Expression of POU2F3 Transcription Factor Control Inflammation, Immunological Recruitment and Metastasis of Pancreatic Cancer in Mice

 , ,
, , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Immunohistochemistry
2.3. RT-qPCR
2.4. Immunophenotyping
2.5. Study Approval
2.6. Statistical Analysis
3. Results
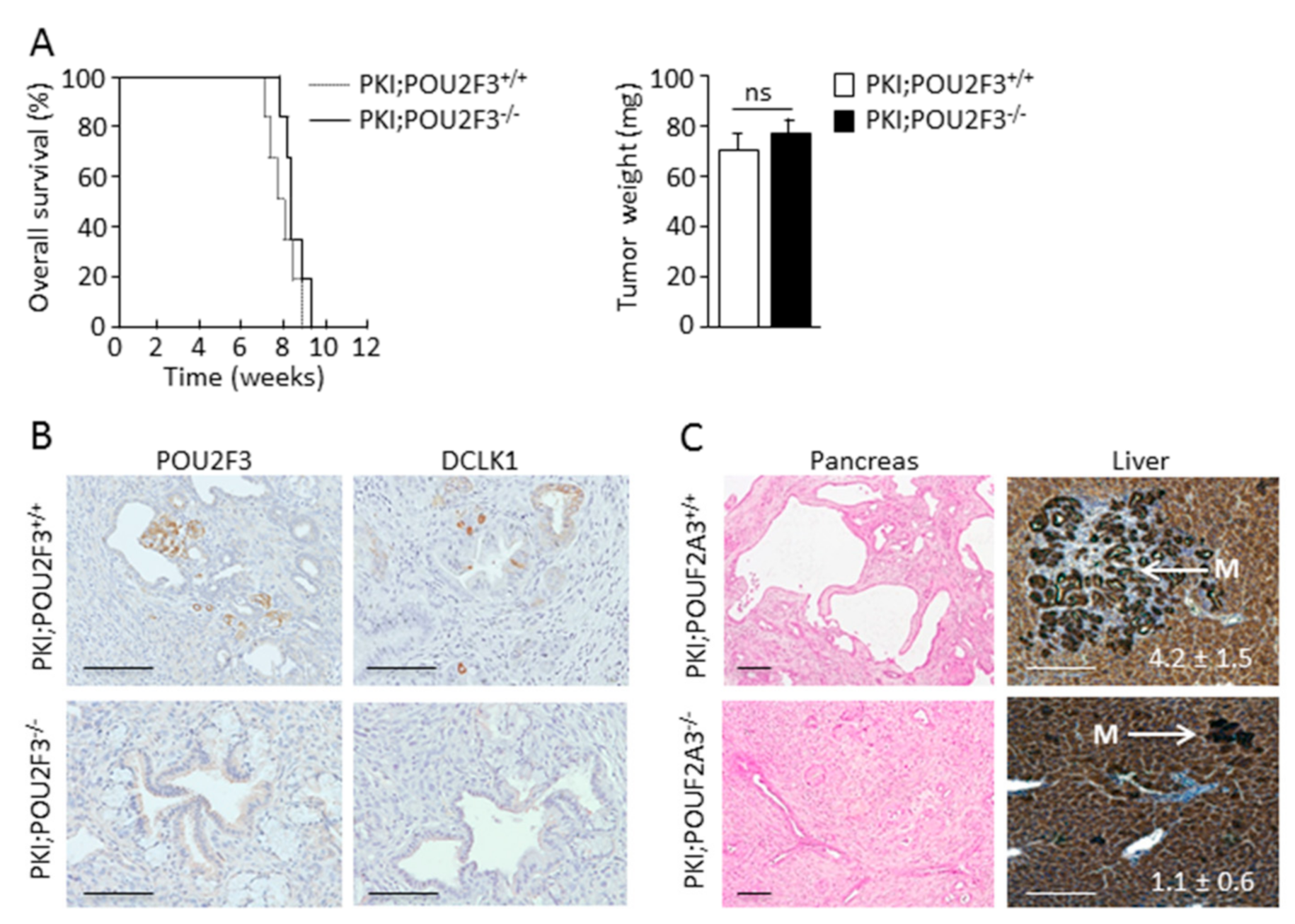
3.1. TUFT Cells Are Not Essential for Pancreatic Carcinogenesis
3.2. POU2F3 Inactivation Decreases Liver Metastasis Development
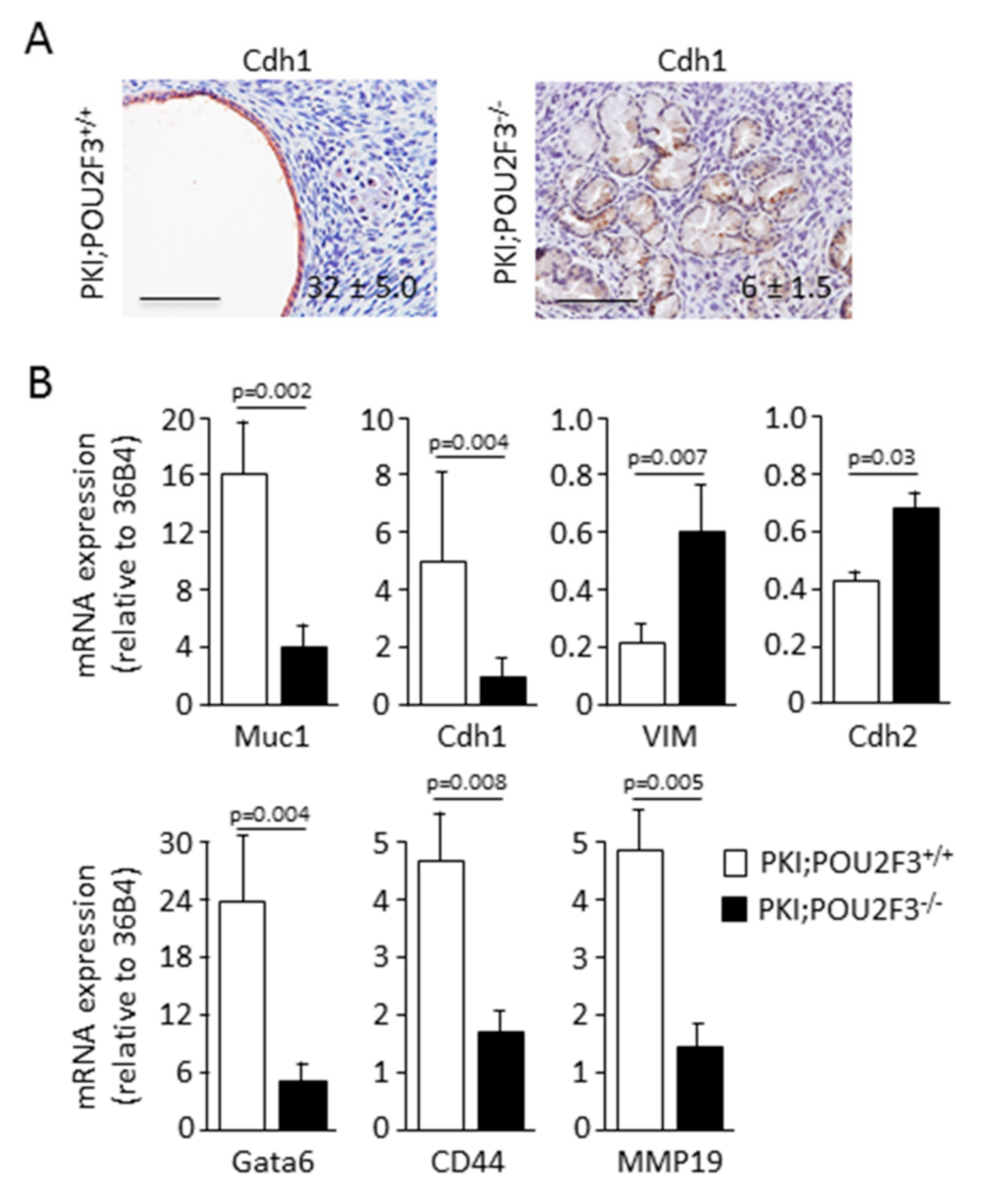
3.3. POU2F3 Inactivation Enhances Epithelial-to-Mesenchymal Transition in PDAC
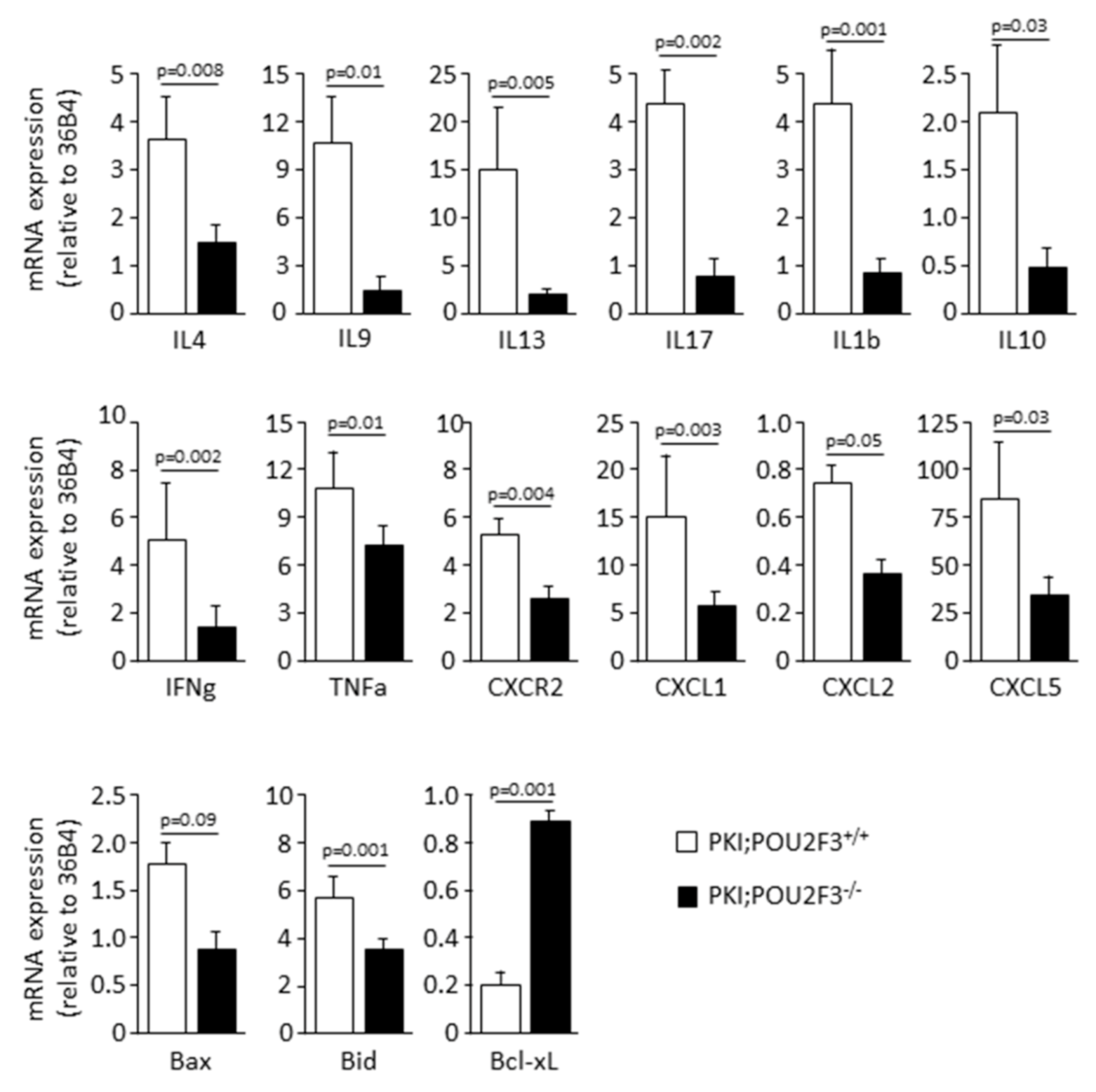
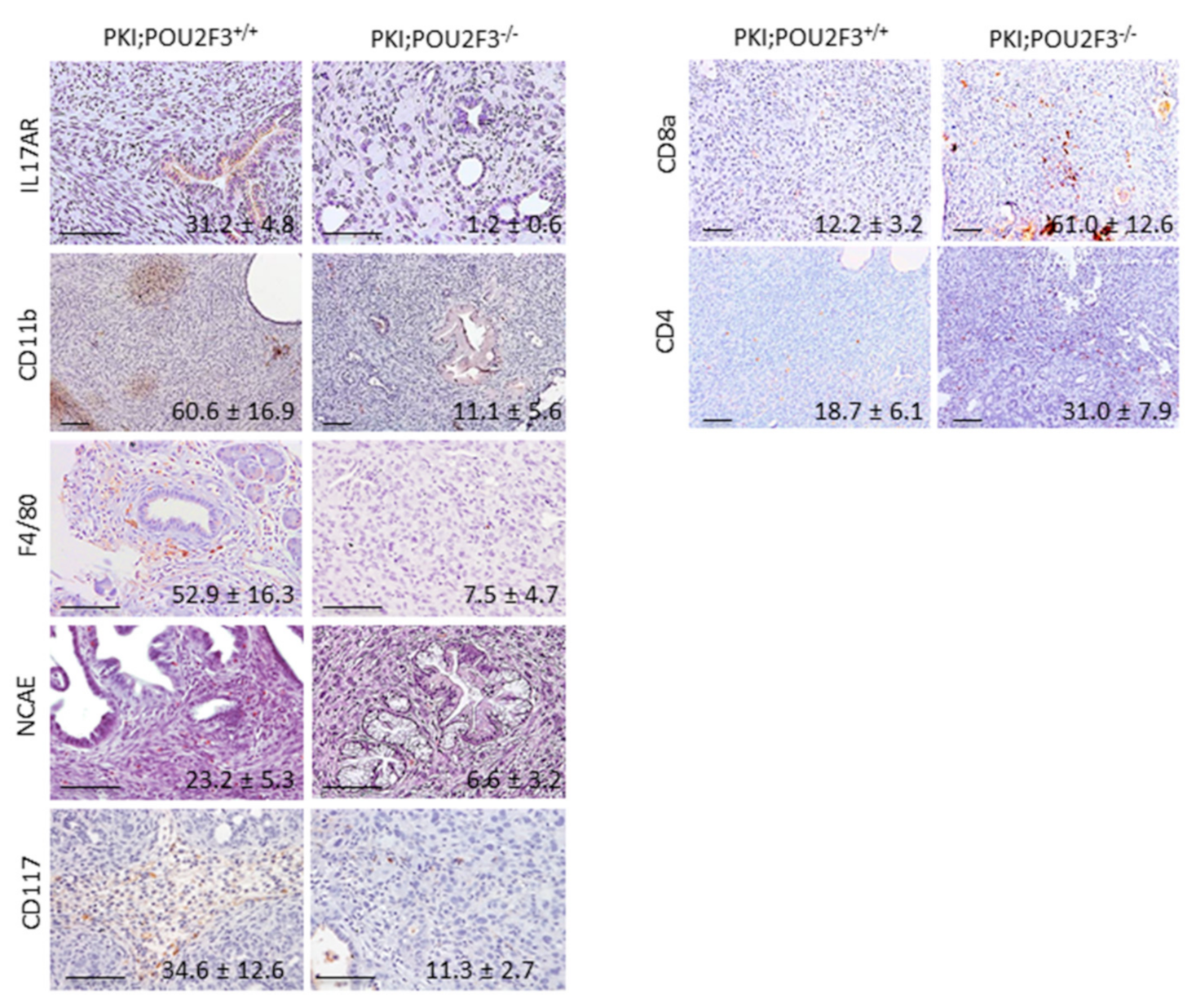
3.4. POU2F3 Inactivation Inhibits the Recruitment Inflammatory Mediators into the PDAC
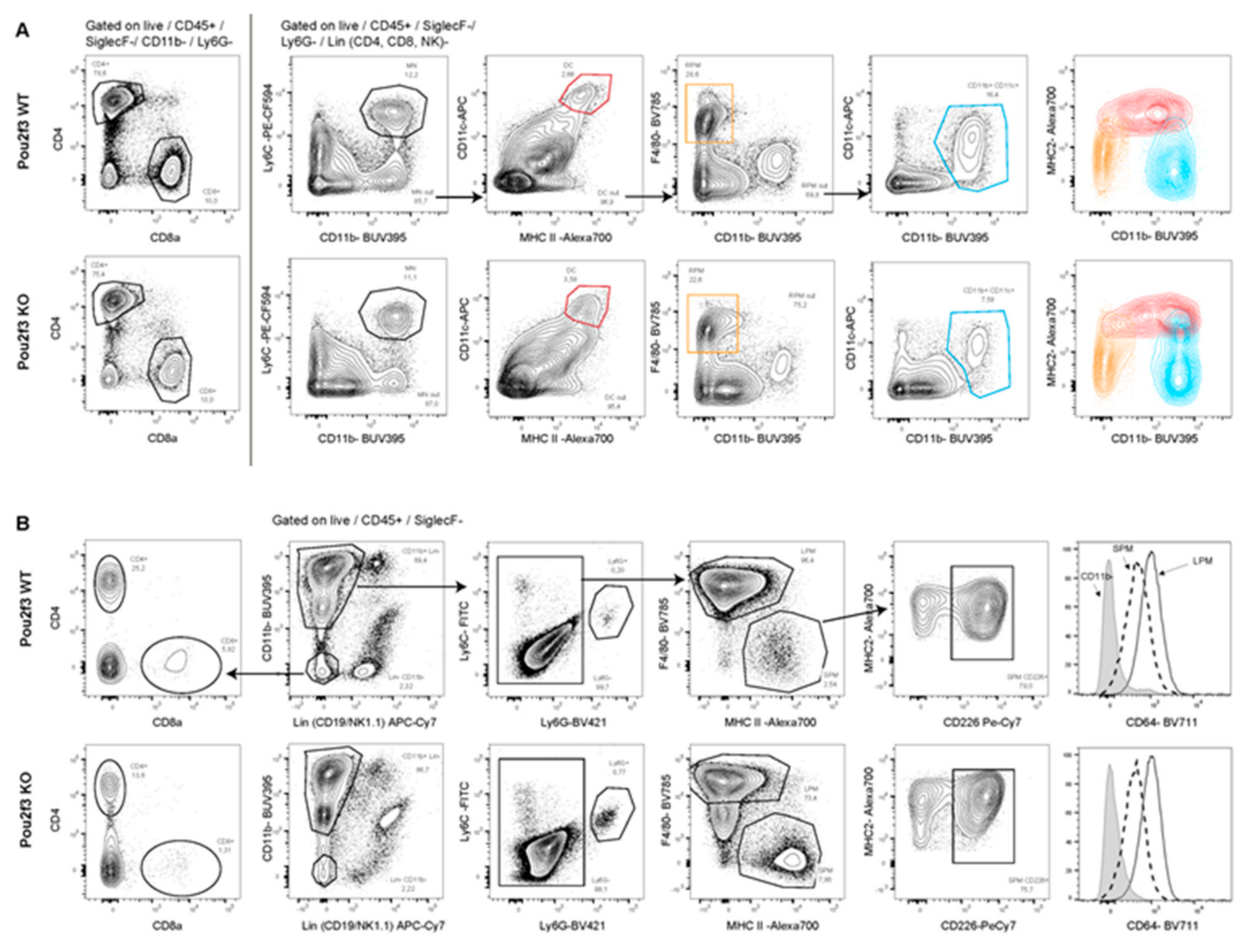
3.5. Systemic POU2F3 Deficiency Has Minimal Effect on Immune Cells Populations
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Du, T.; Bill, K.A.; Ford, J.; Barawi, M.; Hayward, R.D.; Alame, A.; Berri, R.N. The diagnosis and staging of pancreatic cancer: A comparison of endoscopic ultrasound and computed tomography with pancreas protocol. Am. J. Surg. 2018, 215, 472–475. [Google Scholar] [CrossRef] [PubMed]
- Grasso, C.; Jansen, G.; Giovannetti, E. Drug resistance in pancreatic cancer: Impact of altered energy metabolism. Crit. Rev. Oncol. Hematol. 2017, 114, 139–152. [Google Scholar] [CrossRef]
- Lee, B.; Hutchinson, R.; Wong, H.L.; Tie, J.; Putoczki, T.; Tran, B.; Gibbs, P.; Christie, M. Emerging biomarkers for immunomodulatory cancer treatment of upper gastrointestinal, pancreatic and hepatic cancers. Semin. Cancer Biol. 2017. [CrossRef]
- Varghese, A.M. Chimeric antigen receptor (CAR) T and other T cell strategies for pancreas adenocarcinoma. Chin. Clin. Oncol. 2017, 6, 66. [Google Scholar] [CrossRef] [PubMed]
