A Rare De Novo RAI1 Gene Mutation Affecting BDNF-Enhancer-Driven Transcription Activity Associated with Autism and Atypical Smith-Magenis Syndrome Presentation

Abstract
1. Introduction
2. Materials and Methods
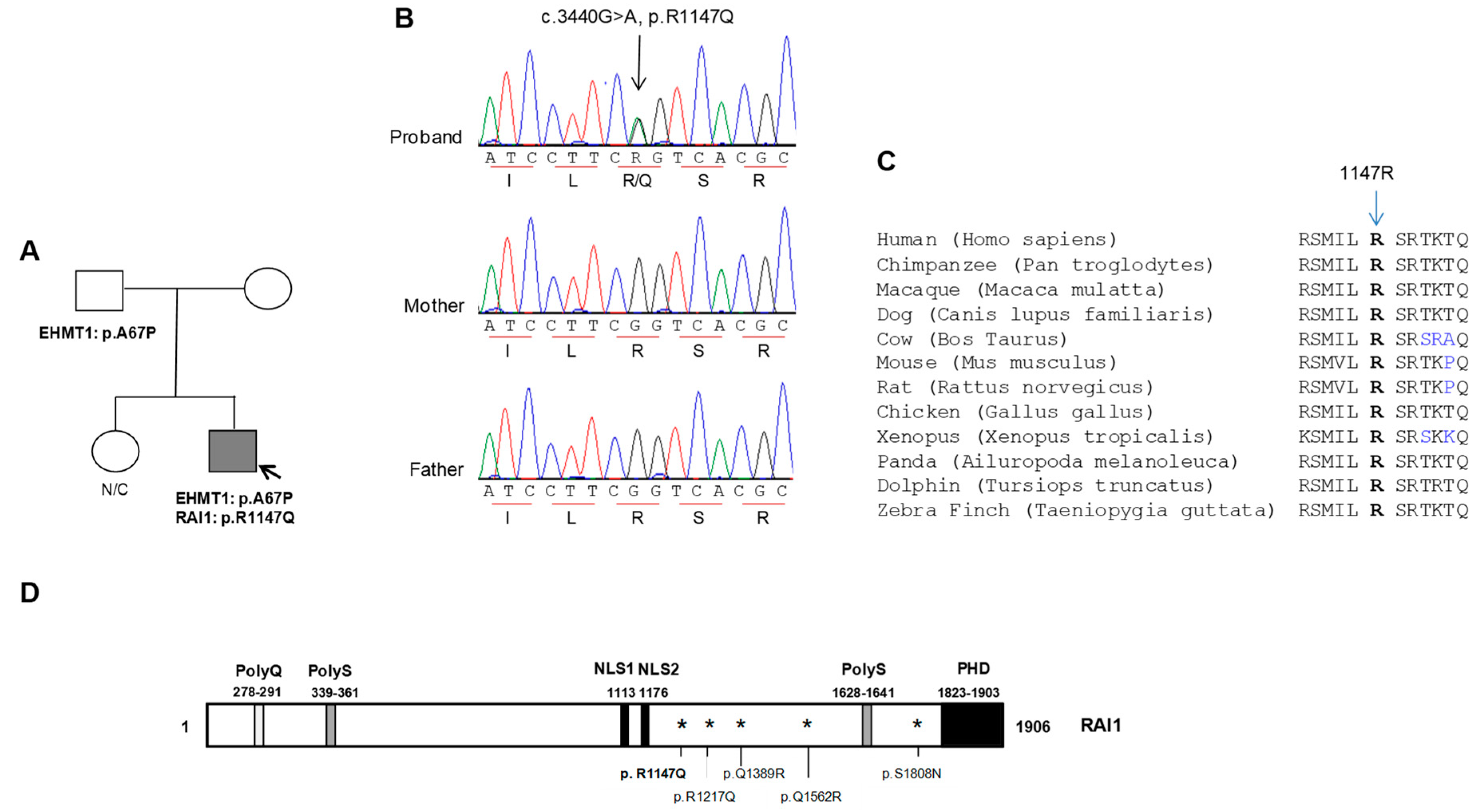
2.1. Patient Report
2.2. Mutation Screening
2.3. Plasmid Constructs
2.4. Cell Culture
2.5. Western Blot and Immunofluorescence Analysis
2.6. Reporter Gene Assays
2.7. Statistical Analysis
2.8. Next Generation RiboZero RNA Sequencing and RNA-Seq Data Analysis
3. Results
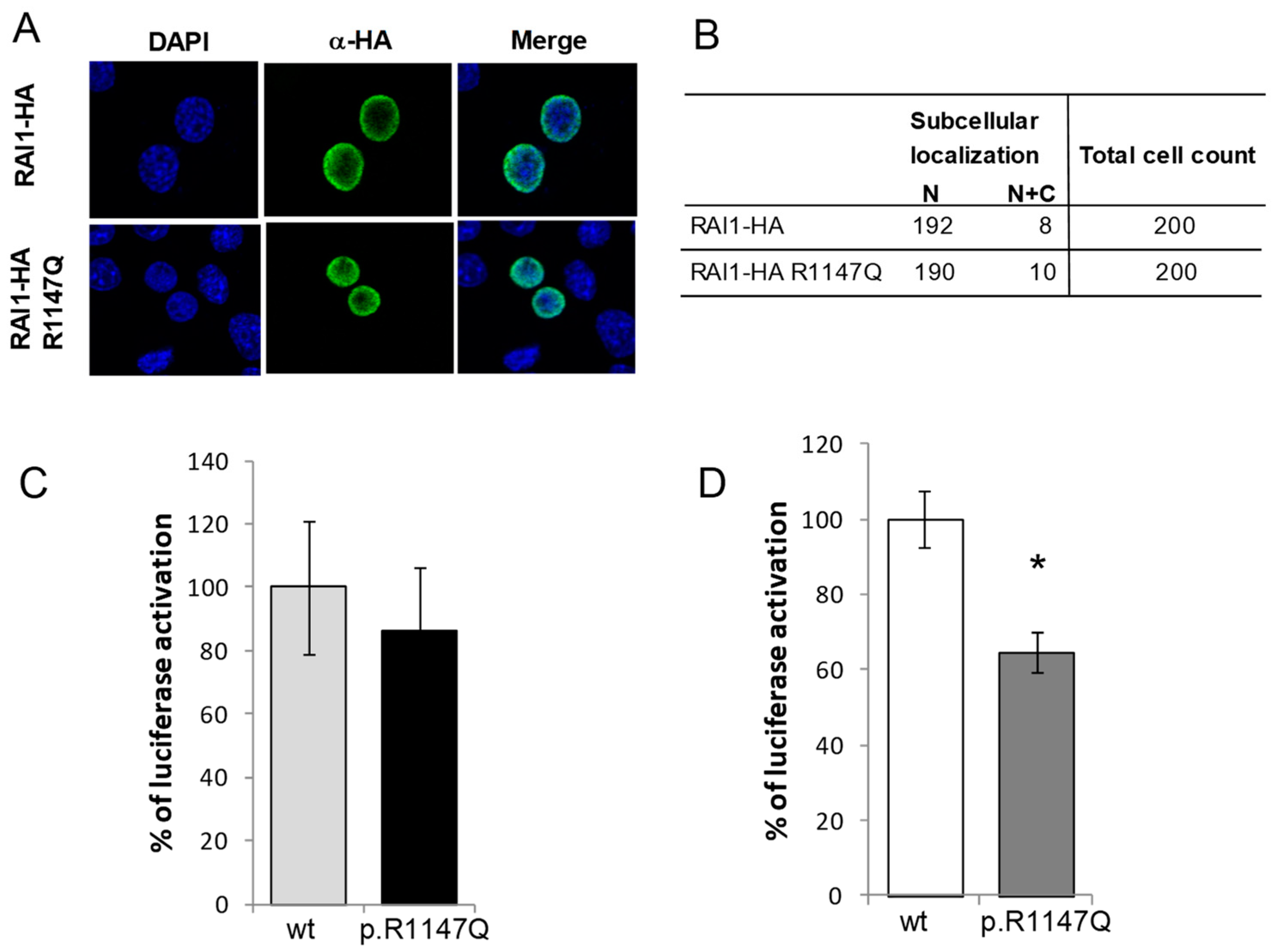
3.1. Functional Characterization of the RAI1-p.R1147Q Mutant Protein
3.2. Additional Variations and Altered Expression of Genes Related with Neurobehavioral Outcomes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Greenberg, F.; Guzzetta, V.; Montes de Oca-Luna, R.; Magenis, R.E.; Smith, A.C.; Richter, S.F.; Kondo, I.; Dobyns, W.B.; Patel, P.I.; Lupski, J.R. Molecular analysis of the Smith-Magenis syndrome: A possible contiguous-gene syndrome associated with del(17)(p11.2). Am. J. Hum. Genet. 1999, 49, 1207–1218. [Google Scholar]
- Smith, A.C.M.; Magenis, R.E.; Elsea, S.H. Overview of Smith-Magenis Syndrome. J. Assoc. Genet. Technol. 2005, 31, 163–167. [Google Scholar] [PubMed]
- Edelman, E.A.; Girirajan, S.; Finucane, B.; Patel, P.I.; Lupski, J.R.; Smith, A.C.; Elsea, S.H. Gender, genotype, and phenotype differences in Smith-Magenis syndrome: A meta-analysis of 105 cases. Clin. Genet. 2007, 71, 540–550. [Google Scholar] [CrossRef] [PubMed]
- Gropman, A.L.; Duncan, W.C.; Smith, A.C. Neurologic and developmental features of the Smith-Magenis syndrome (del 17p11.2). Pediatr. Neurol. 2006, 34, 337–350. [Google Scholar] [CrossRef] [PubMed]
- Laje, G.; Morse, R.; Richter, W.; Ball, J.; Pao, M.; Smith, A.C. Autism spectrum features in Smith-Magenis syndrome. Am. J. Med. Genet. C Semin. Med. Genet. 2010, 154C, 456–462. [Google Scholar] [CrossRef] [PubMed]
- Wolters, P.L.; Gropman, A.L.; Martin, S.C.; Smith, M.R.; Hildenbrand, H.L.; Brewer, C.C.; Smith, A.C. Neurodevelopment of children under 3 years of age with Smith-Magenis syndrome. Pediatr. Neurol. 2009, 41, 250–258. [Google Scholar] [CrossRef] [PubMed]
