq/tree/main/fqlzt/src/aux/lzt_core, accessed on 10 May 2022). These classes encapsulate the space-optimized array of trie nodes, the sub-arrays with node information, and the backing on-disk array structure, respectively.
2.3. Construction of sFASTQ Format
2.3.1. Organization of Streams into Tries and Subdirectories
We separately extract each stream from a FASTQ file and produce three temporary files. The entries in temporary files are prefixed with the ordinal number of the record they are extracted from. The temporary files are used as the input for the LZ trie construction. We therefore construct separate succinct structures for each stream, and store them in the respective subdirectories of the top-level sFASTQ directory. If not specified otherwise, the top-level directory that stores the sFASTQ format for an input FASTQ file fname.fastq is named fname.sfastq. The subdirectories that hold compressed representations of the header, sequence, and quality streams are named fname.head.sfastq, fname.seq.sfastq, and fname.qual.sfastq, respectively. Once the sFASTQ directory structure is constructed, the names of the directories and files should not be changed. At the end of the LZ trie construction for a given stream, the corresponding temporary file is erased. The temporary files are essentially data streams with added prefixes and are cumulatively larger than the input FASTQ dataset. The finished compressed representation of the first processed stream is stored on the disk, while all of the temporary files are still present. Although the original FASTQ file is not required once the temporary files are constructed, our software does not delete it. As a result, the additional disk space necessary for sFASTQ construction, on top of the space occupied by the FASTQ file, is approximately one and a half times the size of the input file.
2.3.2. Paired End Files
When the input consists of two paired end FASTQ files, the temporary files are constructed in an interleaved manner. The corresponding records from both input files are stored consecutively, with reverse sequences complemented, and prefixed with the same ordinal number. However, in order to differentiate between the forward and reverse input, a different character is appended to the prefix number. The added characters are “F” for forward and “R” for reverse input. When reading the paired end dataset from sFASTQ, the output is in the interleaved format. The complementing of reverse sequences leads to more repeated sequence parts, and is a standard trick used to improve the FASTQ data compression. If not specified otherwise, the top-level sFASTQ directory that stores paired end FASTQ files fname1.fastq and fname2.fastq is named fname1.FR.sfastq. The infix FR. is also added to the names of all subdirectories.
Our software expects paired end files to be paired through the order of the records, i.e., the paired forward and reverse records must be at the same position in both files. If they are not properly paired before starting the sFASTQ construction, they should be ordered using a tool like
fastq-pair [
12]. If the numbers of records in forward and reverse files are not equal, SFQ ignores the missing lines in temporary files. This means that there will be a series of a single direction records at the end of the sFASTQ interleaved output.
2.3.3. Multiple Subtries
With the worst case memory requirement of 55 bytes per input symbol, the available RAM becomes a limiting factor in construction of LZ tries. We solve this problem by splitting the input temporary files into smaller blocks of data. The amount of available RAM is automatically determined, or the user can define the limit on memory usage. In this way, the largest possible size of the input is calculated. The lines are read from temporary files until this limit is reached and a trie is constructed for the subset of the input. After the first LZ trie is produced, the reading from temporary file continues, up to the defined size limit, and the next part is processed. Multiple LZ subtries for a stream are stored in separate subdirectories with the corresponding numbers added to the subdirectory names. An example of sFASTQ top-level and subdirectories structure and naming, for the paired end input files fname1.fastq and fname2.fastq, is:
fname1.FR.sfastq/
/fname1.FR.head.sfastq.1/
/fname1.FR.head.sfastq.2/
/fname1.FR.qual.sfastq.1/
/fname1.FR.qual.sfastq.2/
/fname1.FR.qual.sfastq.3/
/fname1.FR.seq.sfastq.1/
/fname1.FR.seq.sfastq.2/
/fname1.FR.seq.sfastq.3/
In this example, DNA and quality streams, which are always equal in size, are both processed in three installments. Headers are usually much shorter and more entries can fit into available RAM. As a result, headers produce smaller numbers of subtries. In the reading stage, all subtries for one stream are seamlessly treated as a single structure.
