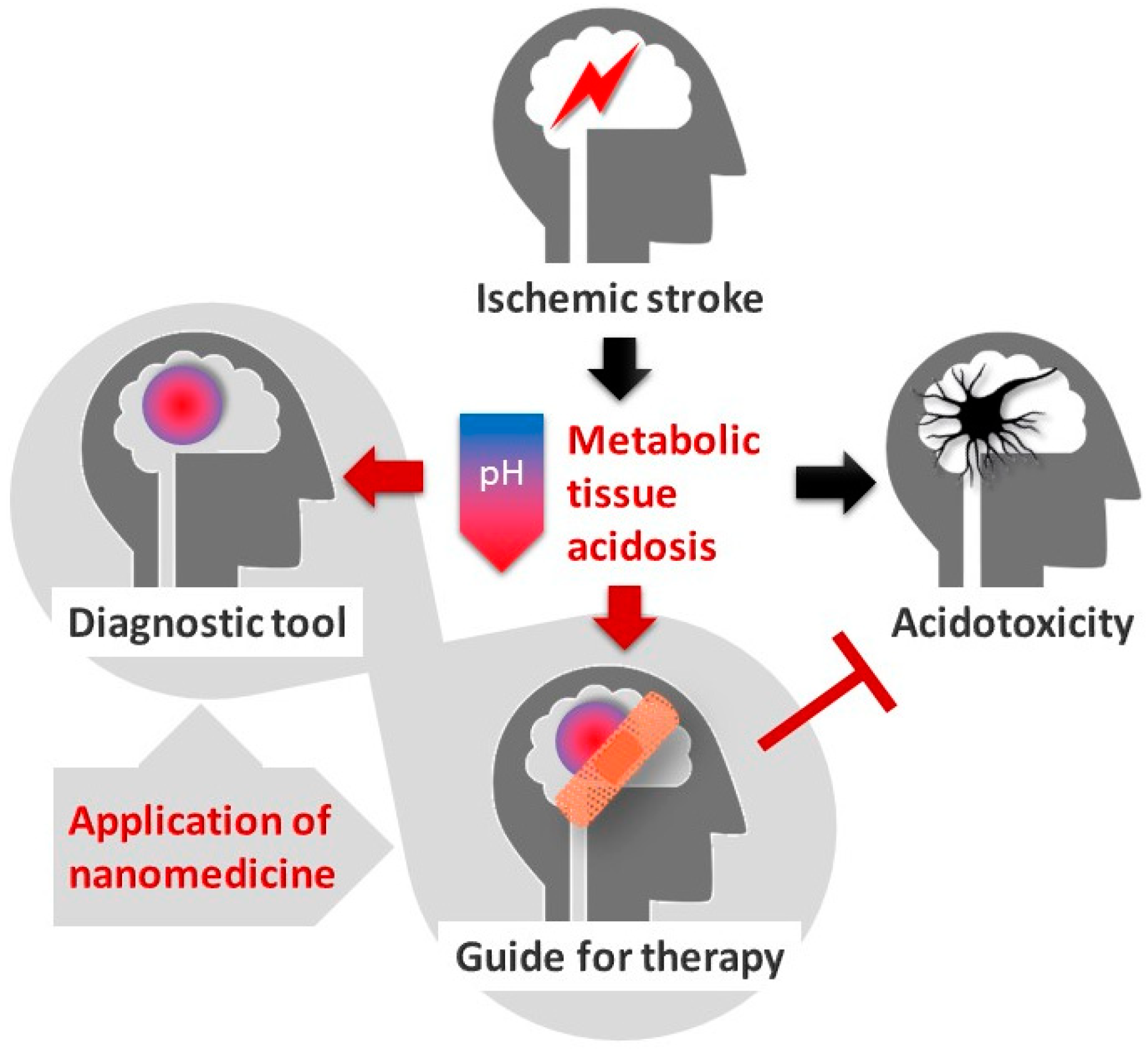
Tissue Acidosis Associated with Ischemic Stroke to Guide Neuroprotective Drug Delivery



{kind=link}
{kind=link}
{kind=link}
{kind=link}
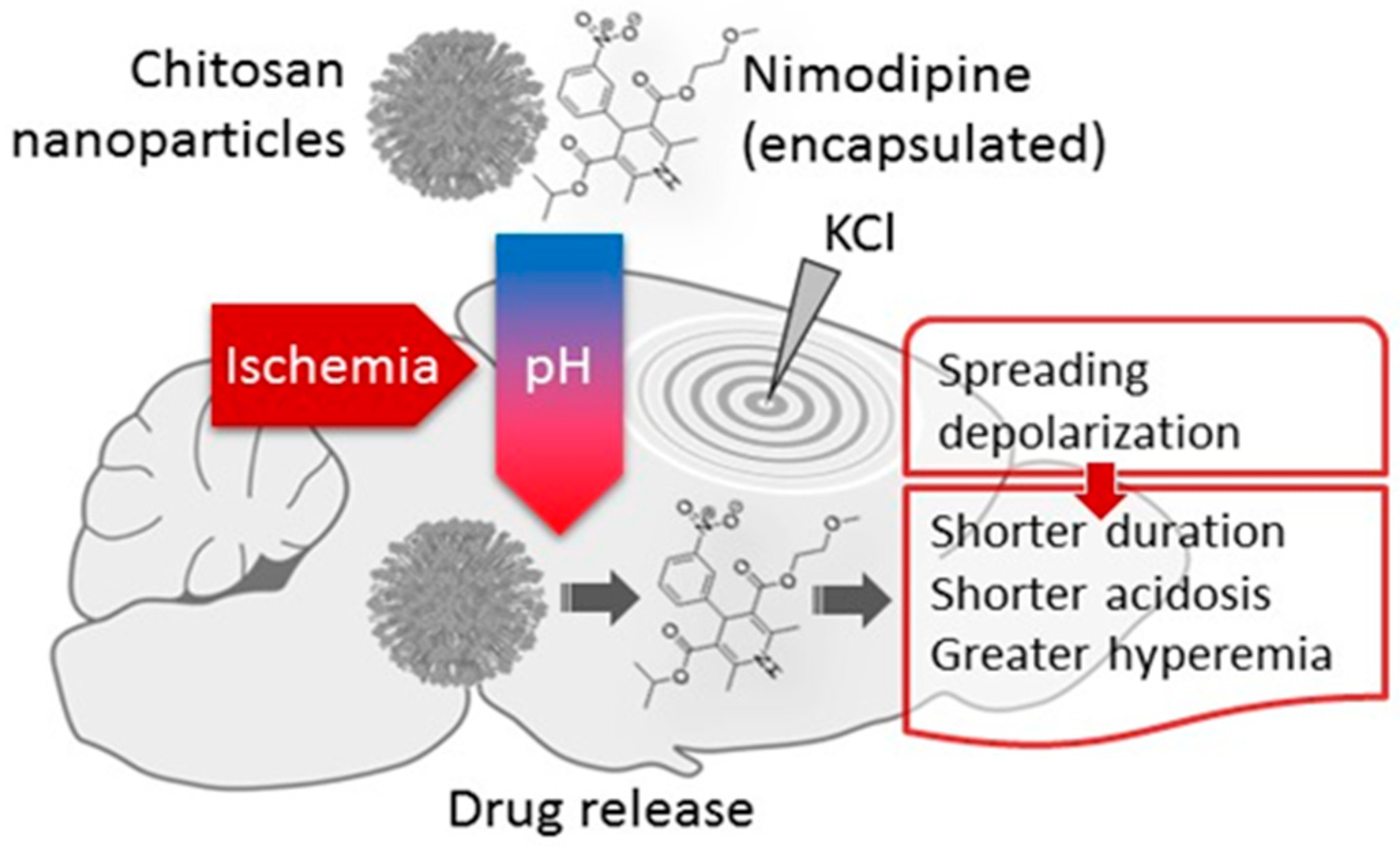{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
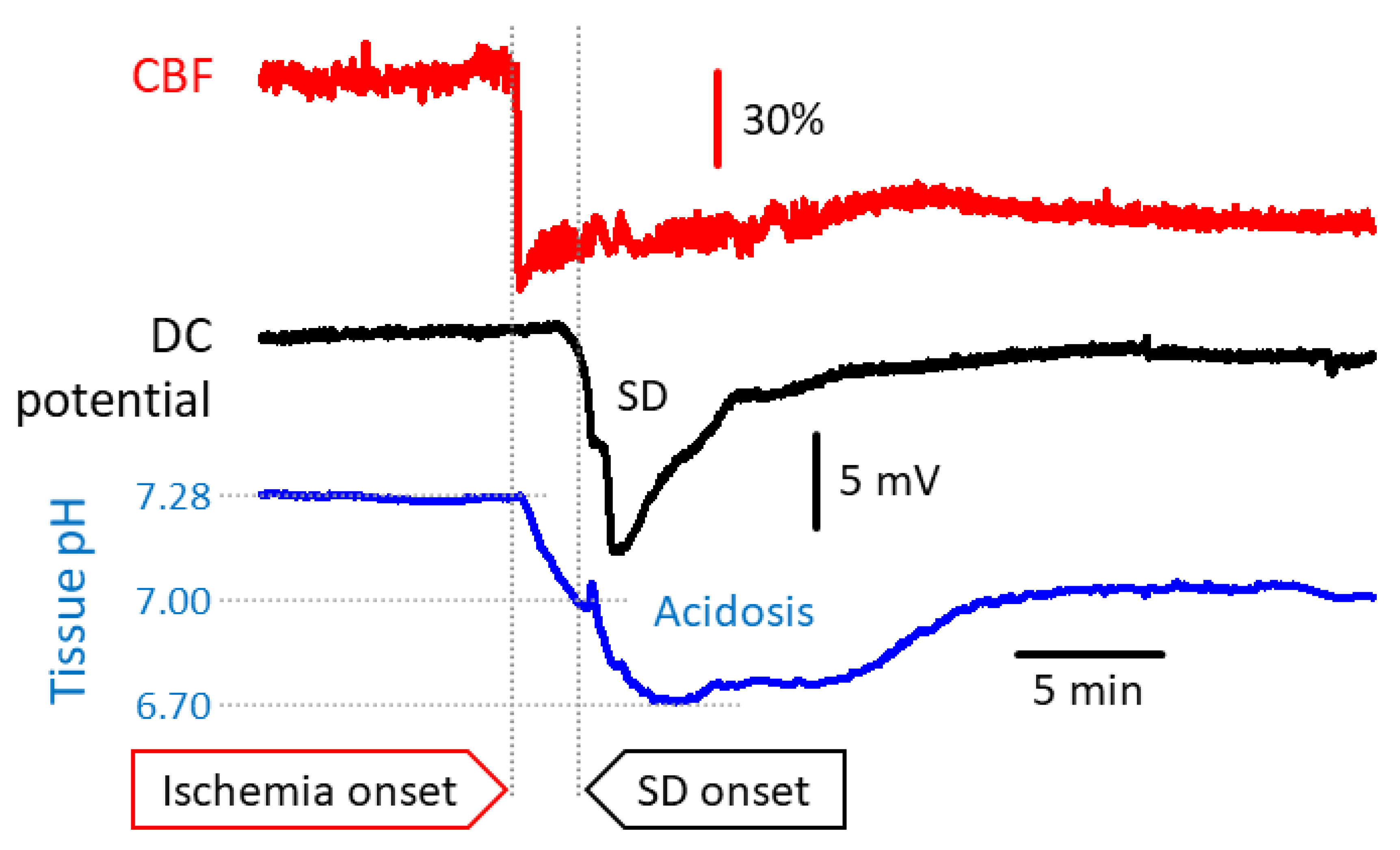
2. Mechanistic Insight into Ischemic Tissue Acidosis and Acidotoxicity
2.1. Mechanisms to Cause Cerebral Ischemic Tissue Acidosis
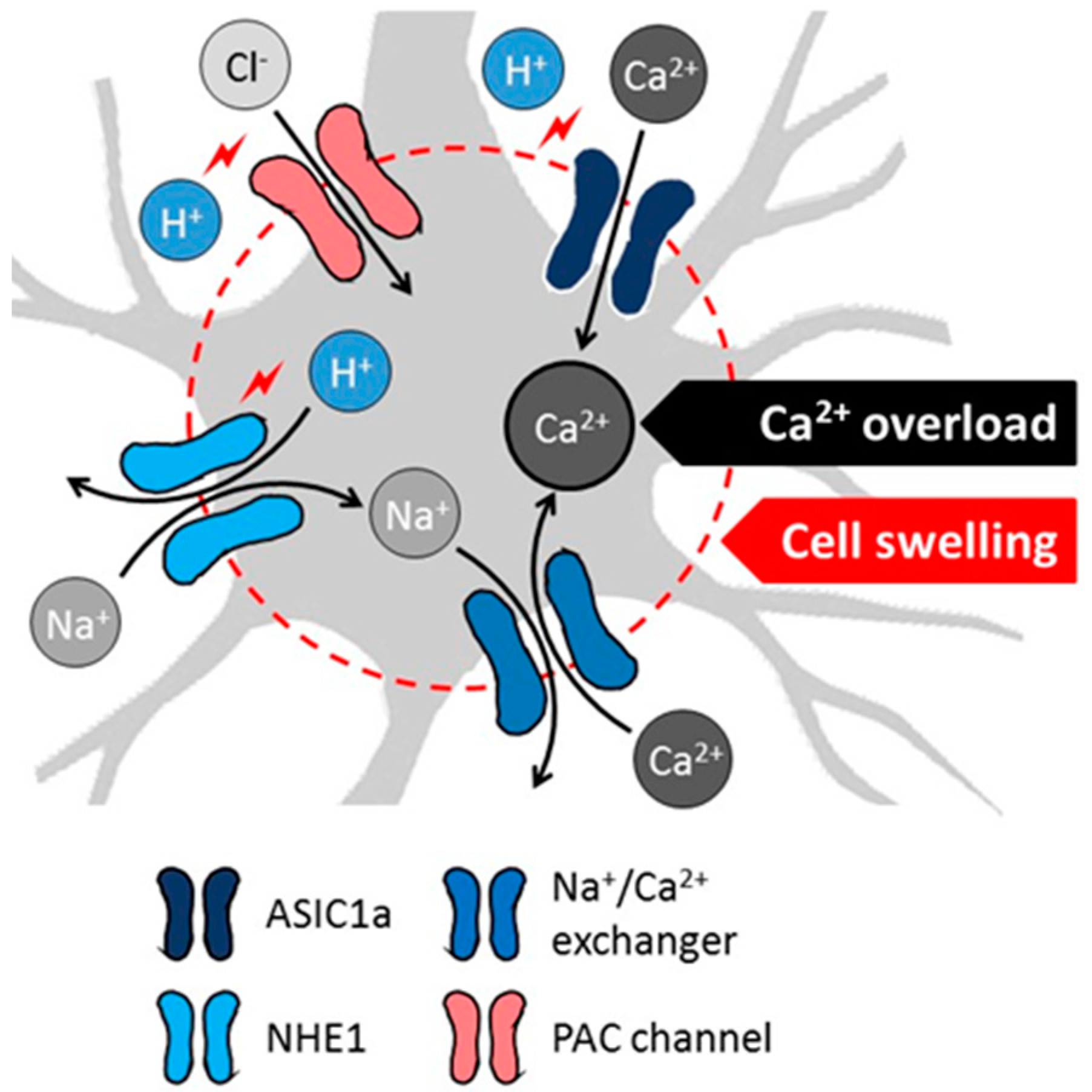
2.2. Mechanistic Insight to Acidosis-Linked Neuronal Injury in Cerebral Ischemia
3. Cerebral Tissue Acidosis in Theranostics and Nanomedicine
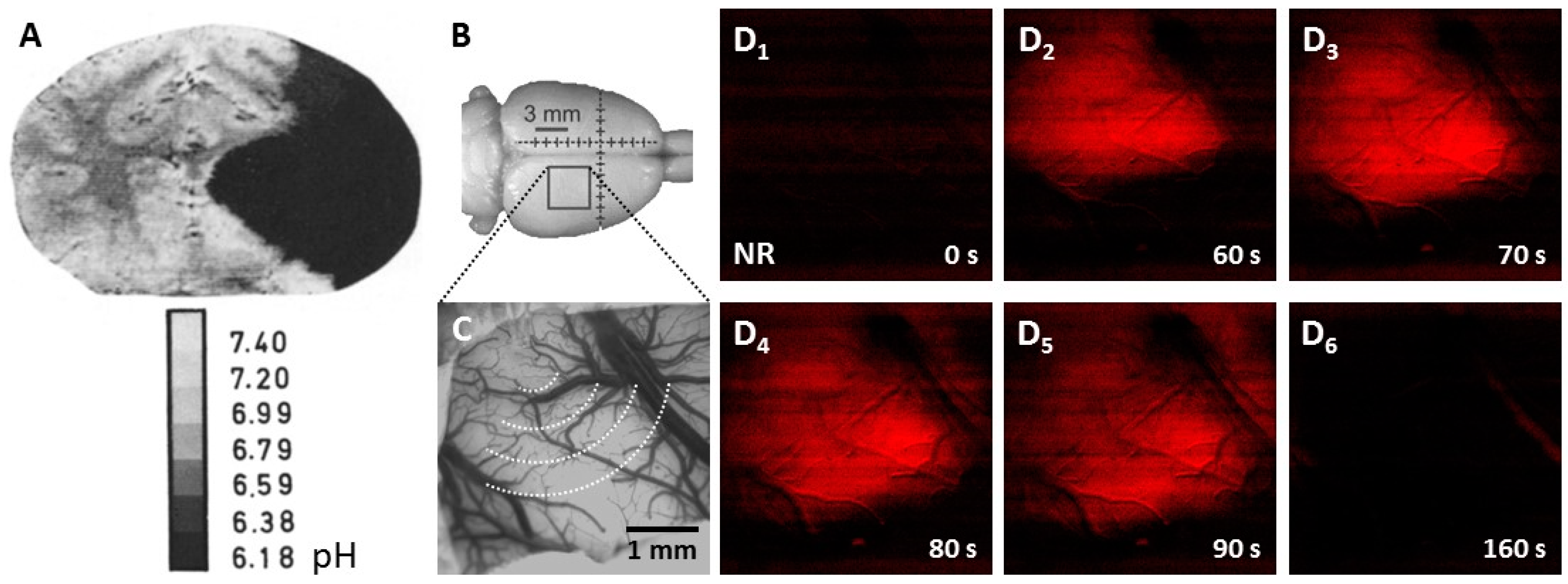
3.1. Tissue Acidosis to Identify Regions at Risk of Ischemic Damage
3.2. Tissue Acidosis to Guide Neuroprotective Intervention in Ischemic Stroke
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Bamford, J.; Sandercock, P.; Dennis, M.; Burn, J.; Warlow, C. Classification and natural history of clinical identifiable subtypes of cerebral infarction. Lancet 1991, 337, 1521–1526. [Google Scholar] [CrossRef]
- Astrup, J.; Symon, L.; Branston, N.M.; Lassen, N.A. Cortical evoked potential and extracellular K+ and H+ at critical levels of brain ischemia. Stroke 1977, 8, 51–57. [Google Scholar] [CrossRef] [PubMed]
- Astrup, J.; Siesjö, B.K.; Symon, L. Thresholds in cerebral ischemia—the ischemic penumbra. Stroke 1981, 12, 723–725. [Google Scholar] [CrossRef] [PubMed]
- Lo, E.H. A new penumbra: Transitioning from injury into repair after stroke. Nat. Med. 2008, 14, 497–500. [Google Scholar] [CrossRef]
- Paciaroni, M.; Caso, V.; Agnelli, G. The concept of ischemic penumbra in acute stroke and therapeutic opportunities. Eur. Neurol. 2009, 61, 321–330. [Google Scholar] [CrossRef]
- Leigh, R.; Knutsson, L.; Zhou, J.; van Zijl, P.C. Imaging the physiological evolution of the ischemic penumbra in acute ischemic stroke. J. Cereb. Blood Flow Metab. 2018, 38, 1500–1516. [Google Scholar] [CrossRef]
- Hossmann, K.A. Viability thresholds and the penumbra of focal ischemia. Ann. Neurol. 1994, 36, 557–565. [Google Scholar] [CrossRef]
- Leao, A.A.P. Spreading depression of activity in the cerebral cortex. J. Neurophys. 1944, 7, 359–390. [Google Scholar] [CrossRef]
- Leao, A.A.P. Further observations on the spreading depression of activity in the cerebral cortex. J. Neurophysiol. 1947, 10, 409–414. [Google Scholar] [CrossRef]
- Hossmann, K.A. Periinfarct depolarizations. Cerebrovasc. Brain Metab. Rev. 1996, 8, 195–208. [Google Scholar]
- Nedergaard, M. Spreading depression as a contributor to ischemic brain damage. Adv. Neurol. 1996, 71, 75–83. [Google Scholar] [PubMed]
- Woitzik, J.; Hecht, N.; Pinczolits, A.; Sandow, N.; Major, S.; Winkler, M.K.; Weber-Carstens, S.; Dohmen, C.; Graf, R.; COSBID Study Group; et al. Propagation of cortical spreading depolarization in the human cortex after malignant stroke. Neurology 2013, 80, 1095–1102. [Google Scholar] [CrossRef] [PubMed]
- Pinczolits, A.; Zdunczyk, A.; Dengler, N.F.; Hecht, N.; Kowoll, C.M.; Dohmen, C.; Graf, R.; Winkler, M.K.; Major, S.; Hartings, J.A.; et al. Standard-sampling microdialysis and spreading depolarizations in patients with malignant hemispheric stroke. J. Cereb. Blood Flow Metab. 2017, 37, 1896–1905. [Google Scholar] [CrossRef] [PubMed]
- Dreier, J.P. The role of spreading depression, spreading depolarization and spreading ischemia in neurological disease. Nat. Med. 2011, 17, 439–447. [Google Scholar] [CrossRef] [PubMed]
- Hartings, J.A.; Shuttleworth, C.W.; Kirov, S.A.; Ayata, C.; Hinzman, J.M.; Foreman, B.; Andrew, R.D.; Boutelle, M.G.; Brennan, K.C.; Carlson, A.P.; et al. The continuum of spreading depolarizations in acute cortical lesion development: Examining Leão’s legacy. J. Cereb. Blood Flow Metab. 2017, 37, 1571–1594. [Google Scholar] [CrossRef]