- Reck, M.; Rabe, K.F. Precision Diagnosis and Treatment for Advanced Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2017, 377, 849–861. [Google Scholar] [CrossRef] [PubMed]
- Eggermont, A.M.; Spatz, A.; Robert, C. Cutaneous melanoma. Lancet 2014, 383, 816–827. [Google Scholar] [CrossRef]
- Aoun, F.; Kourie, H.R.; Sideris, S.; Roumeguere, T.; van Velthoven, R.; Gil, T. Checkpoint inhibitors in bladder and renal cancers: Results and perspectives. Immunotherapy 2015, 7, 1259–1271. [Google Scholar] [CrossRef]
- Wada, K.; Takaori, K.; Traverso, L.W. Screening for Pancreatic Cancer. Surg. Clin. N. Am. 2015, 95, 1041–1052. [Google Scholar] [CrossRef]
- Beuran, M.; Negoi, I.; Paun, S.; Ion, A.D.; Bleotu, C.; Negoi, R.I.; Hostiuc, S. The epithelial to mesenchymal transition in pancreatic cancer: A systematic review. Pancreatol. Off. J. Int. Assoc. Pancreatol. 2015, 15, 217–225. [Google Scholar] [CrossRef]
- Tang, D.; Wang, D.; Yuan, Z.; Xue, X.; Zhang, Y.; An, Y.; Chen, J.; Tu, M.; Lu, Z.; Wei, J.; et al. Persistent activation of pancreatic stellate cells creates a microenvironment favorable for the malignant behavior of pancreatic ductal adenocarcinoma. Int. J. Cancer 2013, 132, 993–1003. [Google Scholar] [CrossRef]
- Achyut, B.R.; Yang, L. Transforming growth factor-beta in the gastrointestinal and hepatic tumor microenvironment. Gastroenterology 2011, 141, 1167–1178. [Google Scholar] [CrossRef] [PubMed]
- Beauchemin, N.; Arabzadeh, A. Carcinoembryonic antigen-related cell adhesion molecules (CEACAMs) in cancer progression and metastasis. Cancer Metastasis Rev. 2013, 32, 643–671. [Google Scholar] [CrossRef] [PubMed]
- Knapinska, A.M.; Estrada, C.A.; Fields, G.B. The Roles of Matrix Metalloproteinases in Pancreatic Cancer. Prog. Mol. Biol. Transl. Sci. 2017, 148, 339–354. [Google Scholar] [CrossRef]