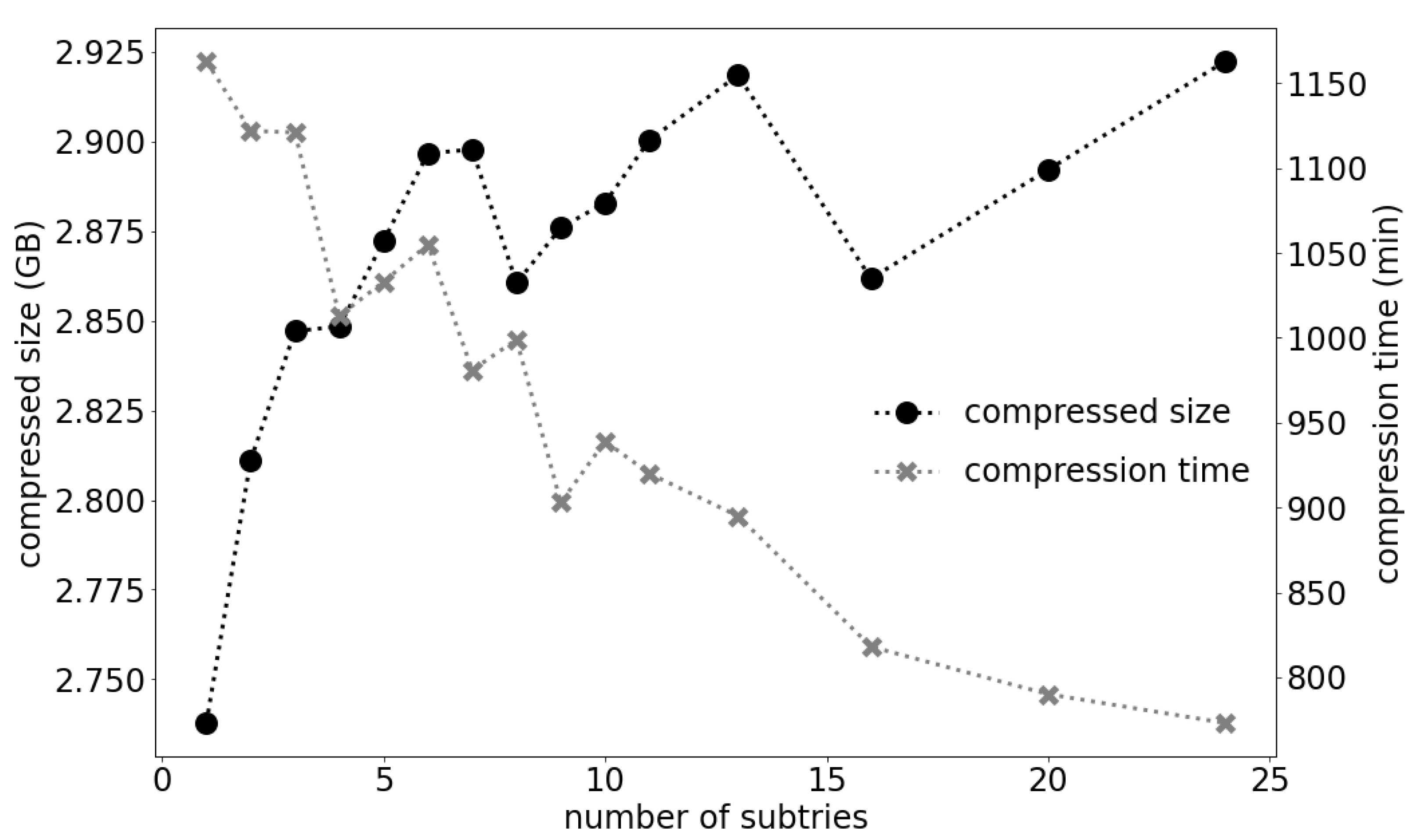
To some extent, the quality and the speed of compression depend on the amount of available RAM. The LZ trie is a global method of compression and, as a rule, the compression is better when a larger part of the input is processed in one installment. On the other hand, the time complexity of the compression procedure is quasilinear, which means that it takes somewhat more time to process the same amount of data in one large installment than in multiple smaller parts. Therefore, with less RAM the compression is worse but faster. However, due to the variations within the data, these dependencies are not monotone. An example is presented in
Figure 1. The dataset used in the experiment was the largest prefix of the H.sapiens2 dataset (details on datasets are given in the Results section) that still produced a single trie with our available RAM. The number of subtries in
Figure 1 relates to the DNA and quality streams. The compression factor noticeably worsens when one LZ trie is split into two to six subtries, but after that oscillates around the same value. On the other hand, the time needed for processing is steadily reduced with the increase in number of subtries. Nevertheless, we would always recommend using the maximal available amount of RAM, as this will, in most cases, lead to the best compression.
