Role of B Cell Lymphoma 2 in the Regulation of Liver Fibrosis in miR-122 Knockout Mice

 ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Dataset Analysis
2.2. Mouse Husbandry and Treatment
2.3. Histological Analysis
2.4. Proliferation Assay
2.5. Luciferase Assay
2.6. Co-Culture of HCC Cells and LX-2 Cells
2.7. Reverse-Transcription Polymerase Chain Reaction (RT-qPCR)
2.8. Immunoblot Analysis
2.9. Statistical Analysis
3. Results
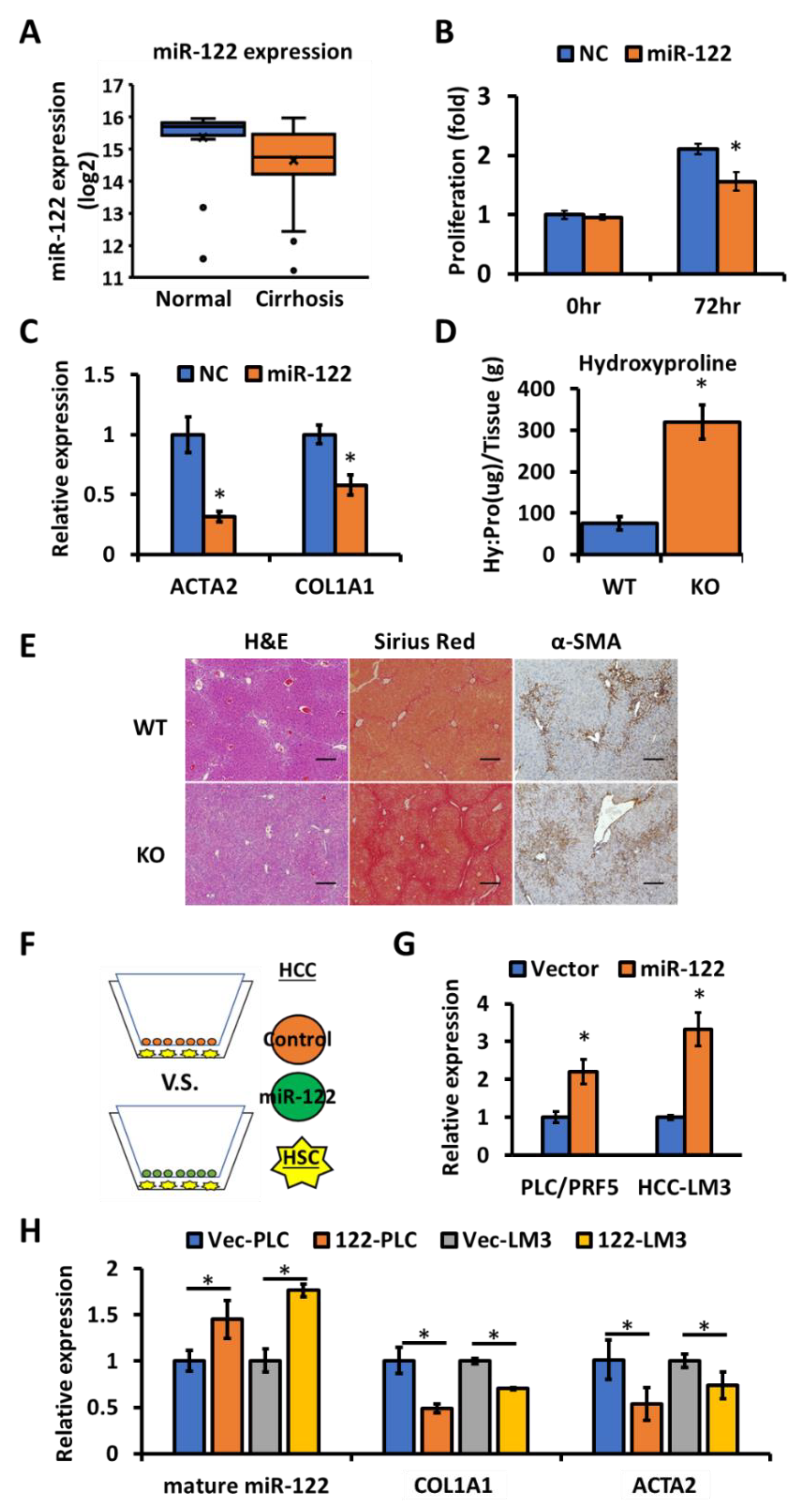
3.1. miR-122 Is a Negative Regulator of Liver Fibrosis
3.2. Extracellular miR-122 Released from Hepatic Cells Modulates Fibrotic Gene Expression in HSCs
3.3. Ago-CLIP Identified Several Profibrotic Targets of Mir-122
3.4. Pro-Fibrotic BCL2 Is Overexpressed in miR-122 KO Mice and Human Cirrhotic Livers
3.5. miR-122 Regulates BCL2 Expression via c-MYC
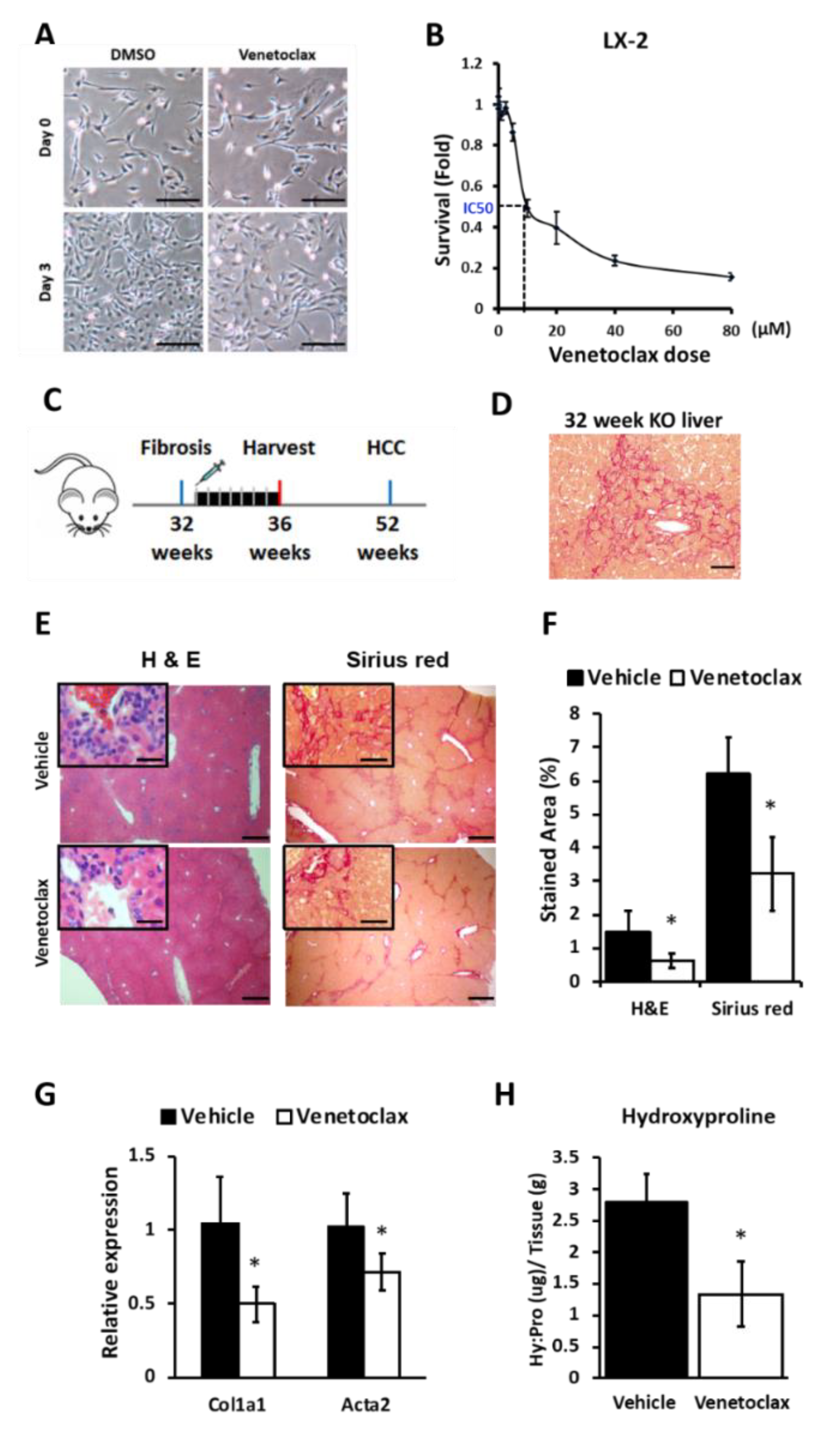
3.6. BCL2 Blockade Reduces Proliferation of Hepatic Stellate Cells (HSCs) and Suppresses Liver Fibrosis in miR-122 KO Mice
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- He, L.; Hannon, G.J. MicroRNAs: Small RNAs with a big role in gene regulation. Nat. Rev. Genet. 2004, 5, 522–531. [Google Scholar] [CrossRef] [PubMed]
- Ha, M.; Kim, V.N. Regulation of microRNA biogenesis. Nat. Rev. Mol. Cell Biol. 2014, 15, 509–524. [Google Scholar] [CrossRef] [PubMed]
- Esau, C.; Davis, S.; Murray, S.F.; Yu, X.X.; Pandey, S.K.; Pear, M.; Watts, L.; Booten, S.L.; Graham, M.; McKay, R.; et al. miR-122 regulation of lipid metabolism revealed by in vivo antisense targeting. Cell Metab. 2006, 3, 87–98. [Google Scholar] [CrossRef] [PubMed]
- Krutzfeldt, J.; Rajewsky, N.; Braich, R.; Rajeev, K.G.; Tuschl, T.; Manoharan, M.; Stoffel, M. Silencing of microRNAs in vivo with ‘antagomirs’. Nature 2005, 438, 685–689. [Google Scholar] [CrossRef]
- Hsu, S.H.; Wang, B.; Kota, J.; Yu, J.; Costinean, S.; Kutay, H.; Yu, L.; Bai, S.; La Perle, K.; Chivukula, R.R.; et al. Essential metabolic, anti-inflammatory, and anti-tumorigenic functions of miR-122 in liver. J. Clin. Investig. 2012, 122, 2871–2883. [Google Scholar] [CrossRef]
- Kim, N.; Kim, H.; Jung, I.; Kim, Y.; Kim, D.; Han, Y.M. Expression profiles of miRNAs in human embryonic stem cells during hepatocyte differentiation. Hepatol. Res. Off. J. Jpn. Soc. Hepatol. 2011, 41, 170–183. [Google Scholar] [CrossRef]
- Hsu, S.H.; Delgado, E.R.; Otero, P.A.; Teng, K.Y.; Kutay, H.; Meehan, K.M.; Moroney, J.B.; Monga, J.K.; Hand, N.J.; Friedman, J.R.; et al. MicroRNA-122 regulates polyploidization in the murine liver. Hepatology 2016, 64, 599–615. [Google Scholar] [CrossRef]