- Von Bornstädt, D.; Houben, T.; Seidel, J.L.; Zheng, Y.; Dilekoz, E.; Qin, T.; Sandoz, N.; Kura, S.; Eikermann-Haerter, K.; Endres, M.; et al. Supply-demand mismatch transients in susceptible peri-infarct hot zones explain the origins of spreading injury depolarizations. Neuron 2015, 85, 1117–1131. [Google Scholar] [CrossRef]
- Dreier, J.P.; Reiffurth, C. The stroke-migraine depolarization continuum. Neuron 2015, 86, 902–922. [Google Scholar] [CrossRef]
- Mutch, W.A.; Hansen, A.J. Extracellular pH changes during spreading depression and cerebral ischemia: Mechanisms of brain pH regulation. J. Cereb. Blood Flow Metab. 1984, 4, 17–27. [Google Scholar] [CrossRef]
- Menyhárt, Á.; Zölei-Szénási, D.; Puskás, T.; Makra, P.; Orsolya, M.T.; Szepes, B.É.; Tóth, R.; Ivánkovits-Kiss, O.; Obrenovitch, T.P.; Bari, F.; et al. Spreading depolarization remarkably exacerbates ischemia-induced tissue acidosis in the young and aged rat brain. Sci. Rep. 2017, 7, 1154. [Google Scholar] [CrossRef]
- Feuerstein, D.; Manning, A.; Hashemi, P.; Bhatia, R.; Fabricius, M.; Tolias, C.; Pahl, C.; Ervine, M.; Strong, A.J.; Boutelle, M.G. Dynamic metabolic response to multiple spreading depolarizations in patients with acute brain injury: An online microdialysis study. J. Cereb. Blood Flow Metab. 2010, 30, 1343–1355. [Google Scholar] [CrossRef]
- Busch, E.; Gyngell, M.L.; Eis, M.; Hoehn-Berlage, M.; Hossmann, K.A. Potassium-induced cortical spreading depressions during focal cerebral ischemia in rats: Contribution to lesion growth assessed by diffusion-weighted NMR and biochemical imaging. J. Cereb. Blood Flow Metab. 1996, 16, 1090–1099. [Google Scholar] [CrossRef] [PubMed]
- Shuttleworth, C.W.; Andrew, R.D.; Akbari, Y.; Ayata, C.; Balu, R.; Brennan, K.C.; Boutelle, M.; Carlson, A.P.; Dreier, J.P.; Fabricius, M.; et al. Which Spreading Depolarizations Are Deleterious to Brain Tissue? Neurocrit. Care 2020, 32, 317–322. [Google Scholar] [CrossRef] [PubMed]
- Dreier, J.P.; Fabricius, M.; Ayata, C.; Sakowitz, O.W.; Shuttleworth, C.W.; Dohmen, C.; Graf, R.; Vajkoczy, P.; Helbok, R.; Suzuki, M.; et al. Recording, analysis, and interpretation of spreading depolarizations in neurointensive care: Review and recommendations of the COSBID research group. J. Cereb. Blood Flow Metab. 2017, 37, 1595–1625. [Google Scholar] [CrossRef] [PubMed]
- Klass, A.; Sánchez-Porras, R.; Santos, E. Systematic review of the pharmacological agents that have been tested against spreading depolarizations. J. Cereb. Blood Flow Metab. 2018, 38, 1149–1179. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Y.; Manwani, B.; Fisher, M. Management of Acute Ischemic Stroke. Am. J. Med. 2019, 132, 286–291. [Google Scholar] [CrossRef]
- Nair, S.B.; Dileep, A.; Rajanikant, G.K. Nanotechnology based diagnostic and therapeutic strategies for neuroscience with special emphasis on ischemic stroke. Curr. Med. Chem. 2012, 19, 744–756. [Google Scholar] [CrossRef]
- Furtado, D.; Björnmalm, M.; Ayton, S.; Bush, A.I.; Kempe, K.; Caruso, F. Overcoming the Blood-Brain Barrier: The Role of Nanomaterials in Treating Neurological Diseases. Adv. Mater. 2018, 30, e1801362. [Google Scholar] [CrossRef]
- González-Nieto, D.; Fernández-Serra, R.; Pérez-Rigueiro, J.; Panetsos, F.; Martinez-Murillo, R.; Guinea, G.V. Biomaterials to Neuroprotect the Stroke Brain: A Large Opportunity for Narrow Time Windows. Cells 2020, 9, 1074. [Google Scholar] [CrossRef]
- Kwon, E.J.; Lo, J.H.; Bhatia, S.N. Smart nanosystems: Bio-inspired technologies that interact with the host environment. Proc. Natl. Acad. Sci. USA 2015, 112, 14460–14466. [Google Scholar] [CrossRef]
- Kandell, R.M.; Waggoner, L.E.; Kwon, E.J. Nanomedicine for Acute Brain Injuries: Insight from Decades of Cancer Nanomedicine. Mol. Pharm. 2020. online ahead of print. [Google Scholar] [CrossRef]
