Structural Insights into the Evolutionarily Conserved BAF Chromatin Remodeling Complex



{kind=link}
Abstract
1. Introduction
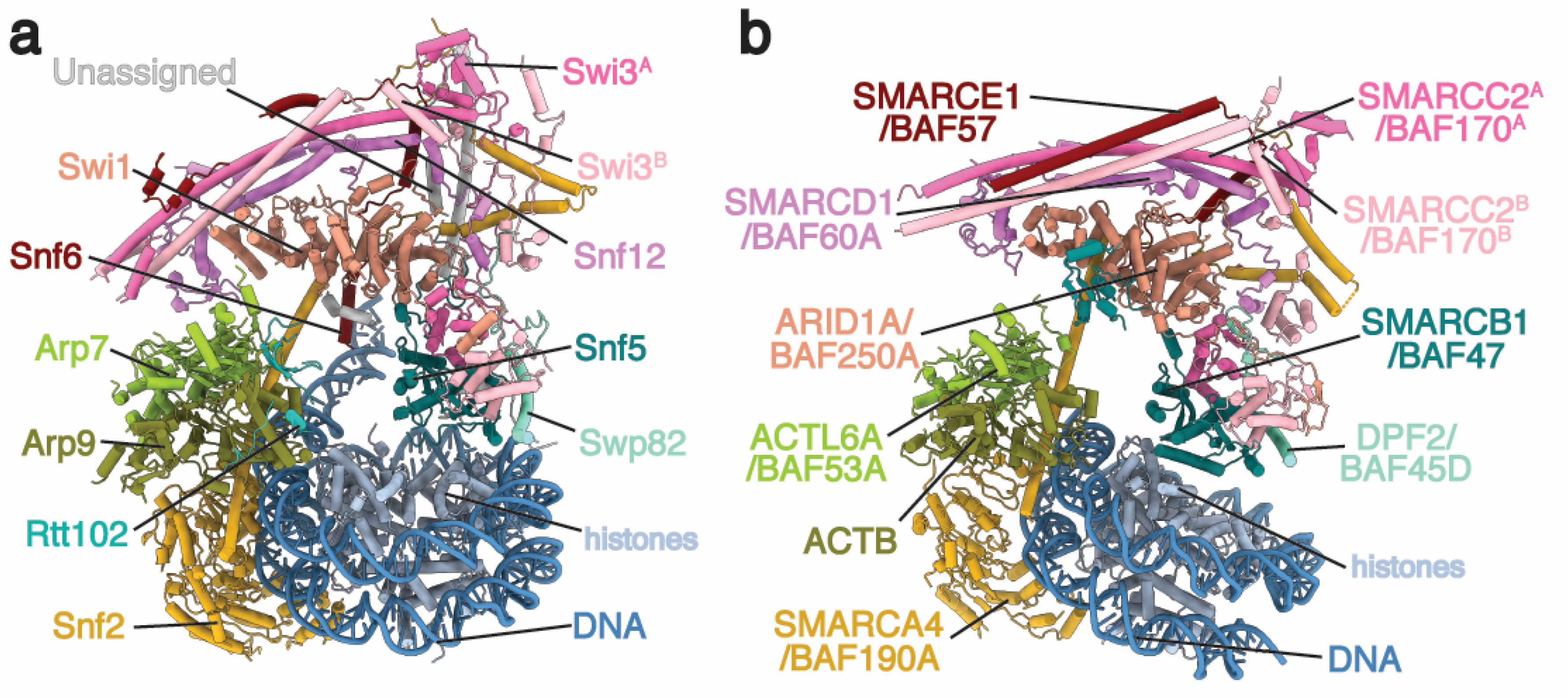
2. Architecture of the BAF Family Remodeler
2.1. ATPase Module
2.2. Arp Module
2.3. Body Module
3. Future Directions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Zhou, C.Y.; Johnson, S.L.; Gamarra, N.I.; Narlikar, G.J. Mechanisms of ATP-Dependent Chromatin Remodeling Motors. Annu. Rev. Biophys. 2016, 45, 153–181. [Google Scholar] [CrossRef]
- Clapier, C.R.; Iwasa, J.; Cairns, B.R.; Peterson, C.L. Mechanisms of action and regulation of ATP-dependent chromatin-remodelling complexes. Nat. Rev. Mol. Cell Biol. 2017, 18, 407–422. [Google Scholar] [CrossRef]
- Kadoch, C.; Hargreaves, D.C.; Hodges, C.; Elias, L.; Ho, L.; Ranish, J.; Crabtree, G.R. Proteomic and bioinformatic analysis of mammalian SWI/SNF complexes identifies extensive roles in human malignancy. Nat. Genet. 2013, 45, 592–601. [Google Scholar] [CrossRef]
- Shain, A.H.; Pollack, J.R. The spectrum of SWI/SNF mutations, ubiquitous in human cancers. PLoS ONE 2013, 8, e55119. [Google Scholar] [CrossRef]
- Arnaud, O.; Le Loarer, F.; Tirode, F. BAFfling pathologies: Alterations of BAF complexes in cancer. Cancer Lett. 2018, 419, 266–279. [Google Scholar] [CrossRef]
- St Pierre, R.; Kadoch, C. Mammalian SWI/SNF complexes in cancer: Emerging therapeutic opportunities. Curr. Opin. Genet. Dev. 2017, 42, 56–67. [Google Scholar] [CrossRef]
- Olivier, M.; Hollstein, M.; Hainaut, P. TP53 mutations in human cancers: Origins, consequences, and clinical use. Cold Spring Harb. Perspect. Biol. 2010, 2, a001008. [Google Scholar] [CrossRef]
- Han, Y.; Reyes, A.A.; Malik, S.; He, Y. Cryo-EM structure of SWI/SNF complex bound to a nucleosome. Nature 2020, 579, 452–455. [Google Scholar] [CrossRef]
- He, S.; Wu, Z.; Tian, Y.; Yu, Z.; Yu, J.; Wang, X.; Li, J.; Liu, B.; Xu, Y. Structure of nucleosome-bound human BAF complex. Science 2020, 367, 875–881. [Google Scholar] [CrossRef]
- Ye, Y.; Wu, H.; Chen, K.; Clapier, C.R.; Verma, N.; Zhang, W.; Deng, H.; Cairns, B.R.; Gao, N.; Chen, Z. Structure of the RSC complex bound to the nucleosome. Science 2019, 366, 838–843. [Google Scholar] [CrossRef]
- Patel, A.B.; Moore, C.M.; Greber, B.J.; Luo, J.; Zukin, S.A.; Ranish, J.; Nogales, E. Architecture of the chromatin remodeler RSC and insights into its nucleosome engagement. Elife 2019, 8, e54449. [Google Scholar] [CrossRef]