- Luna, J.M.; Scheel, T.K.; Danino, T.; Shaw, K.S.; Mele, A.; Fak, J.J.; Nishiuchi, E.; Takacs, C.N.; Catanese, M.T.; de Jong, Y.P.; et al. Hepatitis C virus RNA functionally sequesters miR-122. Cell 2015, 160, 1099–1110. [Google Scholar] [CrossRef]
- Chowdhary, V.; Teng, K.Y.; Thakral, S.; Zhang, B.; Lin, C.H.; Wani, N.; Bruschweiler-Li, L.; Zhang, X.; James, L.; Yang, D.; et al. miRNA-122 Protects Mice and Human Hepatocytes from Acetaminophen Toxicity by Regulating Cytochrome P450 Family 1 Subfamily A Member 2 and Family 2 Subfamily E Member 1 Expression. Am. J. Pathol. 2017, 187, 2758–2774. [Google Scholar] [CrossRef]
- Wang, Y.; Liang, H.; Jin, F.; Yan, X.; Xu, G.; Hu, H.; Liang, G.; Zhan, S.; Hu, X.; Zhao, Q.; et al. Injured liver-released miRNA-122 elicits acute pulmonary inflammation via activating alveolar macrophage TLR7 signaling pathway. Proc. Natl. Acad. Sci. USA 2019, 116, 6162–6171. [Google Scholar] [CrossRef]
- Xu, H.; Xu, S.J.; Xie, S.J.; Zhang, Y.; Yang, J.H.; Zhang, W.Q.; Zheng, M.N.; Zhou, H.; Qu, L.H. MicroRNA-122 supports robust innate immunity in hepatocytes by targeting the RTKs/STAT3 signaling pathway. eLife 2019, 8. [Google Scholar] [CrossRef]
- Coulouarn, C.; Factor, V.M.; Andersen, J.B.; Durkin, M.E.; Thorgeirsson, S.S. Loss of miR-122 expression in liver cancer correlates with suppression of the hepatic phenotype and gain of metastatic properties. Oncogene 2009, 28, 3526–3536. [Google Scholar] [CrossRef] [PubMed]
- Tsai, W.C.; Hsu, P.W.; Lai, T.C.; Chau, G.Y.; Lin, C.W.; Chen, C.M.; Lin, C.D.; Liao, Y.L.; Wang, J.L.; Chau, Y.P.; et al. MicroRNA-122, a tumor suppressor microRNA that regulates intrahepatic metastasis of hepatocellular carcinoma. Hepatology 2009, 49, 1571–1582. [Google Scholar] [CrossRef]
- Bai, S.; Nasser, M.W.; Wang, B.; Hsu, S.H.; Datta, J.; Kutay, H.; Yadav, A.; Nuovo, G.; Kumar, P.; Ghoshal, K. MicroRNA-122 inhibits tumorigenic properties of hepatocellular carcinoma cells and sensitizes these cells to sorafenib. J. Biol. Chem. 2009, 284, 32015–32027. [Google Scholar] [CrossRef] [PubMed]
- Tsai, W.C.; Hsu, S.D.; Hsu, C.S.; Lai, T.C.; Chen, S.J.; Shen, R.; Huang, Y.; Chen, H.C.; Lee, C.H.; Tsai, T.F.; et al. MicroRNA-122 plays a critical role in liver homeostasis and hepatocarcinogenesis. J. Clin. Investig. 2012, 122, 2884–2897. [Google Scholar] [CrossRef] [PubMed]
- Bataller, R.; Brenner, D.A. Liver fibrosis. J. Clin. Investig. 2005, 115, 209–218. [Google Scholar] [CrossRef]
- Friedman, S.L. Liver fibrosis—From bench to bedside. J. Hepatol. 2003, 38 (Suppl. 1), S38–S53. [Google Scholar] [CrossRef]
- Friedman, S.L.; Roll, F.J.; Boyles, J.; Bissell, D.M. Hepatic lipocytes: The principal collagen-producing cells of normal rat liver. Proc. Natl. Acad. Sci. USA 1985, 82, 8681–8685. [Google Scholar] [CrossRef]