- Siesjö, B.K. Pathophysiology and treatment of focal cerebral ischemia Part I: Pathophysiology. J. Neurosurg. 1992, 77, 169–184. [Google Scholar] [CrossRef] [PubMed]
- Rehncrona, S. Brain acidosis. Ann. Emerg. Med. 1985, 14, 770–776. [Google Scholar] [CrossRef]
- Paschen, W.; Djuricic, B.; Mies, G.; Schmidt-Kastner, R.; Linn, F. Lactate and pH in the brain: Association and dissociation in different pathophysiological states. J. Neurochem. 1987, 48, 154–159. [Google Scholar] [CrossRef] [PubMed]
- Katsura, K.; Ekholm, A.; Asplund, B.; Siesjö, B.K. Extracellular pH in the brain during ischemia: Relationship to the severity of lactic acidosis. J. Cereb. Blood Flow Metab. 1991, 11, 597–599. [Google Scholar] [CrossRef]
- Von Hanwehr, R.; Smith, M.L.; Siesjö, B.K. Extra- and intracellular pH during near-complete forebrain ischemia in the rat. J. Neurochem. 1986, 46, 331–339. [Google Scholar] [CrossRef]
- Kraig, R.P.; Pulsinelli, W.A.; Plum, F. Carbonic acid buffer changes during complete brain ischemia. Am. J. Physiol. 1986, 250, R348–R357. [Google Scholar] [CrossRef]
- Plum, F.; Kraig, R.P.; Pulsinelli, W.A. Compartmentation of acid-base balance in brain during complete ischemia. Neurochem. Pathol. 1988, 9, 139–144. [Google Scholar] [CrossRef]
- Boris-Möller, F.; Drakenberg, T.; Elmdén, K.; Forsén, S.; Siesjö, B.K. Evidence against major compartmentalization of H+ in ischemic rat brain tissue. Neurosci. Lett. 1988, 85, 113–118. [Google Scholar] [CrossRef]
- Walz, W.; Mukerji, S. Lactate release from cultured astrocytes and neurons: A comparison. Glia 1988, 1, 366–370. [Google Scholar] [CrossRef]
- Kobatake, K.; Sako, K.; Izawa, M.; Yamamoto, Y.L.; Hakim, A.M. Autoradiographic determination of brain pH following middle cerebral artery occlusion in the rat. Stroke 1984, 15, 540–547. [Google Scholar] [CrossRef]
- Back, T.; Hoehn, M.; Mies, G.; Busch, E.; Schmitz, B.; Kohno, K.; Hossmann, K.A. Penumbral tissue alkalosis in focal cerebral ischemia: Relationship to energy metabolism, blood flow, and steady potential. Ann. Neurol. 2000, 47, 485–492. [Google Scholar] [CrossRef]
- Pignataro, G.; Simon, R.P.; Xiong, Z.G. Prolonged activation of ASIC1a and the time window for neuroprotection in cerebral ischaemia. Brain 2007, 130, 151–158. [Google Scholar] [CrossRef] [PubMed]
- Behar, K.L.; Rothman, D.L.; Hossmann, K.A. NMR spectroscopic investigation of the recovery of energy and acid-base homeostasis in the cat brain after prolonged ischemia. J. Cereb. Blood Flow Metab. 1989, 9, 655–665. [Google Scholar] [CrossRef] [PubMed]
- Scheller, D.; Kolb, J.; Tegtmeier, F. Lactate and pH change in close correlation in the extracellular space of the rat brain during cortical spreading depression. Neurosci. Lett. 1992, 135, 83–86. [Google Scholar] [CrossRef]
- Selman, W.R.; Lust, W.D.; Pundik, S.; Zhou, Y.; Ratcheson, R.A. Compromised metabolic recovery following spontaneous spreading depression in the penumbra. Brain Res. 2004, 999, 167–174. [Google Scholar] [CrossRef] [PubMed]
- Menyhárt, Á.; Zölei-Szénási, D.; Puskás, T.; Makra, P.; Bari, F.; Farkas, E. Age or ischemia uncouples the blood flow response, tissue acidosis, and direct current potential signature of spreading depolarization in the rat brain. Am. J. Physiol. Heart Circ. Physiol. 2017, 313, H328–H337. [Google Scholar] [CrossRef] [PubMed]
- Ay, H.; Koroshetz, W.J.; Vangel, M.; Benner, T.; Melinosky, C.; Zhu, M.; Menezes, N.; Lopez, C.J.; Sorensen, A.G. Conversion of ischemic brain tissue into infarction increases with age. Stroke 2005, 36, 2632–2636. [Google Scholar] [CrossRef]
- Feuerstein, D.; Backes, H.; Gramer, M.; Takagaki, M.; Gabel, P.; Kumagai, T.; Graf, R. Regulation of cerebral metabolism during cortical spreading depression. J. Cereb. Blood Flow Metab. 2016, 36, 1965–1977. [Google Scholar] [CrossRef]