- Bi, W.; Saifi, G.M.; Shaw, C.J.; Walz, K.; Fonseca, P.; Wilson, M.; Potocki, L.; Lupski, J.R. Mutations of RAI1, a PHD-containing protein, in nondeletion patients with Smith-Magenis syndrome. Hum. Genet. 2004, 115, 515–524. [Google Scholar] [CrossRef] [PubMed]
- Bi, W.; Saifi, G.M.; Girirajan, S.; Shi, X.; Szomju, B.; Firth, H.; Magenis, R.E.; Potocki, L.; Elsea, S.H.; Lupski, J.R. RAI1 point mutations, CAG repeat variation, and SNP analysis in non-deletion Smith-Magenis syndrome. Am. J. Med. Genet. A 2006, 140, 2454–2463. [Google Scholar] [CrossRef] [PubMed]
- Dubourg, C.; Bonnet-Brilhault, F.; Toutain, A.; Mignot, C.; Jacquette, A.; Dieux, A.; Gérard, M.; Beaumont-Epinette, M.P.; Julia, S.; Isidor, B.; et al. Identification of Nine New RAI1-Truncating Mutations in Smith-Magenis Syndrome Patients without 17p11.2 Deletions. Mol. Syndromol. 2014, 5, 57–64. [Google Scholar] [CrossRef] [PubMed]
- Girirajan, S.; Elsas, L.J.; Devriendt, K.; Elsea, S.H. RAI1 variations in Smith-Magenis syndrome patients without 17p11.2 deletions. J. Med. Genet. 2005, 42, 820–828. [Google Scholar] [CrossRef] [PubMed]
- Girirajan, S.; Truong, H.T.; Blanchard, C.L.; Elsea, S.H. A functional network module for Smith-Magenis syndrome. Clin. Genet. 2009, 75, 364–374. [Google Scholar] [CrossRef] [PubMed]
- Potocki, L.; Shaw, C.J.; Stankiewicz, P.; Lupski, J.R. Variability in clinical phenotype despite common chromosomal deletion in Smith-Magenis syndrome [del(17)(p11.2p11.2)]. Genet. Med. 2003, 5, 430–434. [Google Scholar] [CrossRef] [PubMed]
- Slager, R.E.; Newton, T.L.; Vlangos, C.N.; Finucane, B.; Elsea, S.H. Mutations in RAI1 associated with Smith-Magenis syndrome. Nat. Genet. 2003, 33, 466–468. [Google Scholar] [CrossRef] [PubMed]
- Truong, H.T.; Dudding, T.; Blanchard, C.L.; Elsea, S.H. Frameshift mutation hotspot identified in Smith-Magenis syndrome: Case report and review of literature. BMC Med. Genet. 2010, 11, 142. [Google Scholar] [CrossRef] [PubMed]
- Vieira, G.H.; Rodriguez, J.D.; Carmona-Mora, P.; Cao, L.; Gamba, B.F.; Carvalho, D.R.; de Rezende Duarte, A.; Santos, S.R.; de Souza, D.H.; DuPont, B.R.; et al. Detection of classical 17p11.2 deletions, an atypical deletion and RAI1 alterations in patients with features suggestive of Smith-Magenis syndrome. Eur. J. Hum. Genet. 2012, 20, 148–154. [Google Scholar] [CrossRef] [PubMed]