- Zofall, M.; Persinger, J.; Kassabov, S.R.; Bartholomew, B. Chromatin remodeling by ISW2 and SWI/SNF requires DNA translocation inside the nucleosome. Nat. Struct. Mol. Biol. 2006, 13, 339–346. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Xia, X.; Tian, Y.; Jia, Q.; Liu, X.; Lu, Y.; Li, M.; Li, X.; Chen, Z. Mechanism of DNA translocation underlying chromatin remodelling by Snf2. Nature 2019, 567, 409–413. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Li, M.; Xia, X.; Li, X.; Chen, Z. Mechanism of chromatin remodelling revealed by the Snf2-nucleosome structure. Nature 2017, 544, 440–445. [Google Scholar] [CrossRef]
- Hassan, A.H.; Awad, S.; Prochasson, P. The Swi2/Snf2 bromodomain is required for the displacement of SAGA and the octamer transfer of SAGA-acetylated nucleosomes. J. Biol. Chem. 2006, 281, 18126–18134. [Google Scholar] [CrossRef]
- Hassan, A.H.; Prochasson, P.; Neely, K.E.; Galasinski, S.C.; Chandy, M.; Carrozza, M.J.; Workman, J.L. Function and selectivity of bromodomains in anchoring chromatin-modifying complexes to promoter nucleosomes. Cell 2002, 111, 369–379. [Google Scholar] [CrossRef]
- Shen, W.; Xu, C.; Huang, W.; Zhang, J.; Carlson, J.E.; Tu, X.; Wu, J.; Shi, Y. Solution structure of human Brg1 bromodomain and its specific binding to acetylated histone tails. Biochemistry 2007, 46, 2100–2110. [Google Scholar] [CrossRef] [PubMed]
- Sen, P.; Vivas, P.; Dechassa, M.L.; Mooney, A.M.; Poirier, M.G.; Bartholomew, B. The SnAC domain of SWI/SNF is a histone anchor required for remodeling. Mol. Cell. Biol. 2013, 33, 360–370. [Google Scholar] [CrossRef] [PubMed]
- Morrison, E.A.; Sanchez, J.C.; Ronan, J.L.; Farrell, D.P.; Varzavand, K.; Johnson, J.K.; Gu, B.X.; Crabtree, G.R.; Musselman, C.A. DNA binding drives the association of BRG1/hBRM bromodomains with nucleosomes. Nat. Commun. 2017, 8, 16080. [Google Scholar] [CrossRef]
- Phelan, M.L.; Sif, S.; Narlikar, G.J.; Kingston, R.E. Reconstitution of a core chromatin remodeling complex from SWI/SNF subunits. Mol. Cell 1999, 3, 247–253. [Google Scholar] [CrossRef]
- Wang, W.; Cote, J.; Xue, Y.; Zhou, S.; Khavari, P.A.; Biggar, S.R.; Muchardt, C.; Kalpana, G.V.; Goff, S.P.; Yaniv, M.; et al. Purification and biochemical heterogeneity of the mammalian SWI-SNF complex. EMBO J. 1996, 15, 5370–5382. [Google Scholar] [CrossRef] [PubMed]
- Pan, J.; McKenzie, Z.M.; D’Avino, A.R.; Mashtalir, N.; Lareau, C.A.; St Pierre, R.; Wang, L.; Shilatifard, A.; Kadoch, C. The ATPase module of mammalian SWI/SNF family complexes mediates subcomplex identity and catalytic activity-independent genomic targeting. Nat. Genet. 2019, 51, 618–626. [Google Scholar] [CrossRef] [PubMed]
- Stanton, B.Z.; Hodges, C.; Calarco, J.P.; Braun, S.M.; Ku, W.L.; Kadoch, C.; Zhao, K.; Crabtree, G.R. Smarca4 ATPase mutations disrupt direct eviction of PRC1 from chromatin. Nat. Genet. 2017, 49, 282–288. [Google Scholar] [CrossRef]
- Strobeck, M.W.; Knudsen, K.E.; Fribourg, A.F.; DeCristofaro, M.F.; Weissman, B.E.; Imbalzano, A.N.; Knudsen, E.S. BRG-1 is required for RB-mediated cell cycle arrest. Proc. Natl. Acad. Sci. USA 2000, 97, 7748–7753. [Google Scholar] [CrossRef]
