The Platelet Fraction Is a Novel Reservoir to Detect Lyme Borrelia in Blood

 , , and
, , and
Simple Summary
Abstract
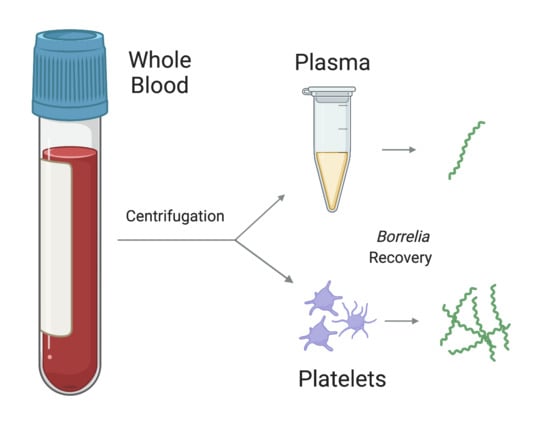
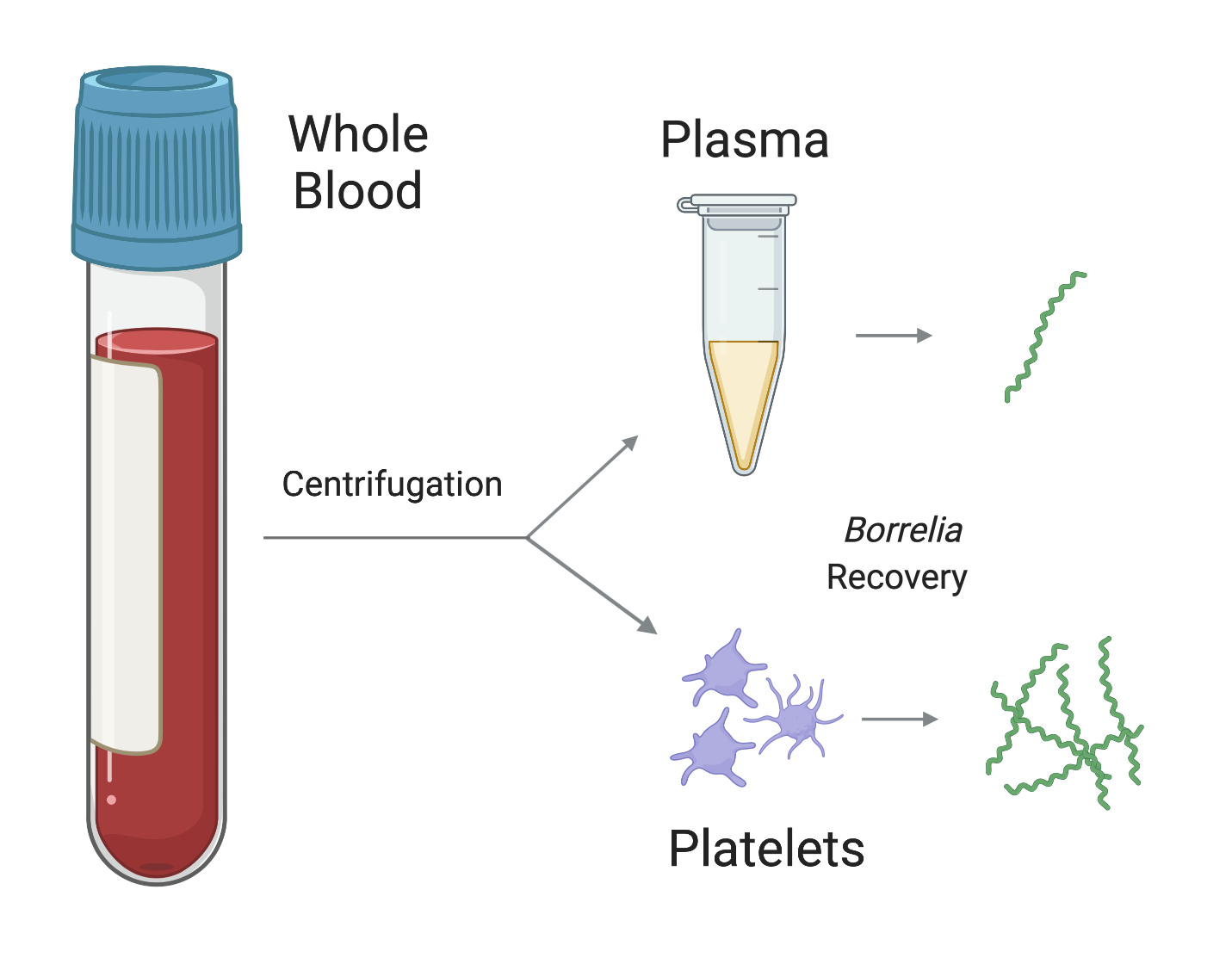
1. Introduction
2. Materials and Methods
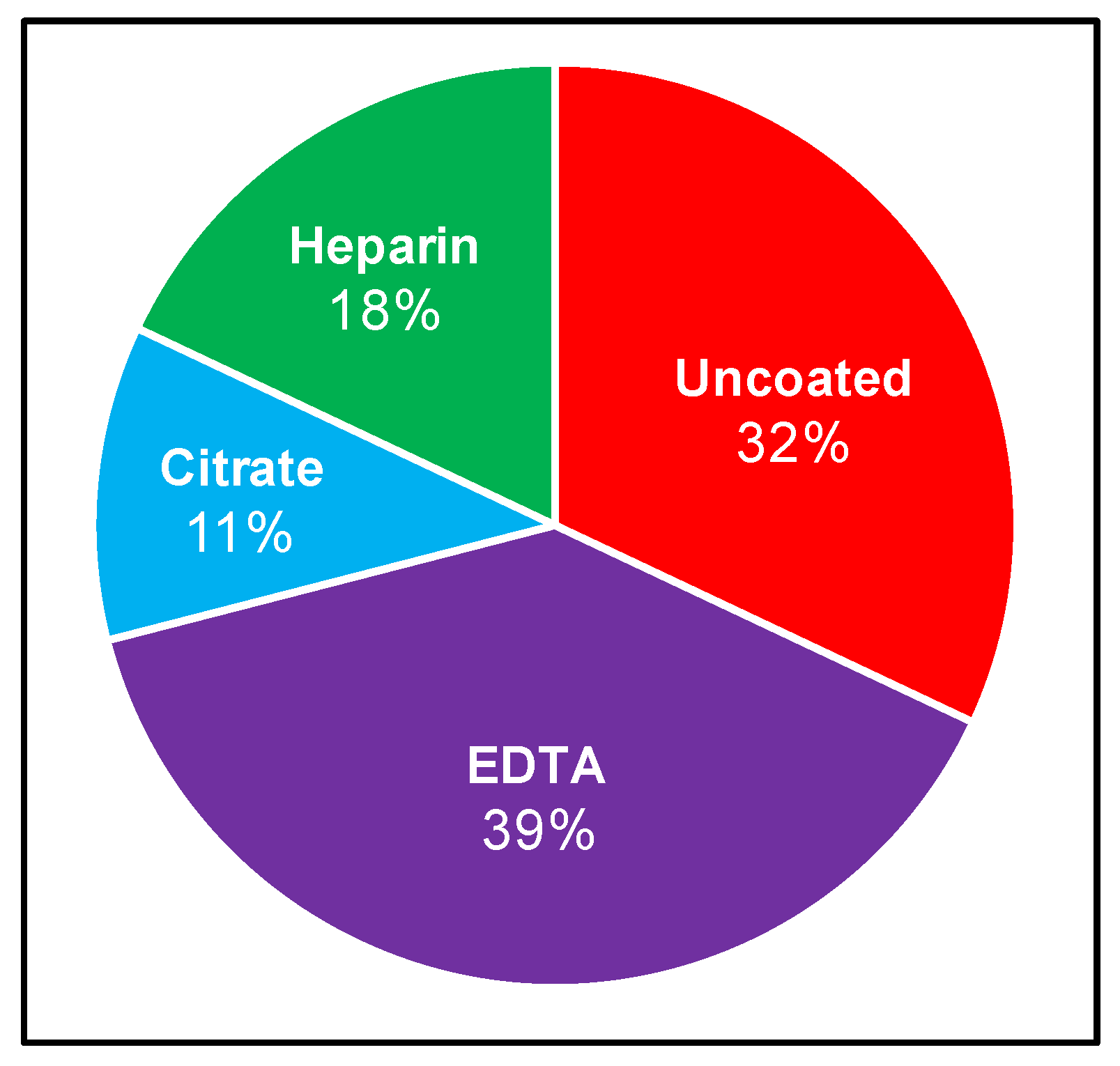
2.1. Comparison of Clinical Culture Protocols
2.2. Bacteria
2.3. Vacutainer Anticoaguant Cell Viability Assessments
2.4. Experimentally Infected Blood Preparation
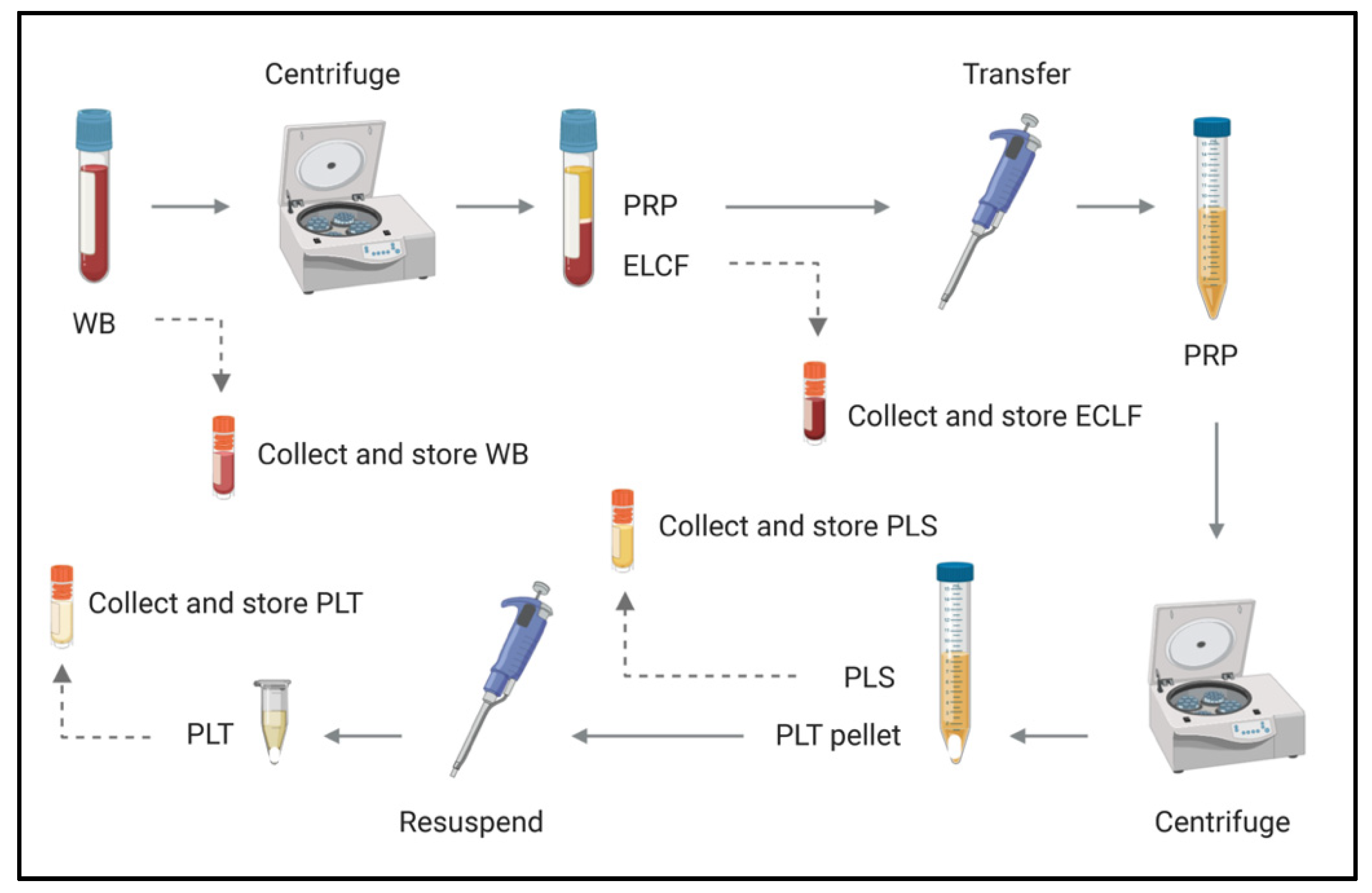
2.5. Blood Fractionation by Centrifugation
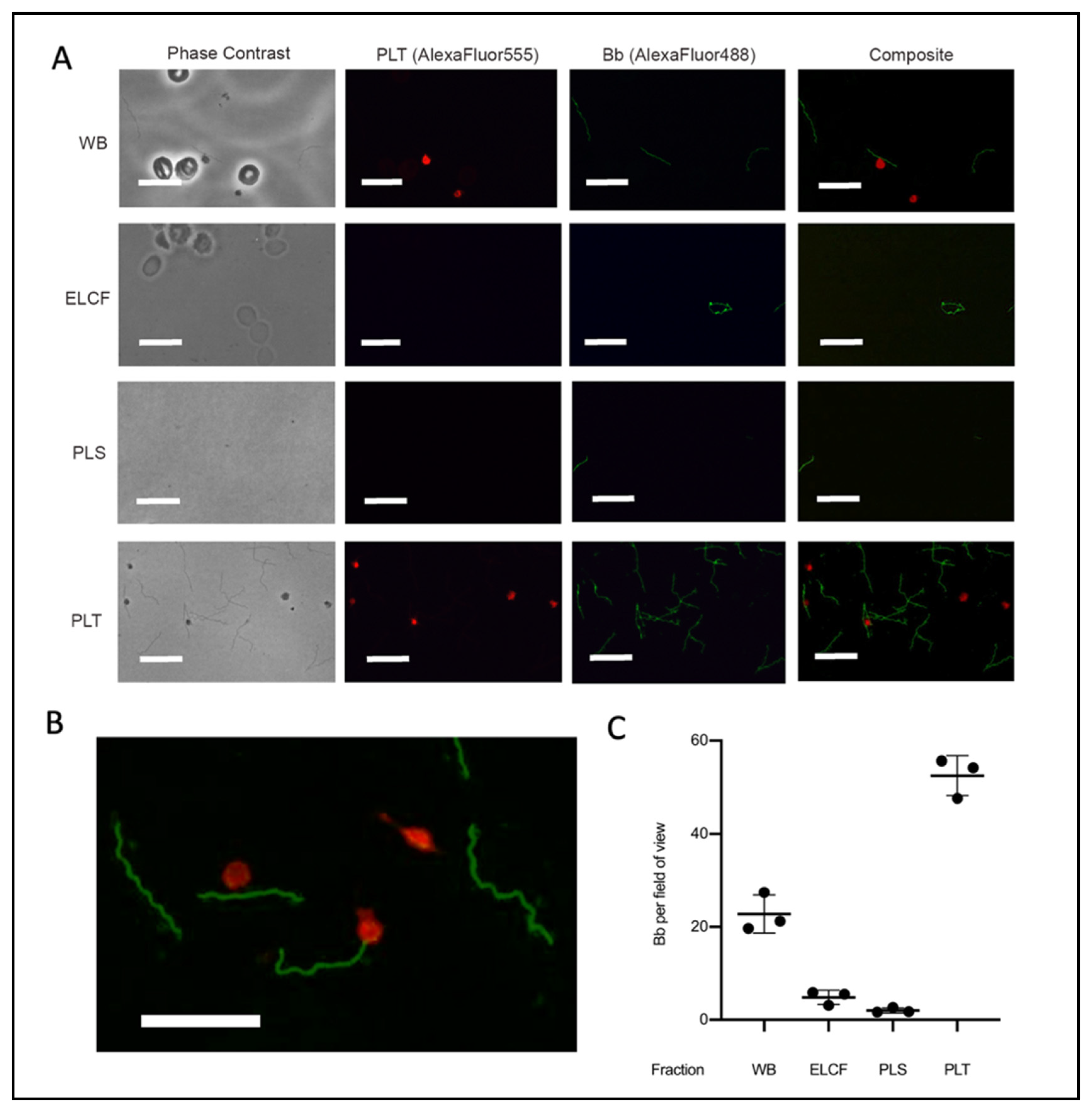
2.6. Immunofluorescent Slide Preparation and Microscopy
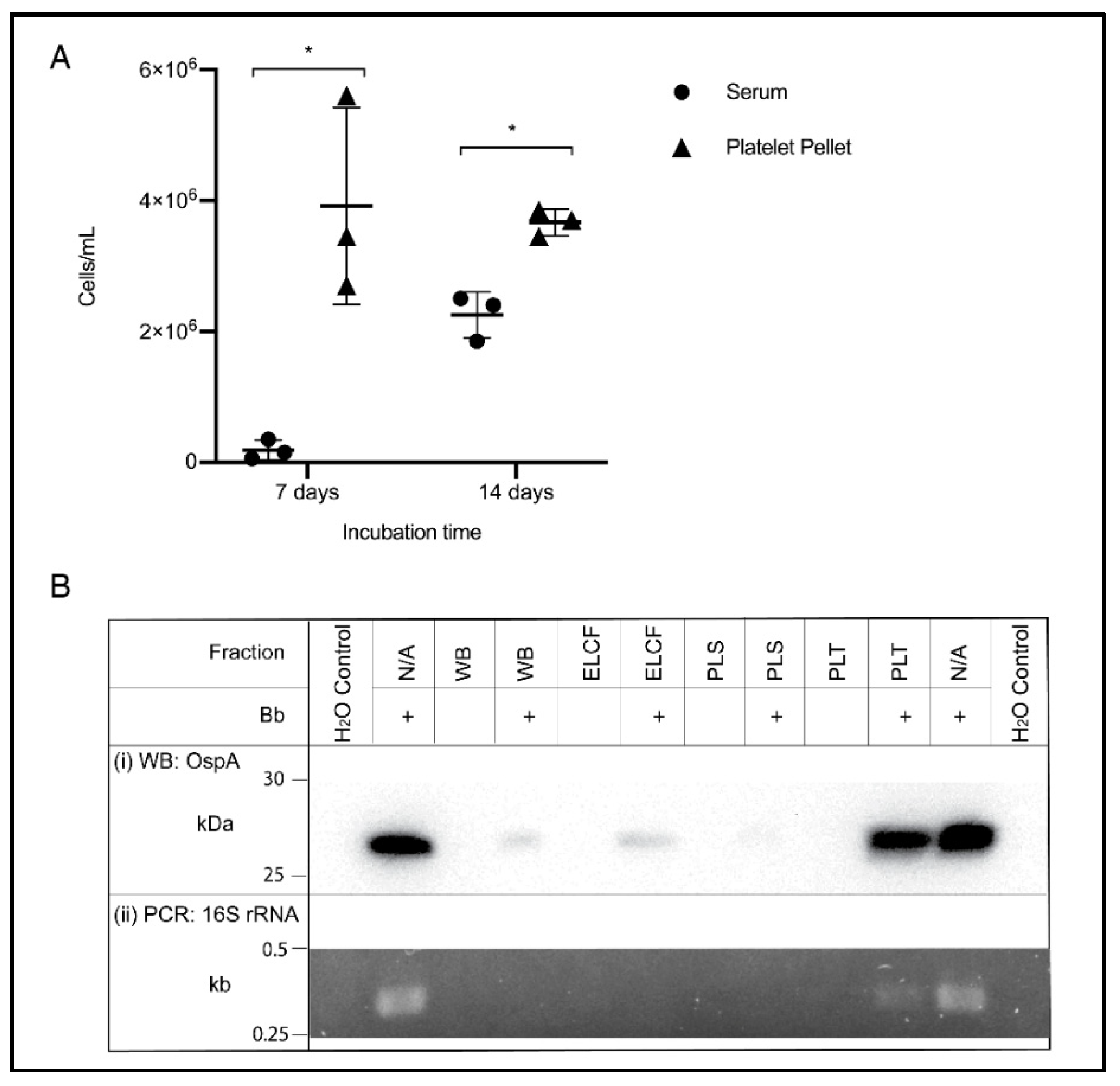
2.7. Culture from Blood Fractions
2.8. Western Blotting
2.9. Polymerase Chain Reaction (PCR)
2.10. Statistical Analyses
3. Results
3.1. Appraisal of Existing Clinical Culture Practices
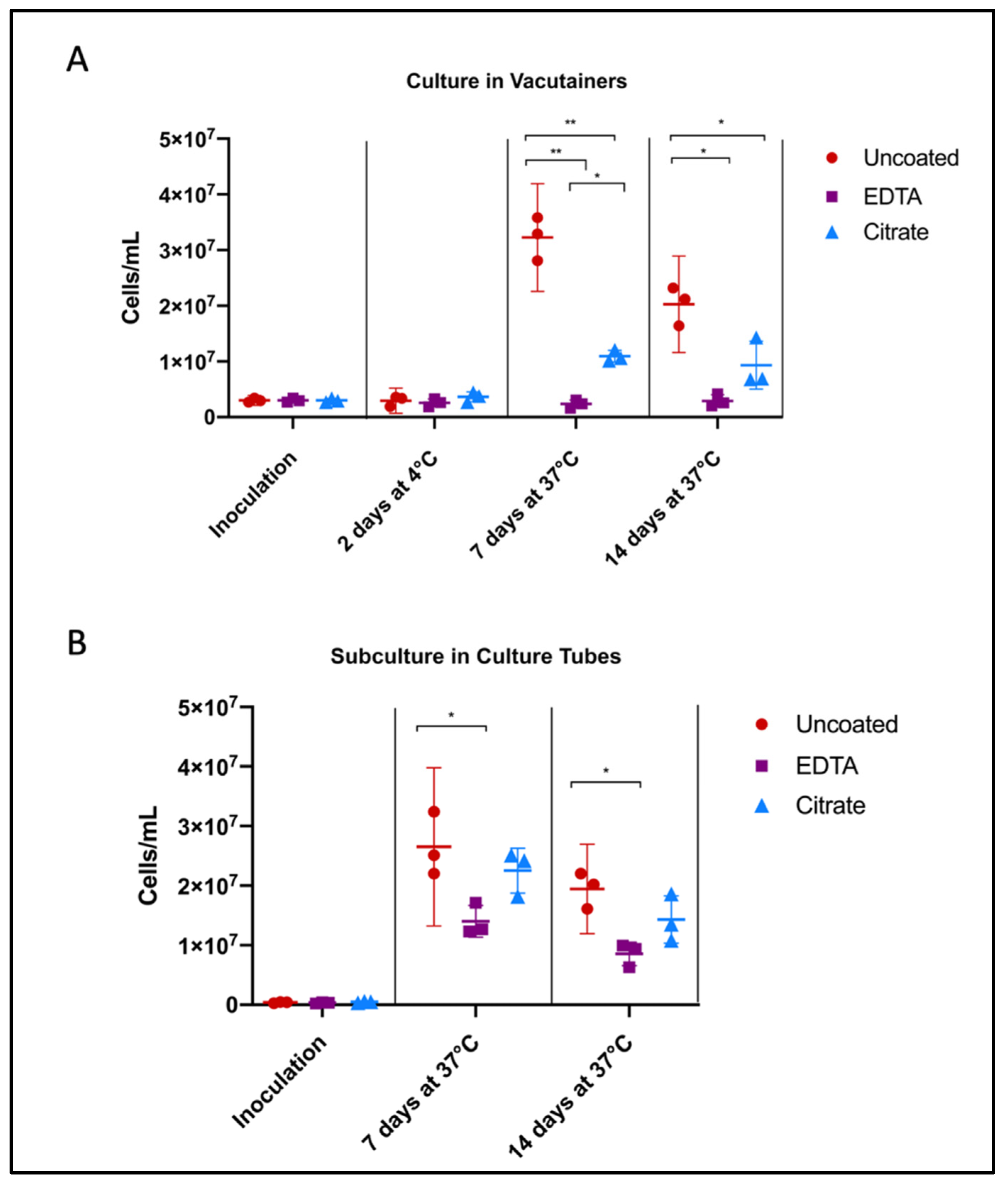
3.2. Effect of Anticoagulants on Borrelia Viability
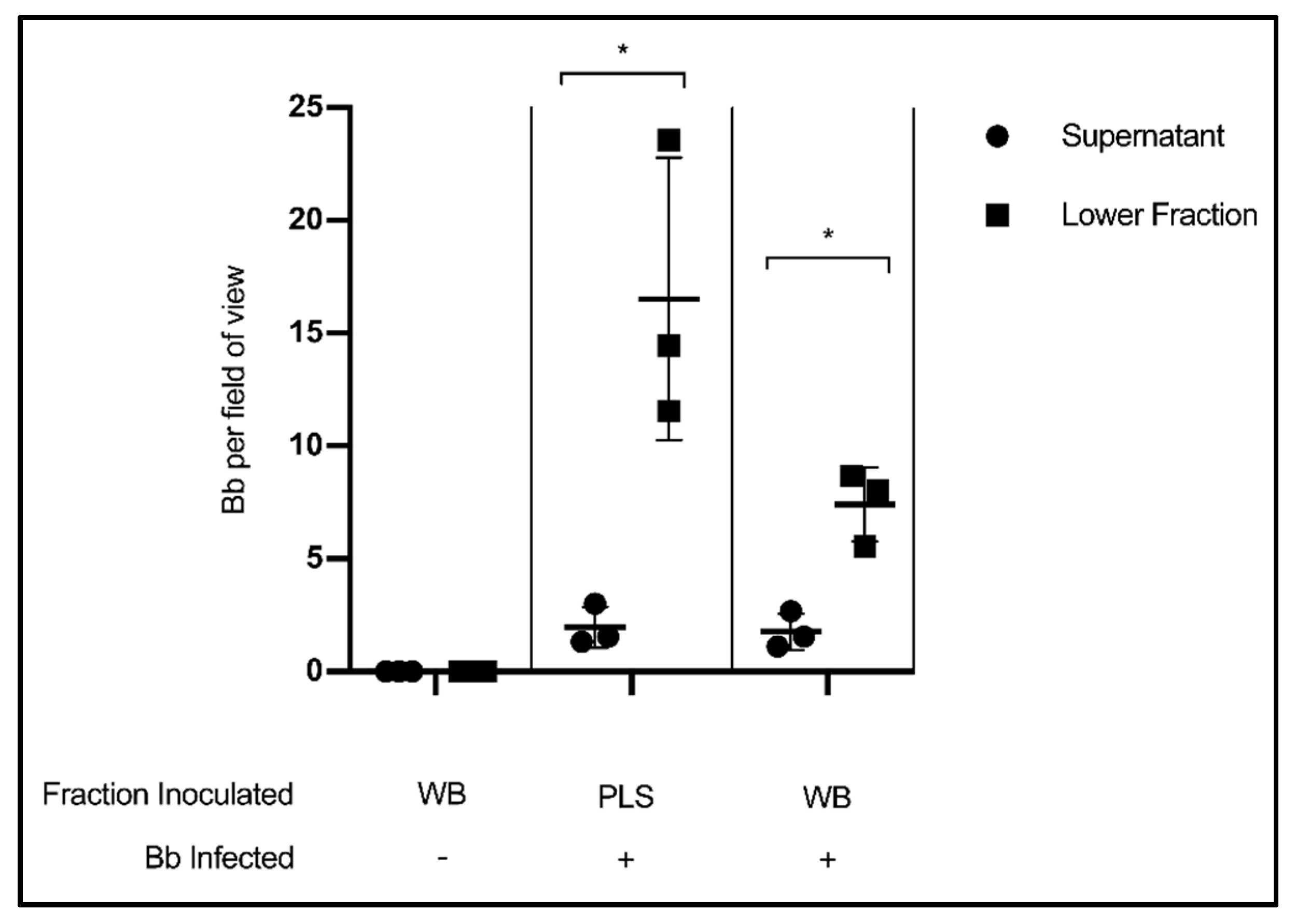
3.3. Fraction Enrichment of Borrelia during Blood Processing
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Inoculation Fraction | Collection Tube | Centrifugation | Citation |
|---|---|---|---|
| Plasma | EDTA | Unspecified | Horn et al. 2020 [40] |
| Serum (+ a few blood cells) | Uncoated | Yes (low speeds) | Middelveen et al. 2018 [9] |
| Serum (+ a few blood cells) | Uncoated | Yes (low speeds) | Middelveen et al. 2015 [41] |
| Serum and whole blood | Uncoated, EDTA and direct into BSK | No | Sapi et al. 2013 [42] |
| Plasma | EDTA | Yes (260× g, 15 min) | Liveris et al. 2012 [43] |
| Plasma | EDTA | Yes (260× g, 15 min) | Liveris et al. 2011 [44] |
| Plasma | Citrate | Yes (800 rpm, 10 min) | Maraspin et al. 2011 [45] |
| Plasma | Citrate | Yes (100× g, 5 min) | Cerar et al., 2008 [46] |
| Plasma | EDTA | Yes (350× g, 15 min) | Coulter et al. 2005 [29] |
| Plasma | EDTA | Unspecified | Wormser et al. 2005 [30] |
| Plasma | EDTA | Yes (260× g, 15 min) | Wormser et al. 2001 [31] |
| Plasma | Citrate | Yes (800 rpm, 5 min) | Arnez et al. 2001 [32] |
| Plasma | EDTA | Unspecified | Marques et al. 2000 [33] |
| Serum and plasma | Uncoated and EDTA | Yes (260× g, 15 min) | Wormser et al. 2000 [34] |
| Serum, plasma and whole blood | Uncoated and heparin | Unspecified | Liveris et al. 1999 [35] |
| Whole blood | EDTA | No | Phillips et al. 1998 [36] |
| Serum and whole blood | Heparin | Yes (serum—1100× g, 10 min) | Wormser et al. 1998 [37] |