- Hamada, S.; Masamune, A.; Shimosegawa, T. Inflammation and pancreatic cancer: Disease promoter and new therapeutic target. J. Gastroenterol. 2014, 49, 605–617. [Google Scholar] [CrossRef] [PubMed]
- Steele, C.W.; Jamieson, N.B.; Evans, T.R.; McKay, C.J.; Sansom, O.J.; Morton, J.P.; Carter, C.R. Exploiting inflammation for therapeutic gain in pancreatic cancer. Br. J. Cancer 2013, 108, 997–1003. [Google Scholar] [CrossRef]
- Guerra, C.; Schuhmacher, A.J.; Canamero, M.; Grippo, P.J.; Verdaguer, L.; Perez-Gallego, L.; Dubus, P.; Sandgren, E.P.; Barbacid, M. Chronic pancreatitis is essential for induction of pancreatic ductal adenocarcinoma by K-Ras oncogenes in adult mice. Cancer Cell 2007, 11, 291–302. [Google Scholar] [CrossRef]
- Guerra, C.; Collado, M.; Navas, C.; Schuhmacher, A.J.; Hernandez-Porras, I.; Canamero, M.; Rodriguez-Justo, M.; Serrano, M.; Barbacid, M. Pancreatitis-induced inflammation contributes to pancreatic cancer by inhibiting oncogene-induced senescence. Cancer Cell 2011, 19, 728–739. [Google Scholar] [CrossRef]
- Gerbe, F.; Legraverend, C.; Jay, P. The intestinal epithelium tuft cells: Specification and function. Cell. Mol. Life Sci. CMLS 2012, 69, 2907–2917. [Google Scholar] [CrossRef]
- Gerbe, F.; van Es, J.H.; Makrini, L.; Brulin, B.; Mellitzer, G.; Robine, S.; Romagnolo, B.; Shroyer, N.F.; Bourgaux, J.F.; Pignodel, C.; et al. Distinct ATOH1 and Neurog3 requirements define tuft cells as a new secretory cell type in the intestinal epithelium. J. Cell Biol. 2011, 192, 767–780. [Google Scholar] [CrossRef]
- Liou, G.Y.; Bastea, L.; Fleming, A.; Doppler, H.; Edenfield, B.H.; Dawson, D.W.; Zhang, L.; Bardeesy, N.; Storz, P. The Presence of Interleukin-13 at Pancreatic ADM/PanIN Lesions Alters Macrophage Populations and Mediates Pancreatic Tumorigenesis. Cell Rep. 2017, 19, 1322–1333. [Google Scholar] [CrossRef]