- Dunaief, J.L.; Strober, B.E.; Guha, S.; Khavari, P.A.; Alin, K.; Luban, J.; Begemann, M.; Crabtree, G.R.; Goff, S.P. The retinoblastoma protein and BRG1 form a complex and cooperate to induce cell cycle arrest. Cell 1994, 79, 119–130. [Google Scholar] [CrossRef]
- Reisman, D.N.; Strobeck, M.W.; Betz, B.L.; Sciariotta, J.; Funkhouser, W.; Jr Murchardt, C.; Yaniv, M.; Sherman, L.S.; Knudsen, E.S.; Weissman, B.E. Concomitant down-regulation of BRM and BRG1 in human tumor cell lines: Differential effects on RB-mediated growth arrest vs CD44 expression. Oncogene 2002, 21, 1196–1207. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Bultman, S.; Gebuhr, T.; Yee, D.; La Mantia, C.; Nicholson, J.; Gilliam, A.; Randazzo, F.; Metzger, D.; Chambon, P.; Crabtree, G.; et al. A Brg1 null mutation in the mouse reveals functional differences among mammalian SWI/SNF complexes. Mol. Cell 2000, 6, 1287–1295. [Google Scholar] [CrossRef]
- Bultman, S.J.; Herschkowitz, J.I.; Godfrey, V.; Gebuhr, T.C.; Yaniv, M.; Perou, C.M.; Magnuson, T. Characterization of mammary tumors from Brg1 heterozygous mice. Oncogene 2008, 27, 460–468. [Google Scholar] [CrossRef]
- Karnezis, A.N.; Wang, Y.; Ramos, P.; Hendricks, W.P.; Oliva, E.; D’Angelo, E.; Prat, J.; Nucci, M.R.; Nielsen, T.O.; Chow, C.; et al. Dual loss of the SWI/SNF complex ATPases SMARCA4/BRG1 and SMARCA2/BRM is highly sensitive and specific for small cell carcinoma of the ovary, hypercalcaemic type. J. Pathol. 2016, 238, 389–400. [Google Scholar] [CrossRef]
- Witkowski, L.; Carrot-Zhang, J.; Albrecht, S.; Fahiminiya, S.; Hamel, N.; Tomiak, E.; Grynspan, D.; Saloustros, E.; Nadaf, J.; Rivera, B.; et al. Germline and somatic SMARCA4 mutations characterize small cell carcinoma of the ovary, hypercalcemic type. Nat. Genet. 2014, 46, 438–443. [Google Scholar] [CrossRef]
- Jelinic, P.; Mueller, J.J.; Olvera, N.; Dao, F.; Scott, S.N.; Shah, R.; Gao, J.; Schultz, N.; Gonen, M.; Soslow, R.A.; et al. Recurrent SMARCA4 mutations in small cell carcinoma of the ovary. Nat. Genet. 2014, 46, 424–426. [Google Scholar] [CrossRef] [PubMed]
- Le Loarer, F.; Watson, S.; Pierron, G.; de Montpreville, V.T.; Ballet, S.; Firmin, N.; Auguste, A.; Pissaloux, D.; Boyault, S.; Paindavoine, S.; et al. SMARCA4 inactivation defines a group of undifferentiated thoracic malignancies transcriptionally related to BAF-deficient sarcomas. Nat. Genet. 2015, 47, 1200–1205. [Google Scholar] [CrossRef] [PubMed]
- Medina, P.P.; Carretero, J.; Fraga, M.F.; Esteller, M.; Sidransky, D.; Sanchez-Cespedes, M. Genetic and epigenetic screening for gene alterations of the chromatin-remodeling factor, SMARCA4/BRG1, in lung tumors. Genes Chromosomes Cancer 2004, 41, 170–177. [Google Scholar] [CrossRef] [PubMed]