| Serum, whole blood, plasma, buffy coat and erythrocytes | Uncoated and EDTA | Yes (260× g, 15 min) | Goodman et al. 1995 [22] |
| Whole blood | Uncoated (directly into BSK) | No | Berger et al. 1994 [38] |
| Serum and whole blood | Uncoated and heparin | No | Wallach et al. 1993 [39] |
| Serum and whole blood | Uncoated and heparin | Yes (serum—500× g, 10 min) | Nadelman 1990 [14] |
References
- Nelson, C.A.; Saha, S.; Kugeler, K.J.; Delorey, M.J.; Shankar, M.B.; Hinckley, A.F.; Mead, P.S. Incidence of Clinician-Diagnosed Lyme Disease, United States, 2005–2010. Emerg. Infect. Dis. 2015, 21, 1625–1631. [Google Scholar]
- Schutzer, S.E.; Body, B.A.; Boyle, J.; Branson, B.M.; Dattwyler, R.J.; Fikrig, E.; Gerald, N.J.; Gomes-Solecki, M.; Kintrup, M.; Ledizet, M.; et al. Direct Diagnostic Tests for Lyme Disease. Clin. Infect. Dis. 2019, 68, 1052–1057. [Google Scholar]
- Feder, H.M., Jr.; Abeles, M.; Bernstein, M.; Whitaker-Worth, D.; Grant-Kels, J.M. Diagnosis, treatment, and prognosis of erythema migrans and Lyme arthritis. Clin. Dermatol. 2006, 24, 509–520. [Google Scholar] [CrossRef]
- Godar, D.A.; Laniosz, V.; Wetter, D.A. Lyme Disease Update for the General Dermatologist. Am. J. Clin. Dermatol. 2015, 16, 5–18. [Google Scholar]
- Fix, A.D.; Peña, C.A.; Strickland, G.T. Racial differences in reported lyme disease incidence. Am. J. Epidemiol. 2000, 152, 756–759. [Google Scholar] [CrossRef]
- Goddard, J. Not All Erythema Migrans Lesions Are Lyme Disease. Am. J. Med. 2017, 130, 231–233. [Google Scholar] [CrossRef]
- Kannangara, D.W.; Patel, P. Report of non-lyme, erythema migrans rashes from New Jersey with a review of possible role of tick salivary toxins. Vector Borne Zoonotic Dis. 2018, 18, 641–652. [Google Scholar] [CrossRef]
- Aucott, J.N.; Rebman, A.W.; Crowder, L.A.; Kortte, K.B. Post-treatment Lyme disease syndrome symptomatology and the impact on life functioning: Is there something here? Qual. Life Res. 2013, 22, 75–84. [Google Scholar]
- Middelveen, M.; Sapi, E.; Burke, J.; Filush, K.; Franco, A.; Fesler, M.; Stricker, R. Persistent Borrelia Infection in Patients with Ongoing Symptoms of Lyme Disease. Healthcare 2018, 6, 33–51. [Google Scholar]
- Bamm, V.V.; Ko, J.T.; Mainprize, I.L.; Sanderson, V.P.; Wills, M.K.B. Lyme Disease Frontiers: Reconciling Borrelia Biology and Clinical Conundrums. Pathogens 2019, 8, 299. [Google Scholar]