- DelGiorno, K.E.; Chung, C.Y.; Vavinskaya, V.; Maurer, H.C.; Novak, S.W.; Lytle, N.K.; Ma, Z.; Giraddi, R.R.; Wang, D.; Fang, L.; et al. Tuft Cells Inhibit Pancreatic Tumorigenesis in Mice by Producing Prostaglandin D2. Gastroenterology 2020. [CrossRef] [PubMed]
- Saqui-Salces, M.; Keeley, T.M.; Grosse, A.S.; Qiao, X.T.; El-Zaatari, M.; Gumucio, D.L.; Samuelson, L.C.; Merchant, J.L. Gastric tuft cells express DCLK1 and are expanded in hyperplasia. Histochem. Cell Biol. 2011, 136, 191–204. [Google Scholar] [CrossRef] [PubMed]
- Gerbe, F.; Sidot, E.; Smyth, D.J.; Ohmoto, M.; Matsumoto, I.; Dardalhon, V.; Cesses, P.; Garnier, L.; Pouzolles, M.; Brulin, B.; et al. Intestinal epithelial tuft cells initiate type 2 mucosal immunity to helminth parasites. Nature 2016, 529, 226–230. [Google Scholar] [CrossRef]
- Huang, Y.H.; Klingbeil, O.; He, X.Y.; Wu, X.S.; Arun, G.; Lu, B.; Somerville, T.D.D.; Milazzo, J.P.; Wilkinson, J.E.; Demerdash, O.E.; et al. POU2F3 is a master regulator of a tuft cell-like variant of small cell lung cancer. Genes Dev. 2018, 32, 915–928. [Google Scholar] [CrossRef] [PubMed]
- Westphalen, C.B.; Takemoto, Y.; Tanaka, T.; Macchini, M.; Jiang, Z.; Renz, B.W.; Chen, X.; Ormanns, S.; Nagar, K.; Tailor, Y.; et al. Dclk1 Defines Quiescent Pancreatic Progenitors that Promote Injury-Induced Regeneration and Tumorigenesis. Cell Stem Cell 2016, 18, 441–455. [Google Scholar] [CrossRef] [PubMed]
- Ito, H.; Tanaka, S.; Akiyama, Y.; Shimada, S.; Adikrisna, R.; Matsumura, S.; Aihara, A.; Mitsunori, Y.; Ban, D.; Ochiai, T.; et al. Dominant Expression of DCLK1 in Human Pancreatic Cancer Stem Cells Accelerates Tumor Invasion and Metastasis. PLoS ONE 2016, 11, e0146564. [Google Scholar] [CrossRef] [PubMed]