- Sun, A.; Tawfik, O.; Gayed, B.; Thrasher, J.B.; Hoestje, S.; Li, C.; Li, B. Aberrant expression of SWI/SNF catalytic subunits BRG1/BRM is associated with tumor development and increased invasiveness in prostate cancers. Prostate 2007, 67, 203–213. [Google Scholar] [CrossRef]
- Hodges, H.C.; Stanton, B.Z.; Cermakova, K.; Chang, C.Y.; Miller, E.L.; Kirkland, J.G.; Ku, W.L.; Veverka, V.; Zhao, K.; Crabtree, G.R. Dominant-negative SMARCA4 mutants alter the accessibility landscape of tissue-unrestricted enhancers. Nat. Struct. Mol. Biol. 2018, 25, 61–72. [Google Scholar] [CrossRef]
- Bartlett, C.; Orvis, T.J.; Rosson, G.S.; Weissman, B.E. BRG1 mutations found in human cancer cell lines inactivate Rb-mediated cell-cycle arrest. J. Cell. Physiol. 2011, 226, 1989–1997. [Google Scholar] [CrossRef]
- Mashtalir, N.; D’Avino, A.R.; Michel, B.C.; Luo, J.; Pan, J.; Otto, J.E.; Zullow, H.J.; McKenzie, Z.M.; Kubiak, R.L.; St Pierre, R.; et al. Modular Organization and Assembly of SWI/SNF Family Chromatin Remodeling Complexes. Cell 2018, 175, 1272–1288. [Google Scholar] [CrossRef]
- Trotter, K.W.; Fan, H.Y.; Ivey, M.L.; Kingston, R.E.; Archer, T.K. The HSA domain of BRG1 mediates critical interactions required for glucocorticoid receptor-dependent transcriptional activation in vivo. Mol. Cell. Biol. 2008, 28, 1413–1426. [Google Scholar] [CrossRef]
- Zhao, K.; Wang, W.; Rando, O.J.; Xue, Y.; Swiderek, K.; Kuo, A.; Crabtree, G.R. Rapid and phosphoinositol-dependent binding of the SWI/SNF-like BAF complex to chromatin after T lymphocyte receptor signaling. Cell 1998, 95, 625–636. [Google Scholar] [CrossRef]
- Shen, X.; Ranallo, R.; Choi, E.; Wu, C. Involvement of actin-related proteins in ATP-dependent chromatin remodeling. Mol. Cell 2003, 12, 147–155. [Google Scholar] [CrossRef]
- Son, E.Y.; Crabtree, G.R. The role of BAF (mSWI/SNF) complexes in mammalian neural development. Am. J. Med. Genet. C Semin. Med. Genet. 2014, 166, 333–349. [Google Scholar] [CrossRef]
- Goutham, A. In silico screening of cancer-associated mutations in the HSA domain of BRG1 and its role in affecting the Arp-HSA sub-complex of SWI/SNF. Comput. Biol. Chem. 2018, 77, 109–115. [Google Scholar]
- Jones, S.; Li, M.; Parsons, D.W.; Zhang, X.; Wesseling, J.; Kristel, P.; Schmidt, M.K.; Markowitz, S.; Yan, H.; Bigner, D.; et al. Somatic mutations in the chromatin remodeling gene ARID1A occur in several tumor types. Hum. Mutat. 2012, 33, 100–103. [Google Scholar] [CrossRef] [PubMed]