- Vilboux, T.; Ciccone, C.; Blancato, J.K.; Cox, G.F.; Deshpande, C.; Introne, W.J.; Gahl, W.A.; Smith, A.C.; Huizing, M. Molecular analysis of the Retinoic Acid Induced 1 gene (RAI1) in patients with suspected Smith-Magenis syndrome without the 17p11.2 deletion. PLoS ONE 2011, 6, e22861. [Google Scholar] [CrossRef] [PubMed]
- Ricard, G.; Molina, J.; Chrast, J.; Gu, W.; Gheldof, N.; Pradervand, S.; Schütz, F.; Young, J.I.; Lupski, J.R.; Reymond, A.; et al. Phenotypic consequences of copy number variation: Insights from Smith-Magenis and Potocki-Lupski syndrome mouse models. PLoS Biol. 2010, 8, e1000543. [Google Scholar] [CrossRef] [PubMed]
- Rao, N.R.; Abad, C.; Perez, I.C.; Srivastava, A.K.; Young, J.I.; Walz, K. Rai1 haploinsufficiency is associated with social abnormalities in mice. Biology 2017, 6, 25. [Google Scholar] [CrossRef] [PubMed]
- Walz, K.; Spencer, C.; Kaasik, K.; Lee, C.C.; Lupski, J.R.; Paylor, R. Behavioral characterization of mouse models for Smith-Magenis syndrome and dup(17)(p11.2p11.2). Hum. Mol. Genet. 2004, 13, 367–378. [Google Scholar] [CrossRef] [PubMed]
- Hiroi, N. Critical reappraisal of mechanistic links of copy number variants to dimensional constructs of neuropsychiatric disorders in mouse models. Psychiatry Clin. Neurosci. 2018, 72, 301–321. [Google Scholar] [CrossRef] [PubMed]
- Fragoso, Y.D.; Stoney, P.N.; Shearer, K.D.; Sementilli, A.; Nanescu, S.E.; Sementilli, P.; McCaffery, P. Expression in the human brain of retinoic acid induced 1, a protein associated with neurobehavioural disorders. Brain Struct. Funct. 2015, 220, 1195–1203. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.H.; Guenthner, C.J.; Xu, J.; Nguyen, T.; Schwarz, L.A.; Wilkinson, A.W.; Gozani, O.; Chang, H.Y.; Shamloo, M.; Luo, L. Molecular and Neural Functions of Rai1, the Causal Gene for Smith-Magenis Syndrome. Neuron 2016, 92, 392–406. [Google Scholar] [CrossRef] [PubMed]
- Tahir, R.; Kennedy, A.; Elsea, S.H.; Dickinson, A.J. Retinoic acid induced-1 (Rai1) regulates craniofacial and brain development in Xenopus. Mech. Dev. 2014, 133, 91–104. [Google Scholar] [CrossRef] [PubMed]