- Gabele, E.; Brenner, D.A.; Rippe, R.A. Liver fibrosis: Signals leading to the amplification of the fibrogenic hepatic stellate cell. Front. Biosci. J. Virtual Libr. 2003, 8, d69–d77. [Google Scholar]
- Higuchi, H.; Gores, G.J. Mechanisms of liver injury: An overview. Curr. Mol. Med. 2003, 3, 483–490. [Google Scholar] [CrossRef]
- Canbay, A.; Friedman, S.; Gores, G.J. Apoptosis: The nexus of liver injury and fibrosis. Hepatology 2004, 39, 273–278. [Google Scholar] [CrossRef] [PubMed]
- Naito, M.; Hasegawa, G.; Ebe, Y.; Yamamoto, T. Differentiation and function of Kupffer cells. Med. Electron. Microsc. Off. J. Clin. Electron Microsc. Soc. Jpn. 2004, 37, 16–28. [Google Scholar] [CrossRef] [PubMed]
- Thurman, R.G., II. Alcoholic liver injury involves activation of Kupffer cells by endotoxin. Am. J. Physiol. 1998, 275, G605–G611. [Google Scholar] [CrossRef] [PubMed]
- Casini, A.; Ceni, E.; Salzano, R.; Biondi, P.; Parola, M.; Galli, A.; Foschi, M.; Caligiuri, A.; Pinzani, M.; Surrenti, C. Neutrophil-derived superoxide anion induces lipid peroxidation and stimulates collagen synthesis in human hepatic stellate cells: Role of nitric oxide. Hepatology 1997, 25, 361–367. [Google Scholar] [CrossRef]
- Schuppan, D.; Afdhal, N.H. Liver cirrhosis. Lancet 2008, 371, 838–851. [Google Scholar] [CrossRef]
- Tsochatzis, E.A.; Bosch, J.; Burroughs, A.K. Liver cirrhosis. Lancet 2014, 383, 1749–1761. [Google Scholar] [CrossRef]
- Li, J.; Ghazwani, M.; Zhang, Y.; Lu, J.; Li, J.; Fan, J.; Gandhi, C.R.; Li, S. miR-122 regulates collagen production via targeting hepatic stellate cells and suppressing P4HA1 expression. J. Hepatol. 2013, 58, 522–528. [Google Scholar] [CrossRef]
- Teng, K.Y.; Ghoshal, K. Role of Noncoding RNAs as Biomarker and Therapeutic Targets for Liver Fibrosis. Gene Expr. 2015, 16, 155–162. [Google Scholar] [CrossRef]
- Tsujimoto, Y.; Gorham, J.; Cossman, J.; Jaffe, E.; Croce, C.M. The t(14;18) chromosome translocations involved in B-cell neoplasms result from mistakes in VDJ joining. Science 1985, 229, 1390–1393. [Google Scholar] [CrossRef]
- Monaghan, P.; Robertson, D.; Amos, T.A.; Dyer, M.J.; Mason, D.Y.; Greaves, M.F. Ultrastructural localization of bcl-2 protein. J. Histochem. Cytochem. Off. J. Histochem. Soc. 1992, 40, 1819–1825. [Google Scholar] [CrossRef]
- Delbridge, A.R.; Grabow, S.; Strasser, A.; Vaux, D.L. Thirty years of BCL-2: Translating cell death discoveries into novel cancer therapies. Nat. Rev. Cancer 2016, 16, 99–109. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, C.; Barbier, O.; Smalling, R.; Tsuchiya, H.; Lee, S.; Delker, D.; Zou, A.; Hagedorn, C.H.; Wang, L. Bcl2 is a critical regulator of bile acid homeostasis by dictating Shp and lncRNA H19 function. Sci. Rep. 2016, 6, 20559. [Google Scholar] [CrossRef]