- Cajuso, T.; Hanninen, U.A.; Kondelin, J.; Gylfe, A.E.; Tanskanen, T.; Katainen, R.; Pitkanen, E.; Ristolainen, H.; Kaasinen, E.; Taipale, M.; et al. Exome sequencing reveals frequent inactivating mutations in ARID1A, ARID1B, ARID2 and ARID4A in microsatellite unstable colorectal cancer. Int. J. Cancer 2014, 135, 611–623. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.N.; Roberts, C.W. ARID1A mutations in cancer: Another epigenetic tumor suppressor? Cancer Discov. 2013, 3, 35–43. [Google Scholar] [CrossRef]
- Guan, B.; Gao, M.; Wu, C.H.; Wang, T.L.; Shih Ie, M. Functional analysis of in-frame indel ARID1A mutations reveals new regulatory mechanisms of its tumor suppressor functions. Neoplasia 2012, 14, 986–993. [Google Scholar] [CrossRef]
- Zang, Z.J.; Cutcutache, I.; Poon, S.L.; Zhang, S.L.; McPherson, J.R.; Tao, J.; Rajasegaran, V.; Heng, H.L.; Deng, N.; Gan, A.; et al. Exome sequencing of gastric adenocarcinoma identifies recurrent somatic mutations in cell adhesion and chromatin remodeling genes. Nat. Genet. 2012, 44, 570–574. [Google Scholar] [CrossRef]
- Mamo, A.; Cavallone, L.; Tuzmen, S.; Chabot, C.; Ferrario, C.; Hassan, S.; Edgren, H.; Kallioniemi, O.; Aleynikova, O.; Przybytkowski, E.; et al. An integrated genomic approach identifies ARID1A as a candidate tumor-suppressor gene in breast cancer. Oncogene 2012, 31, 2090–2100. [Google Scholar] [CrossRef]
- Guan, B.; Wang, T.L.; Shih Ie, M. ARID1A, a factor that promotes formation of SWI/SNF-mediated chromatin remodeling, is a tumor suppressor in gynecologic cancers. Cancer Res. 2011, 71, 6718–6727. [Google Scholar] [CrossRef]
- Wilsker, D.; Patsialou, A.; Zumbrun, S.D.; Kim, S.; Chen, Y.; Dallas, P.B.; Moran, E. The DNA-binding properties of the ARID-containing subunits of yeast and mammalian SWI/SNF complexes. Nucleic Acids Res. 2004, 32, 1345–1353. [Google Scholar] [CrossRef]
- Nagl, N.G.; Wang, X., Jr.; Patsialou, A.; Van Scoy, M.; Moran, E. Distinct mammalian SWI/SNF chromatin remodeling complexes with opposing roles in cell-cycle control. EMBO J. 2007, 26, 752–763. [Google Scholar] [CrossRef]
- Valencia, A.M.; Collings, C.K.; Dao, H.T.; St Pierre, R.; Cheng, Y.C.; Huang, J.; Sun, Z.Y.; Seo, H.S.; Mashtalir, N.; Comstock, D.E.; et al. Recurrent SMARCB1 Mutations Reveal a Nucleosome Acidic Patch Interaction Site That Potentiates mSWI/SNF Complex Chromatin Remodeling. Cell 2019, 179, 1342–1356. [Google Scholar] [CrossRef] [PubMed]
- Versteege, I.; Sevenet, N.; Lange, J.; Rousseau-Merck, M.F.; Ambros, P.; Handgretinger, R.; Aurias, A.; Delattre, O. Truncating mutations of hSNF5/INI1 in aggressive paediatric cancer. Nature 1998, 394, 203–206. [Google Scholar] [CrossRef]
- Roberts, C.W.; Galusha, S.A.; McMenamin, M.E.; Fletcher, C.D.; Orkin, S.H. Haploinsufficiency of Snf5 (integrase interactor 1) predisposes to malignant rhabdoid tumors in mice. Proc. Natl. Acad. Sci. USA 2000, 97, 13796–13800. [Google Scholar] [CrossRef] [PubMed]