- Branda, J.A.; Body, B.A.; Boyle, J.; Branson, B.M.; Dattwyler, R.J.; Fikrig, E.; Gerald, N.J.; Gomes-Solecki, M.; Kintrup, M.; Ledizet, M.; et al. Advances in Serodiagnostic Testing for Lyme Disease Are at Hand. Clin. Infect. Dis. 2017, 66, 1133–1139. [Google Scholar] [CrossRef]
- Rebman, A.W.; Crowder, L.A.; Kirkpatrick, A.; Aucott, J.N. Characteristics of seroconversion and implications for diagnosis of post-treatment Lyme disease syndrome: Acute and convalescent serology among a prospective cohort of early Lyme disease patients. Clin. Rheumatol. 2014, 34, 585–589. [Google Scholar] [CrossRef]
- Fallon, B.A.; Pavlicova, M.; Coffino, S.W.; Brenner, C. A Comparison of Lyme Disease Serologic Test Results From 4 Laboratories in Patients with Persistent Symptoms After Antibiotic Treatment. Clin. Infect. Dis. 2014, 59, 1705–1710. [Google Scholar] [CrossRef] [PubMed]
- Nadelman, R.B.; Pavia, C.S.; Magnarelli, L.A.; Wormser, G.P. Isolation of Borrelia burgdorferi from the blood of seven patients with lyme disease. Am. J. Med. 1990, 88, 21–26. [Google Scholar] [CrossRef]
- Goodman, J.L.; Jurkovich, P.; Kramber, J.M.; Johnson, R.C. Molecular detection of persistent Borrelia burgdorferi in the urine of patients with active Lyme disease. Infect. Immun. 1991, 59, 269–278. [Google Scholar] [CrossRef]
- Dorward, D.W.; Schwan, T.G.; Garon, C.F. Immune capture and detection of Borrelia burgdorferi antigens in urine, blood, or tissues from infected ticks, mice, dogs, and humans. J. Clin. Microbiol. 1991, 29, 1162–1170. [Google Scholar] [CrossRef]
- Galbe, J.L.; Guy, E.; Zapatero, J.M.; Peerschke, E.I.B.; Benach, J.L. Vascular clearance of Borrelia burgdorferi in rats. Microb. Pathog. 1993, 14, 187–201. [Google Scholar] [CrossRef]
- Pritt, B.S.; Mead, P.S.; Johnson, D.K.H.; Neitzel, D.F.; Respicio-Kingry, L.B.; Davis, J.P.; Schiffman, E.; Sloan, L.M.; Schriefer, M.E.; Replogle, A.J.; et al. Identification of a novel pathogenic Borrelia species causing Lyme borreliosis with unusually high spirochaetaemia: A descriptive study. Lancet Infect. Dis. 2016, 16, 556–564. [Google Scholar] [CrossRef]
- Waddell, L.A.; Greig, J.; Mascarenhas, M.; Harding, S.; Lindsay, R.; Ogden, N. The Accuracy of Diagnostic Tests for Lyme Disease in Humans, A Systematic Review and Meta-Analysis of North American Research. PLoS ONE 2016, 11, e0168613. [Google Scholar] [CrossRef] [PubMed]
- Wormser, G.P.; Brisson, D.; Liveris, D.; Hanincova, K.; Sandigursky, S.; Nowakowski, J.; Nadelman, R.B.; Ludin, S.; Schwartz, I. Borrelia burgdorferi genotype predicts the capacity for hematogenous dissemination during early Lyme disease. J. Infect. Dis. 2008, 198, 1358–1364. [Google Scholar] [CrossRef]