- Nedergaard, M.; Goldman, S.A.; Desai, S.; Pulsinelli, W.A. Acid-induced death in neurons and glia. J. Neurosci. 1991, 11, 2489–2497. [Google Scholar] [CrossRef]
- Chesler, M. Failure and function of intracellular pH regulation in acute hypoxic-ischemic injury of astrocytes. Glia 2005, 50, 398–406. [Google Scholar] [CrossRef]
- Siesjö, B.K.; Katsura, K.I.; Kristián, T.; Li, P.A.; Siesjö, P. Molecular Mechanisms of Acidosis-Mediated Damage. Acta Neurochirurgica Suppl. 1996, 66, 8–14. [Google Scholar] [CrossRef]
- Busa, W.B.; Nuccitelli, R. Metabolic regulation via intracellular pH. Am. J. Physiol. 1984, 246, R409–R438. [Google Scholar] [CrossRef] [PubMed]
- Hillered, L.; Ernster, L.; Siesjö, B.K. Influence of in vitro lactic acidosis and hypercapnia on respiratory activity of isolated rat brain mitochondria. J. Cereb. Blood Flow Metab. 1984, 4, 430–437. [Google Scholar] [CrossRef]
- Siesjö, B.K.; Bendek, G.; Koide, T.; Westerberg, E.; Wieloch, T. Influence of acidosis on lipid peroxidation in brain tissues in vitro. J. Cereb. Blood Flow Metab. 1985, 5, 253–258. [Google Scholar] [CrossRef] [PubMed]
- Frøyland, E.; Skjaeret, C.; Wright, M.S.; Dalen, M.L.; Cvancarova, M.; Kasi, C.; Rootwelt, T. Inflammatory receptors and pathways in human NT2-N neurons during hypoxia and reoxygenation. Impact of acidosis. Brain Res. 2008, 1217, 37–49. [Google Scholar] [CrossRef] [PubMed]
- Wemmie, J.A.; Price, M.P.; Welsh, M.J. Acid-sensing ion channels: Advances, questions and therapeutic opportunities. Trends Neurosci. 2006, 29, 578–586. [Google Scholar] [CrossRef] [PubMed]
- Leng, T.; Shi, Y.; Xiong, Z.G.; Sun, D. Proton-sensitive cation channels and ion exchangers in ischemic brain injury: New therapeutic targets for stroke? Prog. Neurobiol. 2014, 115, 189–209. [Google Scholar] [CrossRef]
- Xiong, Z.G.; Zhu, X.M.; Chu, X.P.; Minami, M.; Hey, J.; Wei, W.L.; MacDonald, J.F.; Wemmie, J.A.; Price, M.P.; Welsh, M.J.; et al. Neuroprotection in ischemia: Blocking calcium-permeable acid-sensing ion channels. Cell 2004, 118, 687–698. [Google Scholar] [CrossRef]
- Yermolaieva, O.; Leonard, A.S.; Schnizler, M.K.; Abboud, F.M.; Welsh, M.J. Extracellular acidosis increases neuronal cell calcium by activating acid-sensing ion channel 1a. Proc. Natl. Acad. Sci. USA 2004, 101, 6752–6757. [Google Scholar] [CrossRef]
- Mari, Y.; Katnik, C.; Cuevas, J. ASIC1a channels are activated by endogenous protons during ischemia and contribute to synergistic potentiation of intracellular Ca(2+) overload during ischemia and acidosis. Cell Calcium. 2010, 48, 70–82. [Google Scholar] [CrossRef]
- Gao, J.; Duan, B.; Wang, D.G.; Deng, X.H.; Zhang, G.Y.; Xu, L.; Xu, T.L. Coupling between NMDA receptor and acid-sensing ion channel contributes to ischemic neuronal death. Neuron 2005, 48, 635–646. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.Z.; Wang, J.J.; Huang, Y.; Liu, F.; Zeng, W.Z.; Li, Y.; Xiong, Z.G.; Zhu, M.X.; Xu, T.L. Tissue acidosis induces neuronal necroptosis via ASIC1a channel independent of its ionic conduction. eLife 2015, 4, e05682. [Google Scholar] [CrossRef]
- Yang, J.; Chen, J.; Del Carmen Vitery, M.; Osei-Owusu, J.; Chu, J.; Yu, H.; Sun, S.; Qiu, Z. PAC, an evolutionarily conserved membrane protein, is a proton-activated chloride channel. Science 2019, 364, 395–399. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.Y.; Shimizu, T.; Numata, T.; Okada, Y. Role of acid-sensitive outwardly rectifying anion channels in acidosis-induced cell death in human epithelial cells. Pflugers Arch. 2007, 454, 223–233. [Google Scholar] [CrossRef] [PubMed]