- Bailey, J.M.; Alsina, J.; Rasheed, Z.A.; McAllister, F.M.; Fu, Y.Y.; Plentz, R.; Zhang, H.; Pasricha, P.J.; Bardeesy, N.; Matsui, W.; et al. DCLK1 marks a morphologically distinct subpopulation of cells with stem cell properties in preinvasive pancreatic cancer. Gastroenterology 2014, 146, 245–256. [Google Scholar] [CrossRef] [PubMed]
- Sureban, S.M.; May, R.; Qu, D.; Weygant, N.; Chandrakesan, P.; Ali, N.; Lightfoot, S.A.; Pantazis, P.; Rao, C.V.; Postier, R.G.; et al. DCLK1 regulates pluripotency and angiogenic factors via microRNA-dependent mechanisms in pancreatic cancer. PLoS ONE 2013, 8, e73940. [Google Scholar] [CrossRef]
- Sureban, S.M.; May, R.; Lightfoot, S.A.; Hoskins, A.B.; Lerner, M.; Brackett, D.J.; Postier, R.G.; Ramanujam, R.; Mohammed, A.; Rao, C.V.; et al. DCAMKL-1 regulates epithelial-mesenchymal transition in human pancreatic cells through a miR-200a-dependent mechanism. Cancer Res. 2011, 71, 2328–2338. [Google Scholar] [CrossRef]
- Cano, C.E.; Hamidi, T.; Garcia, M.N.; Grasso, D.; Loncle, C.; Garcia, S.; Calvo, E.; Lomberk, G.; Dusetti, N.; Bartholin, L.; et al. Genetic inactivation of Nupr1 acts as a dominant suppressor event in a two-hit model of pancreatic carcinogenesis. Gut 2014, 63, 984–995. [Google Scholar] [CrossRef]
- Gu, G.; Dubauskaite, J.; Melton, D.A. Direct evidence for the pancreatic lineage: NGN3+ cells are islet progenitors and are distinct from duct progenitors. Development 2002, 129, 2447–2457. [Google Scholar] [PubMed]
- Aguirre, A.J.; Bardeesy, N.; Sinha, M.; Lopez, L.; Tuveson, D.A.; Horner, J.; Redston, M.S.; DePinho, R.A. Activated Kras and Ink4a/Arf deficiency cooperate to produce metastatic pancreatic ductal adenocarcinoma. Genes Dev. 2003, 17, 3112–3126. [Google Scholar] [CrossRef] [PubMed]
- Bardeesy, N.; Aguirre, A.J.; Chu, G.C.; Cheng, K.H.; Lopez, L.V.; Hezel, A.F.; Feng, B.; Brennan, C.; Weissleder, R.; Mahmood, U.; et al. Both p16(Ink4a) and the p19(Arf)-p53 pathway constrain progression of pancreatic adenocarcinoma in the mouse. Proc. Natl. Acad. Sci. USA 2006, 103, 5947–5952. [Google Scholar] [CrossRef] [PubMed]