- Burns, B.; Schmidt, K.; Williams, S.R.; Kim, S.; Girirajan, S.; Elsea, S.H. Rai1 haploinsufficiency causes reduced Bdnf expression resulting in hyperphagia, obesity and altered fat distribution in mice and humans with no evidence of metabolic syndrome. Hum. Mol. Genet. 2010, 19, 4026–4042. [Google Scholar] [CrossRef] [PubMed]
- Carmona-Mora, P.; Walz, K. Retinoic Acid Induced 1, RAI1 A Dosage Sensitive Gene Related to Neurobehavioral Alterations Including Autistic Behavior. Curr. Genom. 2010, 11, 607. [Google Scholar] [CrossRef] [PubMed]
- Carmona-Mora, P.; Encina, C.A.; Canales, C.P.; Cao, L.; Molina, J.; Kairath, P.; Young, J.I.; Walz, K. Functional and cellular characterization of human Retinoic Acid Induced 1 (RAI1) mutations associated with Smith-Magenis Syndrome. BMC Mol. Biol. 2010, 11, 63. [Google Scholar] [CrossRef] [PubMed]
- Carmona-Mora, P.; Canales, C.P.; Cao, L.; Perez, I.C.; Srivastava, A.K.; Young, J.I.; Walz, K. RAI1 transcription factor activity is impaired in mutants associated with Smith-Magenis Syndrome. PLoS ONE 2012, 7, E45155. [Google Scholar] [CrossRef] [PubMed]
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders: DSM-IV®-TR, 4th ed.; American Psychiatric Publishing, Inc.: Washington, DC, USA, 2000. [Google Scholar]
- Ozonoff, S.; Boodlin-Jones, B.; Solomon, M. Evidence-based assessment of Autism Spectrum Disorder in children and adolescents. J. Clin. Child Adolesc. Psychol. 2005, 4, 523–540. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef] [PubMed]
- Kleefstra, T.; Brunner, H.G.; Amiel, J.; Oudakker, A.R.; Nillesen, W.M.; Magee, A.; Geneviève, D.; Cormier-Daire, V.; van Esch, H.; Fryns, J.P.; et al. Loss-of-function mutations in euchromatin histone methyl transferase 1 (EHMT1) cause the 9q34 subtelomeric deletion syndrome. Am. J. Hum. Genet. 2006, 79, 370–377. [Google Scholar] [CrossRef] [PubMed]
- Langouët, M.; Saadi, A.; Rieunier, G.; Moutton, S.; Siquier-Pernet, K.; Fernet, M.; Nitschke, P.; Munnich, A.; Stern, M.H.; Chaouch, M.; et al. Mutation in TTI2 reveals a role for triple T complex in human brain development. Hum. Mutat. 2013, 34, 1472–1476. [Google Scholar] [CrossRef] [PubMed]