- Novo, E.; Marra, F.; Zamara, E.; Valfre di Bonzo, L.; Monitillo, L.; Cannito, S.; Petrai, I.; Mazzocca, A.; Bonacchi, A.; De Franco, R.S.; et al. Overexpression of Bcl-2 by activated human hepatic stellate cells: Resistance to apoptosis as a mechanism of progressive hepatic fibrogenesis in humans. Gut 2006, 55, 1174–1182. [Google Scholar] [CrossRef] [PubMed]
- Czabotar, P.E.; Lessene, G.; Strasser, A.; Adams, J.M. Control of apoptosis by the BCL-2 protein family: Implications for physiology and therapy. Nat. Rev. Mol. Cell Biol. 2014, 15, 49–63. [Google Scholar] [CrossRef] [PubMed]
- Kluck, R.M.; Bossy-Wetzel, E.; Green, D.R.; Newmeyer, D.D. The release of cytochrome c from mitochondria: A primary site for Bcl-2 regulation of apoptosis. Science 1997, 275, 1132–1136. [Google Scholar] [CrossRef]
- Letai, A.G. Diagnosing and exploiting cancer’s addiction to blocks in apoptosis. Nat. Rev. Cancer 2008, 8, 121–132. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Hsu, S.H.; Wang, X.; Kutay, H.; Bid, H.K.; Yu, J.; Ganju, R.K.; Jacob, S.T.; Yuneva, M.; Ghoshal, K. Reciprocal regulation of microRNA-122 and c-Myc in hepatocellular cancer: Role of E2F1 and transcription factor dimerization partner 2. Hepatology 2014, 59, 555–566. [Google Scholar] [CrossRef] [PubMed]
- Kolesnikov, N.; Hastings, E.; Keays, M.; Melnichuk, O.; Tang, Y.A.; Williams, E.; Dylag, M.; Kurbatova, N.; Brandizi, M.; Burdett, T.; et al. ArrayExpress update--simplifying data submissions. Nucleic Acids Res. 2015, 43, D1113–D1116. [Google Scholar] [CrossRef] [PubMed]
- Pineau, P.; Volinia, S.; McJunkin, K.; Marchio, A.; Battiston, C.; Terris, B.; Mazzaferro, V.; Lowe, S.W.; Croce, C.M.; Dejean, A. miR-221 overexpression contributes to liver tumorigenesis. Proc. Natl. Acad. Sci. USA 2010, 107, 264–269. [Google Scholar] [CrossRef]
- Luna, J.M.; Barajas, J.M.; Teng, K.Y.; Sun, H.L.; Moore, M.J.; Rice, C.M.; Darnell, R.B.; Ghoshal, K. Argonaute CLIP Defines a Deregulated miR-122-Bound Transcriptome that Correlates with Patient Survival in Human Liver Cancer. Mol. Cell 2017, 67, 400–410.e407. [Google Scholar] [CrossRef]
- Wang, M.; Gong, Q.; Zhang, J.; Chen, L.; Zhang, Z.; Lu, L.; Yu, D.; Han, Y.; Zhang, D.; Chen, P.; et al. Characterization of gene expression profiles in HBV-related liver fibrosis patients and identification of ITGBL1 as a key regulator of fibrogenesis. Sci. Rep. 2017, 7, 43446. [Google Scholar] [CrossRef]