- Schmidt, B.L. PCR in laboratory diagnosis of human Borrelia burgdorferi infections. Clin. Microbiol. Rev. 1997, 10, 185–201. [Google Scholar] [CrossRef]
- Goodman, J.L.; Bradley, J.F.; Ross, A.E.; Goellner, P.; Lagus, A.; Vitale, B.; Berger, B.W.; Luger, S.; Johnson, R.C. Bloodstream invasion in early Lyme disease: Results from a prospective, controlled, blinded study using the polymerase chain reaction. Am. J. Med. 1995, 99, 6–12. [Google Scholar] [CrossRef]
- Liveris, D.; Schwartz, I.; McKenna, D.; Nowakowski, J.; Nadelman, R.B.; DeMarco, J.; Iyer, R.; Cox, M.E.; Holmgren, D.; Wormser, G.P. Quantitation of cell-associated borrelial DNA in the blood of Lyme disease patients with erythema migrans. Eur. J. Clin. Microbiol. Infect. Dis. 2012, 31, 791–795. [Google Scholar] [CrossRef] [PubMed]
- Wormser, G.P. Hematogenous dissemination in early Lyme disease. Wien. Klin. Wochenschr. 2006, 118, 634–637. [Google Scholar] [CrossRef] [PubMed]
- Mosel, M.R.; Carolan, H.E.; Rebman, A.W.; Castro, S.; Massire, C.; Ecker, D.J.; Soloski, M.J.; Aucott, J.N.; Eshoo, M.W. Molecular Testing of Serial Blood Specimens from Patients with Early Lyme Disease during Treatment Reveals Changing Coinfection with Mixtures of Borrelia burgdorferi Genotypes. Antimicrob. Agents Chemother. 2019, 63, 1625–1626. [Google Scholar]
- Branda, J.A.; Lemieux, J.E.; Blair, L.; Ahmed, A.A.; Hong, D.K.; Bercovici, S.; Blauwkamp, T.A.; Hollemon, D.; Ho, C.; Strle, K.; et al. Detection of Borrelia burgdorferi Cell-free DNA in Human Plasma Samples for Improved Diagnosis of Early Lyme Borreliosis. Clin. Infect. Dis. 2020. [Google Scholar] [CrossRef]
- Nilsson, R.J.A.; Balaj, L.; Hulleman, E.; van Rijn, S.; Pegtel, D.M.; Walraven, M.; Widmark, A.; Gerritsen, W.R.; Verheul, H.M.; Vandertop, W.P.; et al. Blood platelets contain tumor-derived RNA biomarkers. Blood 2011, 118, 3680–3683. [Google Scholar] [CrossRef]
- Lee, S.H.; Vigliotti, V.S.; Vigliotti, J.S.; Jones, W.; Pappu, S. Increased Sensitivity and Specificity of Borrelia burgdorferi 16S Ribosomal DNA Detection. Am. J. Clin. Pathol. 2010, 133, 569–576. [Google Scholar] [CrossRef]
- Coulter, P.; Lema, C.; Flayhart, D.; Linhardt, A.S.; Aucott, J.N.; Auwaerter, P.G.; Dumler, J.S. Two-Year Evaluation of Borrelia burgdorferi Culture and Supplemental Tests for Definitive Diagnosis of Lyme Disease. J. Clin. Microbiol. 2005, 43, 5080–5084. [Google Scholar] [CrossRef]