- Létard, P.; Drunat, S.; Vial, Y.; Duerinckx, S.; Ernault, A.; Amram, D.; Arpin, S.; Bertoli, M.; Busa, T.; Ceulemans, B.; et al. Autosomal recessive primary microcephaly due to ASPM mutations: An update. Hum. Mutat. 2018, 39, 319–332. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.W.; Sherman, B.T.; Tan, Q.; Kir, J.; Liu, D.; Bryant, D.; Guo, Y.; Stephens, R.; Baseler, M.W.; Lane, H.C.; et al. DAVID bioinformatics resources: Expanded annotation database and novel algorithms to better extract biology from large gene lists. Nucleic Acids Res. 2007, 35, W169–W175. [Google Scholar] [CrossRef] [PubMed]
- Hayes, S.; Turecki, G.; Brisebois, K.; Lopes-Cendes, I.; Gaspar, C.; Riess, O.; Ranum, L.P.; Pulst, S.M.; Rouleau, G.A. CAG repeat length in RAI1 is associated with age at onset variability in spinocerebellar ataxia type 2 (SCA2). Hum. Mol. Genet. 2000, 9, 1753–1758. [Google Scholar] [CrossRef] [PubMed]
- Joober, R.; Benkelfat, C.; Toulouse, A.; Lafrenière, R.G.; Lal, S.; Ajroud, S.; Turecki, G.; Bloom, D.; Labelle, A.; Lalonde, P.; et al. Analysis of 14 CAG repeat-containing genes in schizophrenia. Am. J. Med. Genet. 1999, 88, 694–699. [Google Scholar] [CrossRef]
- Van der Zwaag, B.; Franke, L.; Poot, M.; Hochstenbach, R.; Spierenburg, H.A.; Vorstman, J.A.; van Daalen, E.; de Jonge, M.V.; Verbeek, N.E.; Brilstra, E.H.; et al. Gene-network analysis identifies susceptibility genes related to glycobiology in autism. PLoS ONE 2009, 4, e5324. [Google Scholar] [CrossRef] [PubMed]
- Thaker, V.V.; Esteves, K.M.; Towne, M.C.; Brownstein, C.A.; James, P.M.; Crowley, L.; Hirschhorn, J.N.; Elsea, S.H.; Beggs, A.H.; Picker, J.; et al. Whole Exome Sequencing Identifies RAI1 Mutation in a Morbidly Obese Child Diagnosed with ROHHAD Syndrome. J. Clin. Endocrinol. Metab. 2015, 100, 1723–1730. [Google Scholar] [CrossRef] [PubMed]
- De Boer, A.; Vermeulen, K.; Egger, J.I.M.; Janzing, J.G.E.; de Leeuw, N.; Veenstra-Knol, H.E.; den Hollander, N.S.; van Bokhoven, H.; Staal, W.; Kleefstra, T. EHMT1 mosaicism in apparently unaffected parents is associated with autism spectrum disorder and neurocognitive dysfunction. Mol. Autism. 2018, 9, 5. [Google Scholar] [CrossRef] [PubMed]
- Angelucci, F.; Brenè, S.; Mathé, A.A. BDNF in schizophrenia, depression and corresponding animal models. Mol. Psychiatry 2005, 10, 345–352. [Google Scholar] [CrossRef] [PubMed]