- Martinelli, P.; Carrillo-de Santa Pau, E.; Cox, T.; Sainz, B., Jr.; Dusetti, N.; Greenhalf, W.; Rinaldi, L.; Costello, E.; Ghaneh, P.; Malats, N.; et al. GATA6 regulates EMT and tumour dissemination, and is a marker of response to adjuvant chemotherapy in pancreatic cancer. Gut 2017, 66, 1665–1676. [Google Scholar] [CrossRef] [PubMed]
- Lomberk, G.; Blum, Y.; Nicolle, R.; Nair, A.; Gaonkar, K.S.; Marisa, L.; Mathison, A.; Sun, Z.; Yan, H.; Elarouci, N.; et al. Distinct epigenetic landscapes underlie the pathobiology of pancreatic cancer subtypes. Nat. Commun. 2018, 9, 1978. [Google Scholar] [CrossRef] [PubMed]
- Zeng, L.; Chen, Y.; Chen, L.; Tang, C. Prognostic value of CD44v9 expression in human cancers: A systematic review and meta-analysis. Medicine 2020, 99, e20428. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Wu, G.; Ye, F.; Chen, G.; Fan, Q.; Dong, H.; Zhu, X.; Wu, C. High expression of MMP19 is associated with poor prognosis in patients with colorectal cancer. BMC Cancer 2019, 19, 448. [Google Scholar] [CrossRef] [PubMed]
- Fan, H.; Yuan, Y.; Wang, J.; Zhou, F.; Zhang, M.; Giercksky, K.E.; Nesland, J.M.; Suo, Z. CD117 expression in operable oesophageal squamous cell carcinomas predicts worse clinical outcome. Histopathology 2013, 62, 1028–1037. [Google Scholar] [CrossRef]
- Zhang, Y.; Zoltan, M.; Riquelme, E.; Xu, H.; Sahin, I.; Castro-Pando, S.; Montiel, M.F.; Chang, K.; Jiang, Z.; Ling, J.; et al. Immune Cell Production of Interleukin 17 Induces Stem Cell Features of Pancreatic Intraepithelial Neoplasia Cells. Gastroenterology 2018, 155, 210–223.e213. [Google Scholar] [CrossRef]
- Inman, K.S.; Francis, A.A.; Murray, N.R. Complex role for the immune system in initiation and progression of pancreatic cancer. World J. Gastroenterol. 2014, 20, 11160–11181. [Google Scholar] [CrossRef]