- Wormser, G.P.; McKenna, D.; Carlin, J.; Nadelman, R.B.; Cavaliere, L.F.; Holmgren, D.; Byrne, D.W.; Nowakowski, J. Brief Communication: Hematogenous Dissemination in Early Lyme Disease. Ann. Intern. Med. 2005, 142, 751. [Google Scholar] [CrossRef]
- Wormser, G.P.; Bittker, S.; Cooper, D.; Nowakowski, J.; Nadelman, R.B.; Pavia, C. Yield of Large-Volume Blood Cultures in Patients with Early Lyme Disease. J. Infect. Dis. 2001, 184, 1070–1072. [Google Scholar] [CrossRef] [PubMed]
- Arnež, M.; Ružić-Sabljić, E.; Ahčan, J.; Radšel-Medvešček, A.; Pleterski-Rigler, D.; Strle, F. Isolation of Borrelia burgdorferi sensu lato from blood of children with solitary erythema migrans. Pediatr. Infect. Dis. J. 2001, 20, 251–255. [Google Scholar] [CrossRef] [PubMed]
- Marques, A.R.; Stock, F.; Gill, V. Evaluation of a new culture medium for Borrelia burgdorferi. J. Clin. Microbiol. 2000, 38, 4239–4241. [Google Scholar] [CrossRef]
- Wormser, G.P.; Bittker, S.; Cooper, D.; Nowakowski, J.; Nadelman, R.B.; Pavia, C. Comparison of the yields of blood cultures using serum or plasma from patients with early Lyme disease. J. Clin. Microbiol. 2000, 38, 1648–1650. [Google Scholar] [CrossRef] [PubMed]
- Liveris, D.; Varde, S.; Iyer, R.; Koenig, S.; Bittker, S.; Cooper, D.; McKenna, D.; Nowakowski, J.; Nadelman, R.B.; Wormser, G.P.; et al. Genetic diversity of Borrelia burgdorferi in lyme disease patients as determined by culture versus direct PCR with clinical specimens. J. Clin. Microbiol. 1999, 37, 565–569. [Google Scholar] [CrossRef] [PubMed]
- Phillips, S.E.; Mattman, L.H.; Hulínská, D.; Moayad, H. A proposal for the reliable culture of Borrelia burgdorferi from patients with chronic lyme disease, even from those previously aggressively treated. Infection 1998, 26, 364–367. [Google Scholar]
- Wormser, G.P.; Nowakowski, J.; Nadelman, R.B.; Bittker, S.; Cooper, D.; Pavia, C. Improving the yield of blood cultures for patients with early lyme disease. J. Clin. Microbiol. 1998, 36, 296–298. [Google Scholar]
- Berger, B.W.; Johnson, R.C.; Kodner, C.; Coleman, L. Cultivation of Borrelia burgdorferi from the blood of two patients with erythema migrans lesions lacking extracutaneous signs and symptoms of Lyme disease. J. Am. Acad. Dermatol. 1994, 30, 48–51. [Google Scholar]