- Cummings, B.B.; Marshall, J.L.; Tukiainen, T.; Lek, M.; Donkervoort, S.; Foley, A.R.; Bolduc, V.; Waddell, L.B.; Sandaradura, S.A.; O'Grady, G.L.; et al. Improving genetic diagnosis in Mendelian disease with transcriptome sequencing. Sci. Transl. Med. 2017, 9, 386. [Google Scholar] [CrossRef] [PubMed]
- Kenny, E.M.; Cormican, P.; Furlong, S.; Heron, E.; Kenny, G.; Fahey, C.; Kelleher, E.; Ennis, S.; Tropea, D.; Anney, R.; et al. Excess of rare novel loss-of-function variants in synaptic genes in schizophrenia and autism spectrum disorders. Mol. Psychiatry 2014, 19, 872–879. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| SMS Patients | |||
|---|---|---|---|
| Phenotypes | % In Common 17p11.2 Deletion [3,5,10,16,26] | % In RAI1 Mutations [3,9,10,16,26] | PROBAND p.R1147Q |
| Craniofacial Abnormalities | 100 | 100 | - |
| Skeletal Abnormalities | |||
| Short stature | 70–80 | 11 | - |
| Scoliosis/vertebral abnormalities | 73 | 40–50 | - |
| Short broad hands/brachydactyly | 85 | 88 | - |
| Otorhinolaryngolocial | |||
| Hoarse voice | 66–80 | 76–86 | - |
| Hearing loss | 60–68 | 11–33 | - |
| Neurological | |||
| Cognitive impairment | 100 | 100 | + |
| Infantile hypotonia | <90 | 50–61 | - |
| Speech delay | >90 | 70 | + & |
| Motor delay | >90 | 60–70 | + & |
| Sleep disturbance | 90 | 100 | + |
| EEG abnormalities | 50–66 | 80 | Not examined |
| Seizures | 11–30 | 16.6–50 | + |
| Behavioral | |||
| Self-hugging | 50–80 | 100 | - |
| Onychotillomania | 25–85 | 80–100 | - |
| Polyembolokoilamania | 25–85 | 75–80 | - |
| Head banging/face slapping | 70 | 90 | - |
| Hand biting | 80 | 60–71 | - |
| Attention seeking | 80–100 | 100 | + |
| Aggressive behavior | 55 | + | |
| Self-injurious behavior | 70–90 | >90 | - |
| Hyperactivity | 80 | 100 | + |
| Autistic features | 90(6) | NR | +(DSM-IV) |
| Other features | |||
| Cardiac defects | 30 | 0 | - |
| Renal/urinary tract defect | 30 | 0 | - |
| Obesity | 18 | 78 | - |
| Overeating | 25 | 81 | + # |
| Gene | log2FoldChange | Description | p Value | padj |
|---|---|---|---|---|
| GNG12 | 1.719707224 | Upregulated in the Patient sample | 4.81 × 10−9 | 5.02 × 10−5 |
| HCP5 | 1.617432091 | Upregulated in the Patient sample | 3.37 × 10−7 | 0.001757018 |
| GGACT | 1.35253954 | Upregulated in the Patient sample | 1.74 × 10−8 | 0.000120784 |