- Yukawa, K.; Yasui, T.; Yamamoto, A.; Shiku, H.; Kishimoto, T.; Kikutani, H. Epoc-1: A POU-domain gene expressed in murine epidermal basal cells and thymic stromal cells. Gene 1993, 133, 163–169. [Google Scholar]
- Notta, F.; Hahn, S.A.; Real, F.X. A genetic roadmap of pancreatic cancer: Still evolving. Gut 2017, 66, 2170–2178. [Google Scholar] [CrossRef] [PubMed]
- Nicolle, R.; Blum, Y.; Marisa, L.; Loncle, C.; Gayet, O.; Moutardier, V.; Turrini, O.; Giovannini, M.; Bian, B.; Bigonnet, M.; et al. Pancreatic Adenocarcinoma Therapeutic Targets Revealed by Tumor-Stroma Cross-Talk Analyses in Patient-Derived Xenografts. Cell Rep. 2017, 21, 2458–2470. [Google Scholar] [CrossRef] [PubMed]
- Ahn, D.H.; Ramanathan, R.K. Targeting the stroma in pancreatic cancer. Chin. Clin. Oncol. 2017, 6, 65. [Google Scholar] [CrossRef]
- Iovanna, J.L.; Closa, D. Factors released by the tumor far microenvironment are decisive for pancreatic adenocarcinoma development and progression. Oncoimmunology 2017, 6, e1358840. [Google Scholar] [CrossRef]
- Vennin, C.; Murphy, K.J.; Morton, J.P.; Cox, T.R.; Pajic, M.; Timpson, P. Reshaping the Tumor Stroma for Treatment of Pancreatic Cancer. Gastroenterology 2018, 154, 820–838. [Google Scholar] [CrossRef] [PubMed]
- Argentiero, A.; De Summa, S.; Di Fonte, R.; Iacobazzi, R.M.; Porcelli, L.; Da Via, M.; Brunetti, O.; Azzariti, A.; Silvestris, N.; Solimando, A.G. Gene Expression Comparison between the Lymph Node-Positive and -Negative Reveals a Peculiar Immune Microenvironment Signature and a Theranostic Role for WNT Targeting in Pancreatic Ductal Adenocarcinoma: A Pilot Study. Cancers 2019, 11. [Google Scholar] [CrossRef]