- Osei-Owusu, J.; Yang, J.; Del Carmen Vitery, M.; Tian, M.; Qiu, Z. PAC proton-activated chloride channel contributes to acid-induced cell death in primary rat cortical neurons. Channels (Austin) 2020, 14, 53–58. [Google Scholar] [CrossRef] [PubMed]
- Staub, F.; Peters, J.; Kempski, O.; Schneider, G.H.; Schürer, L.; Baethmann, A. Swelling of glial cells in lactacidosis and by glutamate: Significance of Cl(-)-transport. Brain Res. 1993, 610, 69–74. [Google Scholar] [CrossRef]
- Nabekura, T.; Morishima, S.; Cover, T.L.; Mori, S.; Kannan, H.; Komune, S.; Okada, Y. Recovery from lactacidosis-induced glial cell swelling with the aid of exogenous anion channels. Glia 2003, 41, 247–259. [Google Scholar] [CrossRef]
- Manhas, N.; Shi, Y.; Taunton, J.; Sun, D. p90 Activation contributes to cerebral Ischemic damage via phosphorylation of Na+/H+ exchanger isoform 1. J. Neurochem. 2010, 114, 1476–1486. [Google Scholar] [CrossRef]
- Kintner, D.B.; Chen, X.; Currie, J.; Chanana, V.; Ferrazzano, P.; Baba, A.; Matsuda, T.; Cohen, M.; Orlowski, J.; Chiu, S.Y.; et al. Excessive Na+/H+ exchange in disruption of dendritic Na+ and Ca2+ homeostasis and mitochondrial dysfunction following in vitro ischemia. J. Biol. Chem. 2010, 285, 35155–35168. [Google Scholar] [CrossRef]
- Luo, J.; Chen, H.; Kintner, D.B.; Shull, G.E.; Sun, D. Decreased neuronal death in Na+/H+ exchanger isoform 1-null mice after in vitro and in vivo ischemia. J. Neurosci. 2005, 25, 11256–11268. [Google Scholar] [CrossRef]
- Wang, Y.; Luo, J.; Chen, X.; Chen, H.; Cramer, S.W.; Sun, D. Gene inactivation of Na+/H+ exchanger isoform 1 attenuates apoptosis and mitochondrial damage following transient focal cerebral ischemia. Eur. J. Neurosci. 2008, 28, 51–61. [Google Scholar] [CrossRef] [PubMed]
- Kang, B.S.; Choi, B.Y.; Kho, A.R.; Lee, S.H.; Hong, D.K.; Jeong, J.H.; Kang, D.H.; Park, M.K.; Suh, S.W. An Inhibitor of the Sodium-Hydrogen Exchanger-1 (NHE-1), Amiloride, Reduced Zinc Accumulation and Hippocampal Neuronal Death after Ischemia. Int. J. Mol. Sci. 2020, 21, 4232. [Google Scholar] [CrossRef] [PubMed]
- Wemmie, J.A.; Taugher, R.J.; Kreple, C.J. Acid-sensing ion channels in pain and disease. Nat. Rev. Neurosci. 2013, 14, 461–471. [Google Scholar] [CrossRef] [PubMed]
- Thirugnanachandran, T.; Ma, H.; Singhal, S.; Slater, L.A.; Davis, S.M.; Donnan, G.A.; Phan, T. Refining the ischemic penumbra with topography. Int. J. Stroke 2018, 13, 277–284. [Google Scholar] [CrossRef]
- Oishi, M.; Nagasaki, Y. Stimuli-responsive smart nanogels for cancer diagnostics and therapy. Nanomedicine (Lond.) 2010, 5, 451–468. [Google Scholar] [CrossRef]
- Liu, J.; Huang, Y.; Kumar, A.; Tan, A.; Jin, S.; Mozhi, A.; Liang, X.J. pH-sensitive nano-systems for drug delivery in cancer therapy. Biotechnol. Adv. 2014, 32, 693–710. [Google Scholar] [CrossRef]
- Tóth, M.O.; Menyhárt, Á.; Varga, V.É.; Hantosi, D.; Ivánkovits-Kiss, O.; Varga, D.P.; Szabó, Í.; Janovák, L.; Dékány, I.; Farkas, E. Chitosan nanoparticles release nimodipine in response to tissue acidosis to attenuate spreading depolarization evoked during forebrain ischemia. Neuropharmacology 2020, 162, 107850. [Google Scholar] [CrossRef]
- Janovák, L.; Turcsányi, Á.; Bozó, É.; Deák, Á.; Mérai, L.; Sebők, D.; Juhász, Á.; Csapó, E.; Abdelghafour, M.M.; Farkas, E.; et al. Preparation of novel tissue acidosis-responsive chitosan drug nanoparticles: Characterization and in vitro release properties of Ca2+ channel blocker nimodipine drug molecules. Eur. J. Pharm. Sci. 2018, 123, 79–88. [Google Scholar] [CrossRef]
- Kogure, K.; Alonso, O.F.; Martinez, E. A topographic measurement of brain pH. Brain Res. 1980, 195, 95–109. [Google Scholar] [CrossRef]
- Csiba, L.; Paschen, W.; Hossmann, K.A. A topographic quantitative method for measuring brain tissue pH under physiological and pathophysiological conditions. Brain Res. 1983, 289, 334–337. [Google Scholar] [CrossRef]