- Wallach, F.R.; Forni, A.L.; Hariprashad, J.; Stoeckle, M.Y.; Steinberg, C.R.; Fisher, L.; Malawista, S.E.; Murray, H.W. Circulating Borrelia burgdotferi in Patients with Acute Lyme Disease: Results of Blood Cultures and Serum DNA Analysis. J. Infect. Dis. 1993, 168, 1541–1543. [Google Scholar]
- Horn, E.J.; Dempsey, G.; Schotthoefer, A.M.; Prisco, U.L.; McArdle, M.; Gervasi, S.S.; Golightly, M.; De Luca, C.; Evans, M.; Pritt, B.S.; et al. The Lyme Disease Biobank – Characterization of 550 Patient and Control Samples from the East Coast and Upper Midwest of the United States. J. Clin. Microbiol. 2020, 58, 1–12. [Google Scholar] [CrossRef]
- Middelveen, M.J.; Bandoski, C.; Burke, J.; Sapi, E.; Filush, K.R.; Wang, Y.; Franco, A.; Mayne, P.J.; Stricker, R.B. Exploring the association between Morgellons disease and Lyme disease: Identification of Borrelia burgdorferi in Morgellons disease patients. BMC Dermatol. 2015, 15, 1–14. [Google Scholar] [CrossRef][Green Version]
- Sapi, E.; Pabbati, N.; Datar, A.; Davies, E.M.; Rattelle, A.; Kuo, B.A. Improved culture conditions for the growth and detection of Borrelia from human serum. Int. J. Med. Sci. 2013, 10, 362–376. [Google Scholar] [CrossRef]
- Liveris, D.; Schwartz, I.; McKenna, D.; Nowakowski, J.; Nadelman, R.; DeMarco, J.; Iyer, R.; Bittker, S.; Cooper, D.; Holmgren, D.; et al. Comparison of five diagnostic modalities for direct detection of Borrelia burgdorferi in patients with early Lyme disease. Diagn. Microbiol. Infect. Dis. 2012, 73, 243–245. [Google Scholar] [CrossRef]
- Liveris, D.; Schwartz, I.; Bittker, S.; Cooper, D.; Iyer, R.; Cox, M.E.; Wormser, G.P. Improving the yield of blood cultures from patients with early lyme disease. J. Clin. Microbiol. 2011, 49, 2166–2168. [Google Scholar] [CrossRef] [PubMed]
- Maraspin, V.; Ogrinc, K.; Ružić-Sabljić, E.; Lotrič-Furlan, S.; Strle, F. Isolation of Borrelia burgdorferi sensu lato from blood of adult patients with borrelial lymphocytoma, Lyme neuroborreliosis, Lyme arthritis and acrodermatitis chronica atrophicans. Infection 2011, 39, 35–40. [Google Scholar] [CrossRef]
- Cerar, T.; Ogrinc, K.; Cimperman, J.; Lotrič-Furlan, S.; Strle, F.; Ružić-Sabljić, E. Validation of cultivation and PCR methods for diagnosis of Lyme neuroborreliosis. J. Clin. Microbiol. 2008, 46, 3375–3379. [Google Scholar] [CrossRef] [PubMed]
- Rudenko, N.; Golovchenko, M.; Vancova, M.; Clark, K.; Grubhoffer, L.; Oliver, J.H., Jr. Isolation of live Borrelia burgdorferi sensu lato spirochaetes from patients with undefined disorders and symptoms not typical for Lyme borreliosis. Clin. Microbiol. Infect. 2016, 22, 267.e9–267.e15. [Google Scholar] [CrossRef] [PubMed]