- Porcelli, L.; Iacobazzi, R.M.; Di Fonte, R.; Serrati, S.; Intini, A.; Solimando, A.G.; Brunetti, O.; Calabrese, A.; Leonetti, F.; Azzariti, A.; et al. CAFs and TGF-beta Signaling Activation by Mast Cells Contribute to Resistance to Gemcitabine/Nabpaclitaxel in Pancreatic Cancer. Cancers 2019, 11. [Google Scholar] [CrossRef]
- Moffitt, R.A.; Marayati, R.; Flate, E.L.; Volmar, K.E.; Loeza, S.G.; Hoadley, K.A.; Rashid, N.U.; Williams, L.A.; Eaton, S.C.; Chung, A.H.; et al. Virtual microdissection identifies distinct tumor- and stroma-specific subtypes of pancreatic ductal adenocarcinoma. Nat. Genet. 2015, 47, 1168–1178. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bintz, J.; Abuelafia, A.M.; Gerbe, F.; Baudoin, E.; Auphan-Anezin, N.; Sidot, E.; Jay, P.; Iovanna, J. Expression of POU2F3 Transcription Factor Control Inflammation, Immunological Recruitment and Metastasis of Pancreatic Cancer in Mice. Biology 2020, 9, 341. https://doi.org/10.3390/biology9100341
Bintz J, Abuelafia AM, Gerbe F, Baudoin E, Auphan-Anezin N, Sidot E, Jay P, Iovanna J. Expression of POU2F3 Transcription Factor Control Inflammation, Immunological Recruitment and Metastasis of Pancreatic Cancer in Mice. Biology. 2020; 9(10):341. https://doi.org/10.3390/biology9100341
Chicago/Turabian StyleBintz, Jennifer, Analía Meilerman Abuelafia, François Gerbe, Elodie Baudoin, Nathalie Auphan-Anezin, Emmanuelle Sidot, Philippe Jay, and Juan Iovanna. 2020. "Expression of POU2F3 Transcription Factor Control Inflammation, Immunological Recruitment and Metastasis of Pancreatic Cancer in Mice" Biology 9, no. 10: 341. https://doi.org/10.3390/biology9100341
APA StyleBintz, J., Abuelafia, A. M., Gerbe, F., Baudoin, E., Auphan-Anezin, N., Sidot, E., Jay, P., & Iovanna, J. (2020). Expression of POU2F3 Transcription Factor Control Inflammation, Immunological Recruitment and Metastasis of Pancreatic Cancer in Mice. Biology, 9(10), 341. https://doi.org/10.3390/biology9100341