2.4. Random Access to Records
The enumeration of the records allows for direct access to any record in a sFASTQ format. Reading is performed by querying the LZ trie with a prefix, which is by design the ID of the record. Output consists of all entries with the same ID, across all streams. If the sFASTQ directory stores paired end files, two full records are retrieved with a single index. When using the sFASTQ directory that holds N records as a flat file, the numbers from 1 to N are silently generated and fed to the LZ trie core.
Our software supports fully random access to the specified records via an auxiliary file that stores the list of record indices. The list is an ASCII file that in each line stores a single index, or a range of indices. The list file can hold any combination of the two. An example of a legitimate input to the random access search in sFASTQ is:
123
200–300
88
40–50
33
220–230
If a value in the list is out of range, the query is ignored. On the implementation level, the IDs are encoded in the quaternary alphabet [A,C,G,T]. This leads to a slightly improved compression of the DNA stream.
2.5. Lossy Compression of Streams
A lot of information in a FASTQ file is redundant and may not be necessary for different applications. As a result, various methods for lossy compression of FASTQ files were advocated for and implemented, in particular regarding the quality values stream [
13]. We have implemented four different methods that we denote with L1 to L4. The actions that were performed to obtain the lossy formats are:
L1: The quality values are binned according to the Illumina 8-level standard [
14].
L2: L1 and the headers are removed.
L3: Headers are removed, repeated sequences are stored only once, and the corresponding quality values are reduced to the average value for each position over all repeated records. The number of repeated sequences is stored and later reported in an artificially generated header at the time of reading.
L4: L3 and the averaged qualities are binned according to the Illumina 8-level standard.
The methods L1 and L2 are two variants of a standard approach based on the observations from [
13], and L3 and L4 combine this with the approach used in ARSDA software [
15], which is particularly effective for FASTQ files with many repeated identical sequences. An example of this is transcriptomic data files. The number of repetitions in the original file is included in the L3 and L4 formats. The initial order of the records is preserved in L1 and L2, and lost in L3 and L4. While the naming of SFQ output folders can be arbitrary, the names of folders for lossy compression will include infix Lx., where x stands for a number from 1 to 4, depending on the method used.
Compared to the traditional approach to FASTQ files compression, sFASTQ format allows the listing of the content without additional resources. The advantage of the sFASTQ format is the random access and the smaller size than that of a flat FASTQ file. The drawback is considerably slower access to the records than in a flat file. However, the random access is at least twice as fast as with BEETL.
3. Results
We have performed experiments with our SFQ software for the construction and usage of sFASTQ format on the publicly available paired end files used in [
3,
4]. Three datasets from the two papers overlap. They are denoted as ERP001775, ERR532393, and SRR554369 in [
4], and as
H.sapiens1,
Metagenomics, and
P.aeruginosa in [
3], respectively. For these datasets we have used the naming from the earlier paper. The
H.sapiens2 is the raw version that is described in the supplementary material of [
3]. This means that the records are not trimmed to the uniform length. For details on how to access the files please see the
Supplement. The experiments were performed on a 3.6 GHz Xeon Gold 5122 processor with 384 GB RAM running Ubuntu 18.04.5 LTS. We did not process the largest file from [
3], 826 GB large
H.sapiens3, as that would take an unrealistically long time with the current implementation and available hardware resources.
The main results are presented in
Table 1, together with the reference compressed sizes obtained by Gzip (with -9 option). In all cases the sFASTQ directory sizes are comparable with those of Gzip files. The difference in the size factor between sFASTQ and Gzip varies from 0.827 to 1.336, with the sFASTQ directory being, on average, 7.7% larger than the Gzip file. With more available RAM the numbers will be more in favor of sFASTQ, sometimes significantly. We base this assumption on the performed partial experiments with
H.sapiens2 on a 6 TB RAM machine, which indicate that the single trie implementation would be approximately 20% smaller than the obtained version with 13 subtries.
Processing times vary depending on the type of the dataset and the achieved compression factor. As an example, H.sapiens2 and ERR194146 datasets require comparable time for processing, although the latter is about twice the size of the former, but H.sapiens2 compresses better. The average time needed for processing one GB of input is about 40 min.