- Mas, V.R.; Maluf, D.G.; Archer, K.J.; Yanek, K.; Kong, X.; Kulik, L.; Freise, C.E.; Olthoff, K.M.; Ghobrial, R.M.; McIver, P.; et al. Genes involved in viral carcinogenesis and tumor initiation in hepatitis C virus-induced hepatocellular carcinoma. Mol. Med. 2009, 15, 85–94. [Google Scholar] [CrossRef] [PubMed]
- Teng, K.Y.; Han, J.; Zhang, X.; Hsu, S.H.; He, S.; Wani, N.A.; Barajas, J.M.; Snyder, L.A.; Frankel, W.L.; Caligiuri, M.A.; et al. Blocking the CCL2-CCR2 Axis Using CCL2-Neutralizing Antibody Is an Effective Therapy for Hepatocellular Cancer in a Mouse Model. Mol. Cancer Ther. 2017, 16, 312–322. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Hui, A.Y.; Albanis, E.; Arthur, M.J.; O’Byrne, S.M.; Blaner, W.S.; Mukherjee, P.; Friedman, S.L.; Eng, F.J. Human hepatic stellate cell lines, LX-1 and LX-2: New tools for analysis of hepatic fibrosis. Gut 2005, 54, 142–151. [Google Scholar] [CrossRef]
- Sun, H.L.; Cui, R.; Zhou, J.; Teng, K.Y.; Hsiao, Y.H.; Nakanishi, K.; Fassan, M.; Luo, Z.; Shi, G.; Tili, E.; et al. ERK Activation Globally Downregulates miRNAs through Phosphorylating Exportin-5. Cancer Cell 2016, 30, 723–736. [Google Scholar] [CrossRef] [PubMed]
- Tsuchida, T.; Friedman, S.L. Mechanisms of hepatic stellate cell activation. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 397–411. [Google Scholar] [CrossRef] [PubMed]
- Lou, G.; Song, X.; Yang, F.; Wu, S.; Wang, J.; Chen, Z.; Liu, Y. Exosomes derived from miR-122-modified adipose tissue-derived MSCs increase chemosensitivity of hepatocellular carcinoma. J. Hematol. Oncol. 2015, 8, 122. [Google Scholar] [CrossRef]
- Povero, D.; Eguchi, A.; Li, H.; Johnson, C.D.; Papouchado, B.G.; Wree, A.; Messer, K.; Feldstein, A.E. Circulating extracellular vesicles with specific proteome and liver microRNAs are potential biomarkers for liver injury in experimental fatty liver disease. PLoS ONE 2014, 9, e113651. [Google Scholar] [CrossRef]
- Bala, S.; Petrasek, J.; Mundkur, S.; Catalano, D.; Levin, I.; Ward, J.; Alao, H.; Kodys, K.; Szabo, G. Circulating microRNAs in exosomes indicate hepatocyte injury and inflammation in alcoholic, drug-induced, and inflammatory liver diseases. Hepatology 2012, 56, 1946–1957. [Google Scholar] [CrossRef]
- Chi, S.W.; Zang, J.B.; Mele, A.; Darnell, R.B. Argonaute HITS-CLIP decodes microRNA-mRNA interaction maps. Nature 2009, 460, 479–486. [Google Scholar] [CrossRef]
- Yin, S.; Fan, Y.; Zhang, H.; Zhao, Z.; Hao, Y.; Li, J.; Sun, C.; Yang, J.; Yang, Z.; Yang, X.; et al. Differential TGFβ pathway targeting by miR-122 in humans and mice affects liver cancer metastasis. Nat. Commun. 2016, 7, 11012. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Puertos, V.Y.; Hernandez-Perez, E.; Nuno-Lambarri, N.; Ventura-Gallegos, J.L.; Lopez-Diazguerrero, N.E.; Robles-Diaz, G.; Gutierrez-Ruiz, M.C.; Konigsberg, M. Bcl-2 overexpression in hepatic stellate cell line CFSC-2G, induces a pro-fibrotic state. J. Gastroenterol. Hepatol. 2010, 25, 1306–1314. [Google Scholar] [CrossRef]