- Chen, G.; Hanson, C.L.; Ebner, T.J. Optical responses evoked by cerebellar surface stimulation in vivo using neutral red. Neuroscience 1998, 84, 645–668. [Google Scholar] [CrossRef]
- Sun, X.; Wang, Y.; Chen, S.; Luo, W.; Li, P.; Luo, Q. Simultaneous monitoring of intracellular pH changes and hemodynamic response during cortical spreading depression by fluorescence-corrected multimodal optical imaging. Neuroimage 2011, 57, 873–884. [Google Scholar] [CrossRef] [PubMed]
- Bo, B.; Li, Y.; Li, W.; Wang, Y.; Tong, S. Optogenetic translocation of protons out of penumbral neurons is protective in a rodent model of focal cerebral ischemia. Brain Stimul. 2020, 13, 881–890. [Google Scholar] [CrossRef] [PubMed]
- Rottenberg, D.A.; Ginos, J.Z.; Kearfott, K.J.; Junck, L.; Bigner, D.D. In vivo measurement of regional brain tissue pH using positron emission tomography. Ann. Neurol. 1984, 15, S98–S102. [Google Scholar] [CrossRef]
- Senda, M.; Alpert, N.M.; Mackay, B.C.; Buxton, R.B.; Correia, J.A.; Weise, S.B.; Ackerman, R.H.; Dorer, D.; Buonanno, F.S. Evaluation of the 11CO2 positron emission tomographic method for measuring brain pH. II. Quantitative pH mapping in patients with ischemic cerebrovascular diseases. J. Cereb. Blood Flow Metab. 1989, 9, 859–873. [Google Scholar] [CrossRef]
- Henry, K.E.; Chaney, A.M.; Nagle, V.L.; Cropper, H.C.; Mozaffari, S.; Slaybaugh, G.; Parang, K.; Andreev, O.; Reshetnyak, Y.K.; James, M.L.; et al. Demarcation of Sepsis-Induced Peripheral and Central Acidosis with pH-Low Insertion Cyclic (pHLIC) Peptide. J. Nucl. Med. 2020, 61, 1361–1368. [Google Scholar] [CrossRef]
- Zhou, J.; van Zijl, P.C.M. Defining an Acidosis-Based Ischemic Penumbra from pH-Weighted MRI. Transl. Stroke Res. 2011, 3, 76–83. [Google Scholar] [CrossRef]
- Zhou, J.; Payen, J.F.; Wilson, D.A.; Traystman, R.J.; van Zijl, P.C.M. Using the amide proton signals of intracellular proteins and peptides to detect pH effects in MRI. Nat. Med. 2003, 9, 1085–1090. [Google Scholar] [CrossRef]
- Harston, G.W.; Tee, Y.K.; Blockley, N.; Okell, T.W.; Thandeswaran, S.; Shaya, G.; Sheerin, F.; Cellerini, M.; Payne, S.; Jezzard, P.; et al. Identifying the ischaemic penumbra using pH-weighted magnetic resonance imaging. Brain 2015, 138, 36–42. [Google Scholar] [CrossRef]
- Yu, L.; Chen, Y.; Chen, M.; Luo, X.; Jiang, S.; Zhang, Y.; Chen, H.; Gong, T.; Zhou, J.; Li, C. Amide Proton Transfer MRI Signal as a Surrogate Biomarker of Ischemic Stroke Recovery in Patients With Supportive Treatment. Front. Neurol. 2019, 10, 104. [Google Scholar] [CrossRef]
- Huang, G.; Zhao, T.; Wang, C.; Nham, K.; Xiong, Y.; Gao, X.; Wang, Y.; Hao, G.; Ge, X.P.; Sun, X.; et al. PET imaging of occult tumours by temporal integration of tumour-acidosis signals from pH-sensitive 64Cu-labelled polymers. Nat. Biomed. Eng. 2020, 4, 314–324. [Google Scholar] [CrossRef] [PubMed]
- Gao, G.H.; Lee, J.W.; Nguyen, M.K.; Im, G.H.; Yang, J.; Heo, H.; Jeon, P.; Park, T.G.; Lee, J.H.; Lee, D.S. pH-responsive polymeric micelle based on PEG-poly(β-amino ester)/(amido amine) as intelligent vehicle for magnetic resonance imaging in detection of cerebral ischemic area. J. Control Release 2011, 155, 11–17. [Google Scholar] [CrossRef] [PubMed]
- Amani, H.; Habibey, R.; Hajmiresmail, S.J.; Latifi, S.; Pazoki-Toroudi, H.; Akhavan, O. Antioxidant nanomaterials in advanced diagnoses and treatments of ischemia reperfusion injuries. J. Mater Chem. B 2017, 5, 9452–9476. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Gao, X. The application of nanoparticles for neuroprotection in acute ischemic stroke. Ther. Deliv. 2017, 8, 915–928. [Google Scholar] [CrossRef] [PubMed]
- Kaviarasi, S.; Yuba, E.; Harada, A.; Krishnan, U.M. Emerging paradigms in nanotechnology for imaging and treatment of cerebral ischemia. J. Control Release 2019, 300, 22–45. [Google Scholar] [CrossRef]