The maximum RAM usage is given as an illustration that the memory footprint of our process is an important factor. As calculated, in the worst case the RAM space needed for sFASTQ construction is approximately 25 times the size of the input dataset. In cases when that exceeded 384 GB, multiple subtries were constructed. The number of subtries in
Table 1 is given for DNA and quality streams. Header strings are shorter than reads and more headers fit in one trie; therefore, the number of subtries is smaller than for the DNA and quality streams.
The results for lossy compression are presented in
Table 2. We have included results for SPRING lossy compression recommended in [
3] that consist of Illumina binning of quality, removal of headers, and not preserving the initial order of records. We have also included the results for the single end transcriptomic dataset SRR1536586. That dataset was used in [
15] to demonstrate the efficiency of the ARSDA approach. The size of this file, when the repeated sequences are represented with only one, and the related qualities are averaged, as reported in [
15], is 0.120 GB, which is reduced to 0.035 GB with Gzip. Therefore, for this transcriptomic dataset, the L4 lossy version of sFASTQ has the best compression factor among the tested methods. The time needed for construction of the L4 version of sFASTQ for the SRR1536586 dataset was under four minutes.
Table 3 presents typical results for the speed of access to the records in the number of single end records (i.e., four lines of data) per second that are reconstructed from the sFASTQ archive. The difference in reading speed between HDD and SSD is marginal, and reading from RAM is about three times faster. The results for RAM exclude the time needed to load sFASTQ into memory. The differences across datasets are due to the different read lengths. On average, 420 KB per second are reconstructed from the disk. The datasets in
Table 3 are all from the paired end sequencing. Reading from a single end sFASTQ is slightly faster since it does not involve reversing and complementing the paired sequences. Reconstruction of records in lossy variants is faster because the headers are omitted, and because binning of the qualities leads to more regularity in the strings; therefore, the decompression procedure is simpler.
Sequential access speed was measured by listing all the records in a dataset. Random access speed was measured for two cases: the random blocks of consecutive records and random individual records. The queries are supplied to SFQ software via the external lists. In the first case the list comprised five randomly placed blocks of 50,000 records, and in the second case the list consisted of 25,000 random record IDs.
The reading speed is somewhat influenced by the number of subtries. Interestingly, for the same dataset the reading speed increases with the number of subtries. In the example from
Figure 1, the reading speed on HDD varies from 2200 records per second for one subtrie, to 2580 records per second for 24 subtries.
An important feature of SFQ is the low memory footprint of the records retrieval stage. For the random access, the amount of used RAM is less than 20 MB, when reading from the disk.
Comparison with BEETL
BEETL-fastq [
6], together with BEETL [
8], the only existing software that supports random access to FASTQ records (SPRING only supports the extraction of a single block of records), does not enable random access through the interface. The main functionality of BEETL-fastq is k-mer indexing and record retrieval. The random access to the records that contain a given k-mer is silently integrated in the system. In order to compare the times needed for the retrieval of a random FASTQ record we have used the following procedure. With trial and error we have determined the k-mers that are, for a given FASTQ file, present in up to approximately
records (higher values caused BEETL to crash). Then, the command:
beetl search -i prefix -k <k-mer>
would produce the searchedKmers_positions file. We have timed the BEETL command that retrieves the sequence part of the records defined in that file:
time beetl extend -i searchedKmers_positions -b prefix --propagate-sequence
-o extend.out
The
extend.out file contains the IDs of the retrieved records and we have transformed that file into a list of records required by SFQ. Finally, we have measured the time needed to retrieve these records from the sFASTQ archive with the
-f s option (retrieving only the sequence stream of the FASTQ record). Even though BEETL employs some parallelization for this task, the random access with SFQ (reading from the disk) was, on average, slightly more than twice as fast. That increases to approximately seven times faster when sFASTQ was loaded into RAM. This is in accordance with our previous findings. In [
11] we have compared the response times of automata and BWT-based structures and automata were several times faster, albeit in a different retrieval task.
Although BEETL software does not provide the interface for random access, that functionality is implied and we expect it could be implemented without too much of a programming effort. Compared to sFASTQ, BEETL archives have the advantage of a shorter construction time and a smaller size. The construction of BEETL archives is performed on multiple cores and, in our experiments, was from 5 to 20 times faster than the single core sFASTQ construction. While the sizes of sFASTQ and BEETL archives are comparable, the BEETL archive includes the complete k-mer index as well. The main advantages of SFQ are the functional interface, faster random access, and negligible memory footprint of the reading stage. In addition, SFQ implements lossy compression of quality scores which is particularly useful with transcriptomics data.