| HAPLN3 | 1.269895566 | Upregulated in the Patient sample | 2.34 × 10−5 | 0.044419951 |
| ICOS | 1.228236573 | Upregulated in the Patient sample | 2.58 × 10−9 | 5.02 × 10−5 |
| TRIM26 | 0.991872296 | Upregulated in the Patient sample | 1.60 × 10−8 | 0.0371298 |
| PKHD1L1 | −0.998015573 | Downregulated in the patient sample | 8.08 × 10−7 | 0.002466581 |
| DST | −1.109285101 | Downregulated in the patient sample | 5.29 × 10−7 | 0.002209546 |
| CCZ1 | −1.313033217 | Downregulated in the patient sample | 2.33 × 10−5 | 0.044419951 |
| MTRNR2L8 | −1.319578464 | Downregulated in the patient sample | 2.83 × 10−6 | 0.007389326 |
| PTCH1 | −1.559860559 | Downregulated in the patient sample | 8.27 × 10−7 | 0.002466581 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abad, C.; Cook, M.M.; Cao, L.; Jones, J.R.; Rao, N.R.; Dukes-Rimsky, L.; Pauly, R.; Skinner, C.; Wang, Y.; Luo, F.; et al. A Rare De Novo RAI1 Gene Mutation Affecting BDNF-Enhancer-Driven Transcription Activity Associated with Autism and Atypical Smith-Magenis Syndrome Presentation. Biology 2018, 7, 31. https://doi.org/10.3390/biology7020031
Abad C, Cook MM, Cao L, Jones JR, Rao NR, Dukes-Rimsky L, Pauly R, Skinner C, Wang Y, Luo F, et al. A Rare De Novo RAI1 Gene Mutation Affecting BDNF-Enhancer-Driven Transcription Activity Associated with Autism and Atypical Smith-Magenis Syndrome Presentation. Biology. 2018; 7(2):31. https://doi.org/10.3390/biology7020031
Chicago/Turabian StyleAbad, Clemer, Melissa M. Cook, Lei Cao, Julie R. Jones, Nalini R. Rao, Lynn Dukes-Rimsky, Rini Pauly, Cindy Skinner, Yunsheng Wang, Feng Luo, and et al. 2018. "A Rare De Novo RAI1 Gene Mutation Affecting BDNF-Enhancer-Driven Transcription Activity Associated with Autism and Atypical Smith-Magenis Syndrome Presentation" Biology 7, no. 2: 31. https://doi.org/10.3390/biology7020031
APA StyleAbad, C., Cook, M. M., Cao, L., Jones, J. R., Rao, N. R., Dukes-Rimsky, L., Pauly, R., Skinner, C., Wang, Y., Luo, F., Stevenson, R. E., Walz, K., & Srivastava, A. K. (2018). A Rare De Novo RAI1 Gene Mutation Affecting BDNF-Enhancer-Driven Transcription Activity Associated with Autism and Atypical Smith-Magenis Syndrome Presentation. Biology, 7(2), 31. https://doi.org/10.3390/biology7020031