- Luo, Z.; Cai, K.; Hu, Y.; Zhao, L.; Liu, P.; Duan, L.; Yang, W. Mesoporous silica nanoparticles end-capped with collagen: Redox-responsive nanoreservoirs for targeted drug delivery. Angew. Chem. Int. Ed. Engl. 2011, 50, 640–643. [Google Scholar] [CrossRef]
- Felber, A.E.; Dufresne, M.H.; Leroux, J.C. pH-sensitive vesicles, polymeric micelles, and nanospheres prepared with polycarboxylates. Adv. Drug Deliv. Rev. 2012, 64, 979–992. [Google Scholar] [CrossRef]
- Zhai, J.; Zhao, M.; Cao, X.; Li, M.; Zhao, M. Metal-Ion-Responsive Bionanocomposite for Selective and Reversible Enzyme Inhibition. J. Am. Chem. Soc. 2018, 140, 16925–16928. [Google Scholar] [CrossRef]
- Thistlethwaite, A.J.; Leeper, D.B.; Moylan, D.J., 3rd; Nerlinger, R.E. pH distribution in human tumors. Int. J. Radiat. Oncol. Biol. Phys. 1985, 11, 1647–1652. [Google Scholar] [CrossRef]
- Martin, G.R.; Jain, R.K. Noninvasive measurement of interstitial pH profiles in normal and neoplastic tissue using fluorescence ratio imaging microscopy. Cancer Res. 1994, 54, 5670–5674. [Google Scholar]
- Engin, K.; Leeper, D.B.; Cater, J.R.; Thistlethwaite, A.J.; Tupchong, L.; McFarlane, J.D. Extracellular pH distribution in human tumours. Int. J. Hyperthermia 1995, 11, 211–216. [Google Scholar] [CrossRef] [PubMed]
- Shenoy, D.; Little, S.; Langer, R.; Amiji, M. Poly(ethylene oxide)-modified poly(beta-amino ester) nanoparticles as a pH-sensitive system for tumor-targeted delivery of hydrophobic drugs: Part 2. In vivo distribution and tumor localization studies. Pharm. Res. 2005, 22, 2107–2114. [Google Scholar] [CrossRef] [PubMed]
- Poon, Z.; Chang, D.; Zhao, X.; Hammond, P.T. Layer-by-layer nanoparticles with a pH-sheddable layer for in vivo targeting of tumor hypoxia. ACS Nano 2011, 5, 4284–4292. [Google Scholar] [CrossRef] [PubMed]
- Allen, G.S.; Ahn, H.S.; Preziosi, T.J.; Battye, R.; Boone, S.C.; Chou, S.N.; Kelly, D.L.; Weir, B.K.; Crabbe, R.A.; Lavik, P.J.; et al. Cerebral arterial spasm--a controlled trial of nimodipine in patients with subarachnoid hemorrhage. N. Engl. J. Med. 1983, 308, 619–624. [Google Scholar] [CrossRef] [PubMed]
- Freedman, D.D.; Waters, D.D. ‘Second generation’ dihydropyridine calcium antagonists. Greater vascular selectivity and some unique applications. Drugs 1987, 34, 578–598. [Google Scholar] [CrossRef] [PubMed]
- Scriabine, A.; Schuurman, T.; Traber, J. Pharmacological basis for the use of nimodipine in central nervous system disorders. FASEB J. 1989, 3, 1799–1806. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Vila, E.; Guillén, F.; Villanueva, J.A.; Matías-Guiu, J.; Bigorra, J.; Gil, P.; Carbonell, A.; Martínez-Lage, J.M. Placebo-controlled trial of nimodipine in the treatment of acute ischemic cerebral infarction. Stroke 1990, 21, 1023–1028. [Google Scholar] [CrossRef]
- Dreier, J.P.; Windmüller, O.; Petzold, G.; Lindauer, U.; Einhäupl, K.M.; Dirnagl, U. Ischemia triggered by red blood cell products in the subarachnoid space is inhibited by nimodipine administration or moderate volume expansion/hemodilution in rats. Neurosurgery 2002, 51, 1457–1465. [Google Scholar] [CrossRef]
- Menyhárt, Á.; Farkas, A.E.; Varga, D.P.; Frank, R.; Tóth, R.; Bálint, A.R.; Makra, P.; Dreier, J.P.; Bari, F.; Krizbai, I.A.; et al. Large-conductance Ca2+-activated potassium channels are potently involved in the inverse neurovascular response to spreading depolarization. Neurobiol. Dis. 2018, 119, 41–52. [Google Scholar] [CrossRef]
- Szabó, Í.; Tóth, M.O.; Török, Z.; Varga, D.P.; Menyhárt, Á.; Frank, R.; Hantosi, D.; Hunya, Á.; Bari, F.; Horváth, I.; et al. The impact of dihydropyridine derivatives on the cerebral blood flow response to somatosensory stimulation and spreading depolarization. Br. J. Pharmacol. 2019, 176, 1222–1234. [Google Scholar] [CrossRef]