- Le Loarer, F.; Zhang, L.; Fletcher, C.D.; Ribeiro, A.; Singer, S.; Italiano, A.; Neuville, A.; Houlier, A.; Chibon, F.; Coindre, J.M.; et al. Consistent SMARCB1 homozygous deletions in epithelioid sarcoma and in a subset of myoepithelial carcinomas can be reliably detected by FISH in archival material. Genes Chromosomes Cancer 2014, 53, 475–486. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, L.M.; Folpe, A.L.; Pawel, B.R.; Judkins, A.R.; Biegel, J.A. Epithelioid sarcoma is associated with a high percentage of SMARCB1 deletions. Mod. Pathol. 2013, 26, 385–392. [Google Scholar] [CrossRef]
- Calderaro, J.; Moroch, J.; Pierron, G.; Pedeutour, F.; Grison, C.; Maille, P.; Soyeux, P.; de la Taille, A.; Couturier, J.; Vieillefond, A.; et al. SMARCB1/INI1 inactivation in renal medullary carcinoma. Histopathology 2012, 61, 428–435. [Google Scholar] [CrossRef]
- Liu, Q.; Galli, S.; Srinivasan, R.; Linehan, W.M.; Tsokos, M.; Merino, M.J. Renal medullary carcinoma: Molecular, immunohistochemistry, and morphologic correlation. Am. J. Surg. Pathol. 2013, 37, 368–374. [Google Scholar] [CrossRef]
- Kadoch, C.; Crabtree, G.R. Reversible disruption of mSWI/SNF (BAF) complexes by the SS18-SSX oncogenic fusion in synovial sarcoma. Cell 2013, 153, 71–85. [Google Scholar] [CrossRef]
- Hulsebos, T.J.; Plomp, A.S.; Wolterman, R.A.; Robanus-Maandag, E.C.; Baas, F.; Wesseling, P. Germline mutation of INI1/SMARCB1 in familial schwannomatosis. Am. J. Hum. Genet. 2007, 80, 805–810. [Google Scholar] [CrossRef]
- Kohashi, K.; Oda, Y.; Yamamoto, H.; Tamiya, S.; Oshiro, Y.; Izumi, T.; Taguchi, T.; Tsuneyoshi, M. SMARCB1/INI1 protein expression in round cell soft tissue sarcomas associated with chromosomal translocations involving EWS: A special reference to SMARCB1/INI1 negative variant extraskeletal myxoid chondrosarcoma. Am. J. Surg. Pathol. 2008, 32, 1168–1174. [Google Scholar] [CrossRef] [PubMed]
- Trobaugh-Lotrario, A.D.; Tomlinson, G.E.; Finegold, M.J.; Gore, L.; Feusner, J.H. Small cell undifferentiated variant of hepatoblastoma: Adverse clinical and molecular features similar to rhabdoid tumors. Pediatric Blood Cancer 2009, 52, 328–334. [Google Scholar] [CrossRef] [PubMed]
- Middeljans, E.; Wan, X.; Jansen, P.W.; Sharma, V.; Stunnenberg, H.G.; Logie, C. SS18 together with animal-specific factors defines human BAF-type SWI/SNF complexes. PLoS ONE 2012, 7, e33834. [Google Scholar] [CrossRef]
- Lee, R.S.; Stewart, C.; Carter, S.L.; Ambrogio, L.; Cibulskis, K.; Sougnez, C.; Lawrence, M.S.; Auclair, D.; Mora, J.; Golub, T.R.; et al. A remarkably simple genome underlies highly malignant pediatric rhabdoid cancers. J. Clin. Investig. 2012, 122, 2983–2988. [Google Scholar] [CrossRef]