- Nevzorova, Y.A.; Hu, W.; Cubero, F.J.; Haas, U.; Freimuth, J.; Tacke, F.; Trautwein, C.; Liedtke, C. Overexpression of c-myc in hepatocytes promotes activation of hepatic stellate cells and facilitates the onset of liver fibrosis. Biochim. Biophys. Acta 2013, 1832, 1765–1775. [Google Scholar] [CrossRef] [PubMed]
- Cao, Z.; Fan-Minogue, H.; Bellovin, D.I.; Yevtodiyenko, A.; Arzeno, J.; Yang, Q.; Gambhir, S.S.; Felsher, D.W. MYC phosphorylation, activation, and tumorigenic potential in hepatocellular carcinoma are regulated by HMG-CoA reductase. Cancer Res. 2011, 71, 2286–2297. [Google Scholar] [CrossRef] [PubMed]
- Stilgenbauer, S.; Eichhorst, B.; Schetelig, J.; Coutre, S.; Seymour, J.F.; Munir, T.; Puvvada, S.D.; Wendtner, C.M.; Roberts, A.W.; Jurczak, W.; et al. Venetoclax in relapsed or refractory chronic lymphocytic leukaemia with 17p deletion: A multicentre, open-label, phase 2 study. Lancet. Oncol. 2016, 17, 768–778. [Google Scholar] [CrossRef]
- Lochmann, T.L.; Floros, K.V.; Naseri, M.; Powell, K.M.; Cook, W.; March, R.J.; Stein, G.T.; Greninger, P.; Maves, Y.K.; Saunders, L.R.; et al. Venetoclax Is Effective in Small-Cell Lung Cancers with High BCL-2 Expression. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2018, 24, 360–369. [Google Scholar] [CrossRef]
- Chen, L.; Chen, R.; Kemper, S.; Charrier, A.; Brigstock, D.R. Suppression of fibrogenic signaling in hepatic stellate cells by Twist1-dependent microRNA-214 expression: Role of exosomes in horizontal transfer of Twist1. Am. J. Physiol. Gastrointest. Liver Physiol. 2015, 309, G491–G499. [Google Scholar] [CrossRef]
- Breitkopf, K.; Haas, S.; Wiercinska, E.; Singer, M.V.; Dooley, S. Anti-TGF-beta strategies for the treatment of chronic liver disease. Alcohol. Clin. Exp. Res. 2005, 29, 121s–131s. [Google Scholar] [CrossRef]
- Ogawa, S.; Ochi, T.; Shimada, H.; Inagaki, K.; Fujita, I.; Nii, A.; Moffat, M.A.; Katragadda, M.; Violand, B.N.; Arch, R.H.; et al. Anti-PDGF-B monoclonal antibody reduces liver fibrosis development. Hepatol. Res. Off. J. Jpn. Soc. Hepatol. 2010, 40, 1128–1141. [Google Scholar] [CrossRef]
- Eischen, C.M.; Packham, G.; Nip, J.; Fee, B.E.; Hiebert, S.W.; Zambetti, G.P.; Cleveland, J.L. Bcl-2 is an apoptotic target suppressed by both c-Myc and E2F-1. Oncogene 2001, 20, 6983–6993. [Google Scholar] [CrossRef]
- McMahon, S.B. MYC and the control of apoptosis. Cold Spring Harb Perspect Med 2014, 4, a014407. [Google Scholar] [CrossRef]