- Sarvaiya, J.; Agrawal, Y.K. Chitosan as a suitable nanocarrier material for anti-Alzheimer drug delivery. Int. J. Biol. Macromol. 2015, 72, 454–465. [Google Scholar] [CrossRef] [PubMed]
- Wilson, B.; Samanta, M.K.; Santhi, K.; Kumar, K.P.S.; Ramasamy, M.; Suresh, B. Chitosan nanoparticles as a new delivery system for the anti-Alzheimer drug tacrine. Nanomedicine 2010, 6, 144–152. [Google Scholar] [CrossRef] [PubMed]
- Aktaş, Y.; Yemisci, M.; Andrieux, K.; Gürsoy, R.N.; Alonso, M.J.; Fernandez-Megia, E.; Novoa-Carballal, R.; Quiñoá, E.; Riguera, R.; Sargon, M.F.; et al. Development and brain delivery of chitosan-PEG nanoparticles functionalized with the monoclonal antibody OX26. Bioconjug Chem. 2005, 16, 1503–1511. [Google Scholar] [CrossRef] [PubMed]
- Yemişci, M.; Gürsoy-Özdemir, Y.; Caban, S.; Bodur, E.; Capan, Y.; Dalkara, T. Transport of a caspase inhibitor across the blood-brain barrier by chitosan nanoparticles. Methods Enzymol. 2012, 508, 253–269. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Andjelkovic, A.V.; Zhu, L.; Yang, T.; Bennett, M.V.L.; Chen, J.; Keep, R.F.; Shi, Y. Blood-brain barrier dysfunction and recovery after ischemic stroke. Prog. Neurobiol. 2018, 163–164, 144–171. [Google Scholar] [CrossRef] [PubMed]
- Gursoy-Ozdemir, Y.; Qiu, J.; Matsuoka, N.; Bolay, H.; Bermpohl, D.; Jin, H.; Wang, X.; Rosenberg, G.A.; Lo, E.H.; Moskowitz, M.A. Cortical spreading depression activates and upregulates MMP-9. J. Clin. Invest. 2004, 113, 1447–1455. [Google Scholar] [CrossRef] [PubMed]
- Cottier, K.E.; Galloway, E.A.; Calabrese, E.C.; Tome, M.E.; Liktor-Busa, E.; Kim, J.; Davis, T.P.; Vanderah, T.W.; Largent-Milnes, T.M. Loss of Blood-Brain Barrier Integrity in a KCl-Induced Model of Episodic Headache Enhances CNS Drug Delivery. eNeuro 2018, 5. [Google Scholar] [CrossRef]
- Sadeghian, H.; Lacoste, B.; Qin, T.; Toussay, X.; Rosa, R.; Oka, F.; Chung, D.Y.; Takizawa, T.; Gu, C.; Ayata, C. Spreading depolarizations trigger caveolin-1-dependent endothelial transcytosis. Ann. Neurol. 2018, 84, 409–423. [Google Scholar] [CrossRef]
- Joshi, S.; Meyers, P.M.; Ornstein, E. Intracarotid delivery of drugs: The potential and the pitfalls. Anesthesiology 2008, 109, 543–564. [Google Scholar] [CrossRef]
- Yu, S.; Xu, X.; Feng, J.; Liu, M.; Hu, K. Chitosan and chitosan coating nanoparticles for the treatment of brain disease. Int. J. Pharm. 2019, 560, 282–293. [Google Scholar] [CrossRef]
- Dodane, V.; Khan, M.A.; Merwin, J.R. Effect of chitosan on epithelial permeability and structure. Int. J. Pharm. 1999, 182, 21–32. [Google Scholar] [CrossRef]
- Smith, J.; Wood, E.; Dornish, M. Effect of chitosan on epithelial cell tight junctions. Pharm. Res. 2004, 21, 43–49. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
M. Tóth, O.; Menyhárt, Á.; Frank, R.; Hantosi, D.; Farkas, E.; Bari, F. Tissue Acidosis Associated with Ischemic Stroke to Guide Neuroprotective Drug Delivery. Biology 2020, 9, 460. https://doi.org/10.3390/biology9120460
M. Tóth O, Menyhárt Á, Frank R, Hantosi D, Farkas E, Bari F. Tissue Acidosis Associated with Ischemic Stroke to Guide Neuroprotective Drug Delivery. Biology. 2020; 9(12):460. https://doi.org/10.3390/biology9120460
Chicago/Turabian StyleM. Tóth, Orsolya, Ákos Menyhárt, Rita Frank, Dóra Hantosi, Eszter Farkas, and Ferenc Bari. 2020. "Tissue Acidosis Associated with Ischemic Stroke to Guide Neuroprotective Drug Delivery" Biology 9, no. 12: 460. https://doi.org/10.3390/biology9120460
APA StyleM. Tóth, O., Menyhárt, Á., Frank, R., Hantosi, D., Farkas, E., & Bari, F. (2020). Tissue Acidosis Associated with Ischemic Stroke to Guide Neuroprotective Drug Delivery. Biology, 9(12), 460. https://doi.org/10.3390/biology9120460