- Marques, A.R.; Yang, X.; Smith, A.A.; Zhuang, X.; Turk, S.; Williams, C.D.; Law, M.A.; Barbour, A.G. Citrate Anticoagulant Improves the burgdorferi Plasma Culture. J. Clin. Microbiol. 2017, 55, 3297–3299. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Lin, Y.P.; Li, L.; Zhang, F.; Linhardt, R.J. Borrelia burgdorferi glycosaminoglycan-binding proteins: A potential target for new therapeutics against Lyme disease. Microbiology 2017, 163, 1759–1766. [Google Scholar] [CrossRef]
- Nolte, O. Nucleic Acid Amplification Based Diagnostic of Lyme (Neuro-)borreliosis—Lost in the Jungle of Methods, Targets, and Assays? Open Neurol. J. 2012, 6, 129–139. [Google Scholar] [CrossRef]
- Sanderson, V. University of Guelph, Guelph, ON, Canada. Unpublished work. 2020. [Google Scholar]
- Koetsveld, J.; Kolyasnikova, N.M.; Wagemakers, A.; Toporkova, M.G.; Sarksyan, D.S.; Oei, A.; Platonov, A.E.; Hovius, J.W. Development and optimization of an in vitro cultivation protocol allows for isolation of Borrelia miyamotoi from patients with hard tick-borne relapsing fever. Clin. Microbiol. Infect. 2017, 23, 480–484. [Google Scholar] [CrossRef]
- Singh, S.K.; Girschick, H.J. Molecular survival strategies of the Lyme disease spirochete. Infect. Dis. 2004, 4, 575–583. [Google Scholar]
- Kersten, A.; Poitschek, C.; Rauch, S.; Aberer, E. Effects of penicillin, ceftriaxone, and doxycycline on morphology of Borrelia burgdorferi. Antimicrob. Agents Chemother. 1995, 39, 1127–1133. [Google Scholar] [CrossRef]
- Murgia, R.; Cinco, M. Induction of cystic forms by different stress conditions in Borrelia burgdorferi. APMIS 2004, 112, 57–62. [Google Scholar] [CrossRef] [PubMed]
- Motaleb, M.A.; Corum, L.; Bono, J.L.; Elias, A.F.; Rosa, P.; Samuels, D.S.; Charon, N.W. Borrelia burgdorferi periplasmic flagella have both skeletal and motility functions. Proc. Natl. Acad. Sci. USA 2000, 97, 10899–10904. [Google Scholar] [CrossRef]
- Dorward, D.W.; Fischer, E.R.; Brooks, D.M. Invasion and Cytopathic Killing of Human Lymphocytes by Spirochetes Causing Lyme Disease. Clin. Infect. Dis. 1997, 25, S2–S8. [Google Scholar] [CrossRef]
- Coburn, J.; Barthold, S.W.; Leong, J.M. Diverse Lyme disease spirochetes bind integrin alpha IIb beta 3 on human platelets. Infect. Immun. 1994, 62, 5559–5567. [Google Scholar] [CrossRef] [PubMed]