- Yip, K.W.; Reed, J.C. Bcl-2 family proteins and cancer. Oncogene 2008, 27, 6398–6406. [Google Scholar] [CrossRef] [PubMed]
- Reed, J.C. Bcl-2-family proteins and hematologic malignancies: History and future prospects. Blood 2008, 111, 3322–3330. [Google Scholar] [CrossRef]
- Tse, C.; Shoemaker, A.R.; Adickes, J.; Anderson, M.G.; Chen, J.; Jin, S.; Johnson, E.F.; Marsh, K.C.; Mitten, M.J.; Nimmer, P.; et al. ABT-263: A potent and orally bioavailable Bcl-2 family inhibitor. Cancer Res. 2008, 68, 3421–3428. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.; Wang, Y.; Shao, L.; Laberge, R.M.; Demaria, M.; Campisi, J.; Janakiraman, K.; Sharpless, N.E.; Ding, S.; Feng, W.; et al. Clearance of senescent cells by ABT263 rejuvenates aged hematopoietic stem cells in mice. Nat. Med. 2016, 22, 78–83. [Google Scholar] [CrossRef] [PubMed]
- Kwon, O.S.; Hong, S.K.; Kwon, S.J.; Go, Y.H.; Oh, E.; Cha, H.J. BCL2 induced by LAMTOR3/MAPK is a druggable target of chemoradioresistance in mesenchymal lung cancer. Cancer Lett. 2017, 403, 48–58. [Google Scholar] [CrossRef] [PubMed]
- Mason, K.D.; Carpinelli, M.R.; Fletcher, J.I.; Collinge, J.E.; Hilton, A.A.; Ellis, S.; Kelly, P.N.; Ekert, P.G.; Metcalf, D.; Roberts, A.W.; et al. Programmed anuclear cell death delimits platelet life span. Cell 2007, 128, 1173–1186. [Google Scholar] [CrossRef]
- Roberts, A.W.; Seymour, J.F.; Brown, J.R.; Wierda, W.G.; Kipps, T.J.; Khaw, S.L.; Carney, D.A.; He, S.Z.; Huang, D.C.; Xiong, H.; et al. Substantial susceptibility of chronic lymphocytic leukemia to BCL2 inhibition: Results of a phase I study of navitoclax in patients with relapsed or refractory disease. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2012, 30, 488–496. [Google Scholar] [CrossRef]
- Souers, A.J.; Leverson, J.D.; Boghaert, E.R.; Ackler, S.L.; Catron, N.D.; Chen, J.; Dayton, B.D.; Ding, H.; Enschede, S.H.; Fairbrother, W.J.; et al. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat. Med. 2013, 19, 202–208. [Google Scholar] [CrossRef]
- Roberts, A.W.; Davids, M.S.; Pagel, J.M.; Kahl, B.S.; Puvvada, S.D.; Gerecitano, J.F.; Kipps, T.J.; Anderson, M.A.; Brown, J.R.; Gressick, L.; et al. Targeting BCL2 with Venetoclax in Relapsed Chronic Lymphocytic Leukemia. N. Engl. J. Med. 2016, 374, 311–322. [Google Scholar] [CrossRef]
- Grever, M.R.; Lucas, D.M.; Dewald, G.W.; Neuberg, D.S.; Reed, J.C.; Kitada, S.; Flinn, I.W.; Tallman, M.S.; Appelbaum, F.R.; Larson, R.A.; et al. Comprehensive assessment of genetic and molecular features predicting outcome in patients with chronic lymphocytic leukemia: Results from the US Intergroup Phase III Trial E2997. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2007, 25, 799–804. [Google Scholar] [CrossRef] [PubMed]