- Crew, A.J.; Clark, J.; Fisher, C.; Gill, S.; Grimer, R.; Chand, A.; Shipley, J.; Gusterson, B.A.; Cooper, C.S. Fusion of SYT to two genes, SSX1 and SSX2, encoding proteins with homology to the Kruppel-associated box in human synovial sarcoma. EMBO J. 1995, 14, 2333–2340. [Google Scholar] [CrossRef] [PubMed]
- Clark, J.; Rocques, P.J.; Crew, A.J.; Gill, S.; Shipley, J.; Chan, A.M.; Gusterson, B.A.; Cooper, C.S. Identification of novel genes, SYT and SSX, involved in the t(X;18)(p11.2;q11.2) translocation found in human synovial sarcoma. Nat. Genet. 1994, 7, 502–508. [Google Scholar] [CrossRef]
- Hiraga, H.; Nojima, T.; Abe, S.; Sawa, H.; Yamashiro, K.; Yamawaki, S.; Kaneda, K.; Nagashima, K. Diagnosis of synovial sarcoma with the reverse transcriptase-polymerase chain reaction: Analyses of 84 soft tissue and bone tumors. Diagn. Mol. Pathol. 1998, 7, 102–110. [Google Scholar] [CrossRef]
- McBride, M.J.; Pulice, J.L.; Beird, H.C.; Ingram, D.R.; D’Avino, A.R.; Shern, J.F.; Charville, G.W.; Hornick, J.L.; Nakayama, R.T.; Garcia-Rivera, E.M.; et al. The SS18-SSX Fusion Oncoprotein Hijacks BAF Complex Targeting and Function to Drive Synovial Sarcoma. Cancer Cell 2018, 33, 1128–1141 e1127. [Google Scholar] [CrossRef]
- Kia, S.K.; Gorski, M.M.; Giannakopoulos, S.; Verrijzer, C.P. SWI/SNF mediates polycomb eviction and epigenetic reprogramming of the INK4b-ARF-INK4a locus. Mol. Cell. Biol. 2008, 28, 3457–3464. [Google Scholar] [CrossRef]
- Wilson, B.G.; Wang, X.; Shen, X.; McKenna, E.S.; Lemieux, M.E.; Cho, Y.J.; Koellhoffer, E.C.; Pomeroy, S.L.; Orkin, S.H.; Roberts, C.W. Epigenetic antagonism between polycomb and SWI/SNF complexes during oncogenic transformation. Cancer Cell 2010, 18, 316–328. [Google Scholar] [CrossRef]
- Nakayama, R.T.; Pulice, J.L.; Valencia, A.M.; McBride, M.J.; McKenzie, Z.M.; Gillespie, M.A.; Ku, W.L.; Teng, M.; Cui, K.; Williams, R.T.; et al. SMARCB1 is required for widespread BAF complex-mediated activation of enhancers and bivalent promoters. Nat. Genet. 2017, 49, 1613–1623. [Google Scholar] [CrossRef] [PubMed]

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marcum, R.D.; Reyes, A.A.; He, Y. Structural Insights into the Evolutionarily Conserved BAF Chromatin Remodeling Complex. Biology 2020, 9, 146. https://doi.org/10.3390/biology9070146
Marcum RD, Reyes AA, He Y. Structural Insights into the Evolutionarily Conserved BAF Chromatin Remodeling Complex. Biology. 2020; 9(7):146. https://doi.org/10.3390/biology9070146
Chicago/Turabian StyleMarcum, Ryan D., Alexis A. Reyes, and Yuan He. 2020. "Structural Insights into the Evolutionarily Conserved BAF Chromatin Remodeling Complex" Biology 9, no. 7: 146. https://doi.org/10.3390/biology9070146
APA StyleMarcum, R. D., Reyes, A. A., & He, Y. (2020). Structural Insights into the Evolutionarily Conserved BAF Chromatin Remodeling Complex. Biology, 9(7), 146. https://doi.org/10.3390/biology9070146