4. Additional Considerations
4.1. On Paralellization
In our experimental setup, the available RAM was the critical resource in producing the sFASTQ directory. For this reason, the current version of our SFQ software works exclusively in a single thread mode. With enough RAM resources, two parallelization schemes can be applied. A straightforward method would be to process three streams separately on three cores, if there is enough RAM to hold all three processes simultaneously in a single trie each, i.e., if the available RAM is at least 55 times larger than the size of the input FASTQ file. Another possibility is to use a cluster of processing nodes and divide the processing of the subtries among them according to the available RAM at each node. Therefore, with the appropriate hardware resources, the production of the sFASTQ format could be performed in an acceptable time even for the largest files.
Large files could be processed in a realistic time on large clusters with parallelization and adequate resources, or slowly on a single core. Although possible, the latter is not economical, unless an important application emerges that requires random access. We expect that the main target field for the current implementation of SFQ/sFASTQ are FASTQ datasets of intermediate sizes that can be constructed overnight and stored in the cloud-based archives. They could later be used for remote random access, or on personal computers with limited disk and RAM resources. Within this framework, one can envision the workflow where only a set of integers would be transferred between the users.
An sFASTQ archive can be used as a flat file since it can be accessed on the disk without memory overhead. If we are prepared to forego this possibility, the parallelization could be applied to the reading stage, too. Multiple cores could be assigned the task of decompressing successive blocks of records, with scheduled access to STDOUT. However, this could lead to a meaningful speed-up only if the sFASTQ folder is loaded into RAM.
4.2. On K-Mer Indexing
The full potential of random access capability can be exploited with an external k-mer index. There exists a substantial body of literature on the construction and usage of the k-mer index. The (non-exhaustive) list of relevant papers includes [
16,
17,
18,
19,
20,
21,
22]. Ref. [
7] presents a survey of the methods, and a more technical overview of the employed data structures is given in [
23]. Most of the works consider k-mers in the context of indexing collections of sequences. Of the listed papers, only [
7,
22] explicitly mention indexing raw reads. However, the inclusion of the read ID data in the index is straightforward.
4.3. On FASTQ Sampling Functionality
An immediate application of SFQ random access capability is the extraction of a subsample of records from a FASTQ file. Random sampling is regularly used in machine learning and statistical analysis, e.g., for bootstraping–repeated sampling of small data subsets. Machine learning procedures rely on division of data into training, validation, and test sets. This process also relies on sampling and is sometimes performed multiple times. The staple of deep learning, a prevalent machine learning technique with promising applications in biology, is the stochastic gradient descent algorithm that depends on repeated sampling of small data subsets.
So far, sampling of FASTQ records has been employed in RNA sequencing applications [
24,
25] and is included as a functionality in FASTQ processing software tools (e.g., [
26]). The existing software tools that offer subsampling functionality work on decompressed FASTQ data and employ a random selection of a fraction of streamed records. In contrast, the unique characteristics of SFQ make it possible to perform sampling through random direct access to FASTQ records on the disk. This means that random subsamples can be obtained quickly, without the need for decompressing the entire FASTQ file (or large chunks of it), and without any memory overhead. The speed and low memory overhead are particularly important when the FASTQ subsampling is used as part of a potential high-resource processing pipeline.
5. Conclusions
We have described sFASTQ, a new method for compressing FASTQ files, and implemented it in SFQ software for the construction and usage of an sFASTQ format that supports variable length reads, pairing of records, and both lossless and lossy compression of quality scores. As the usage of the sFASTQ format has a low memory footprint and does not require additional disk space, parallel downstream processes can be immediately started on multiple cores. This may in fact save time compared to the work-flow that first requires the decompression of multiple files. However, a downstream application should be able to read input from STDIN, i.e., support the standard method of chaining command-line tools on Unix-like systems. The future development of SFQ will include investigation of possible improvement of the compression factor by pairing the automata minimization with other compression methods for streams, and the use of paralellization to reduce the construction time and increase the reading speed.
As an example of a practical application, besides subsampling, random access is important in various scenarios where the privacy of an individual is of concern. We envision the application where, as a part of the medical treatment, a patient is in possession of their raw genomic sequences, and a physician can access and perform the alignment of only the records relevant to the medical case.





{kind=link}