- Coburn, J.; Leong, J.M.; Erban, J.K. Integrin alpha IIb beta 3 mediates binding of the Lyme disease agent Borrelia burgdorferi to human platelets. Proc. Natl. Acad. Sci. USA 1993, 90, 7059–7063. [Google Scholar] [CrossRef]
- Thorp, A.M.; Tonnetti, L. Distribution and survival of Borrelia miyamotoi in human blood components. Transfusion 2016, 56, 705–711. [Google Scholar] [CrossRef]
- Eshoo, M.W.; Crowder, C.C.; Rebman, A.W.; Rounds, M.A.; Matthews, H.E.; Picuri, J.M.; Soloski, M.J.; Ecker, D.J.; Schutzer, S.E.; Aucott, J.N. Direct molecular detection and genotyping of Borrelia burgdorferi from whole blood of patients with early Lyme disease. PLoS ONE 2012, 7, 3–8. [Google Scholar] [CrossRef]
- Bugert, P. (Ed.) DNA and RNA Profiling in Human Blood; Methods in Molecular Biology; Humana Press: Totowa, NJ, USA, 2009; Volume 496, ISBN 978-1-934115-93-0. [Google Scholar]
- Al-Soud, W.A.; Radstrom, P. Purification and Characterization of PCR-Inhibitory Components in Blood Cells. J. Clin. Microbiol. 2001, 39, 485–493. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Weening, E.H.; Faske, J.B.; Höök, M.; Skare, J.T. Invasion of eukaryotic cells by Borrelia burgdorferi requires β1 integrins and Src kinase activity. Infect. Immun. 2011, 79, 1338. [Google Scholar] [CrossRef]






| Component | Avg Bb | Avg Plt | Bb-Plt/Total Bb | Plt-Bb/Total Plt |
|---|---|---|---|---|
| Whole Blood | 22.8 ± 4.1 | 9.9 ± 2.7 | 8.9 ± 2.0% | 20.3 ± 5.4% |
| PLT Fraction | 53.9 ± 4.7 | 22.6 ± 14.8 | 11.3 ± 5.6% | 25.9 ± 7.1% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sanderson, V.P.; Mainprize, I.L.; Verzijlenberg, L.; Khursigara, C.M.; Wills, M.K.B. The Platelet Fraction Is a Novel Reservoir to Detect Lyme Borrelia in Blood. Biology 2020, 9, 366. https://doi.org/10.3390/biology9110366
Sanderson VP, Mainprize IL, Verzijlenberg L, Khursigara CM, Wills MKB. The Platelet Fraction Is a Novel Reservoir to Detect Lyme Borrelia in Blood. Biology. 2020; 9(11):366. https://doi.org/10.3390/biology9110366
Chicago/Turabian StyleSanderson, Victoria P., Iain L. Mainprize, Lisette Verzijlenberg, Cezar M. Khursigara, and Melanie K. B. Wills. 2020. "The Platelet Fraction Is a Novel Reservoir to Detect Lyme Borrelia in Blood" Biology 9, no. 11: 366. https://doi.org/10.3390/biology9110366
APA StyleSanderson, V. P., Mainprize, I. L., Verzijlenberg, L., Khursigara, C. M., & Wills, M. K. B. (2020). The Platelet Fraction Is a Novel Reservoir to Detect Lyme Borrelia in Blood. Biology, 9(11), 366. https://doi.org/10.3390/biology9110366