- Catovsky, D.; Richards, S.; Matutes, E.; Oscier, D.; Dyer, M.; Bezares, R.F.; Pettitt, A.R.; Hamblin, T.; Milligan, D.W.; Child, J.A.; et al. Assessment of fludarabine plus cyclophosphamide for patients with chronic lymphocytic leukaemia (the LRF CLL4 Trial): A randomised controlled trial. Lancet 2007, 370, 230–239. [Google Scholar] [CrossRef]
- Wang, P.; Koyama, Y.; Liu, X.; Xu, J.; Ma, H.Y.; Liang, S.; Kim, I.H.; Brenner, D.A.; Kisseleva, T. Promising Therapy Candidates for Liver Fibrosis. Front. Physiol. 2016, 7, 47. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Annotation | Seed Type | p-Value | WT VS KO (log2) |
|---|---|---|---|---|
| Ctgf | 5’UTR | 7A1 | 0.016988 | −1.518624491 |
| Pdgfrb | CDS | 8mer | 3.75 × −21 | −0.96498004 |
| Col5a1 | CDS | 8mer | 3.26 × −20 | −0.952192613 |
| P4ha1 | 3’UTR | 8mer | 0.001085 | −3.232382621 |
| Mmp2 | 3’UTR | 7A1 | 1.02 × −24 | −1.307276327 |
| Klf6 | 3’UTR | 7m8 | 5.68 × −47 | −1.190961957 |
| Col4a1 | 3’UTR | 6mer | 7.08 × −24 | −1.102863702 |
| Vcam1 | 3’UTR | 7A1 | 4.68 × −6 | −0.979919528 |
| Ngfr | 3’UTR | 7m8 | 7.06 × −12 | −0.954727937 |
| Myh9 | 3’UTR | 6mer | 1.67 × −16 | −0.796204606 |
| Tnfrsf1b | 3’UTR | 6mer | 0.002705 | −0.766674249 |
| Tgfbr1 | 3’UTR | 7A1 | 3.83 × −6 | −0.332025716 |
| Tgfbr1 | 3’UTR | 6mer | 3.83 × −6 | −0.332025716 |
| Treatment | Mouse ID | Score | Summary |
|---|---|---|---|
| Vehicle | 5136 | 3 | 2.6 ± 0.55 |
| 5137 | 3 | ||
| 5138 | 3 | ||
| 5139 | 2 | ||
| 5140 | 2 | ||
| Venetoclax | 5141 | 2 | 1.2 ± 0.45 |
| 5142 | 1 | ||
| 4963 | 1 | ||
| 4964 | 1 | ||
| 4190 | 1 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Teng, K.-Y.; Barajas, J.M.; Hu, P.; Jacob, S.T.; Ghoshal, K. Role of B Cell Lymphoma 2 in the Regulation of Liver Fibrosis in miR-122 Knockout Mice. Biology 2020, 9, 157. https://doi.org/10.3390/biology9070157
Teng K-Y, Barajas JM, Hu P, Jacob ST, Ghoshal K. Role of B Cell Lymphoma 2 in the Regulation of Liver Fibrosis in miR-122 Knockout Mice. Biology. 2020; 9(7):157. https://doi.org/10.3390/biology9070157
Chicago/Turabian StyleTeng, Kun-Yu, Juan M. Barajas, Peng Hu, Samson T. Jacob, and Kalpana Ghoshal. 2020. "Role of B Cell Lymphoma 2 in the Regulation of Liver Fibrosis in miR-122 Knockout Mice" Biology 9, no. 7: 157. https://doi.org/10.3390/biology9070157
APA StyleTeng, K.-Y., Barajas, J. M., Hu, P., Jacob, S. T., & Ghoshal, K. (2020). Role of B Cell Lymphoma 2 in the Regulation of Liver Fibrosis in miR-122 Knockout Mice. Biology, 9(7), 157. https://doi.org/10.3390/biology9070157