
Multi-Facial, Non-Peptidic α-Helix Mimetics

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
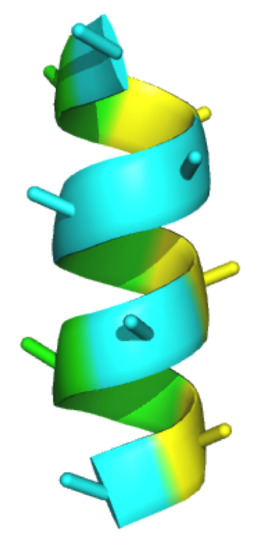
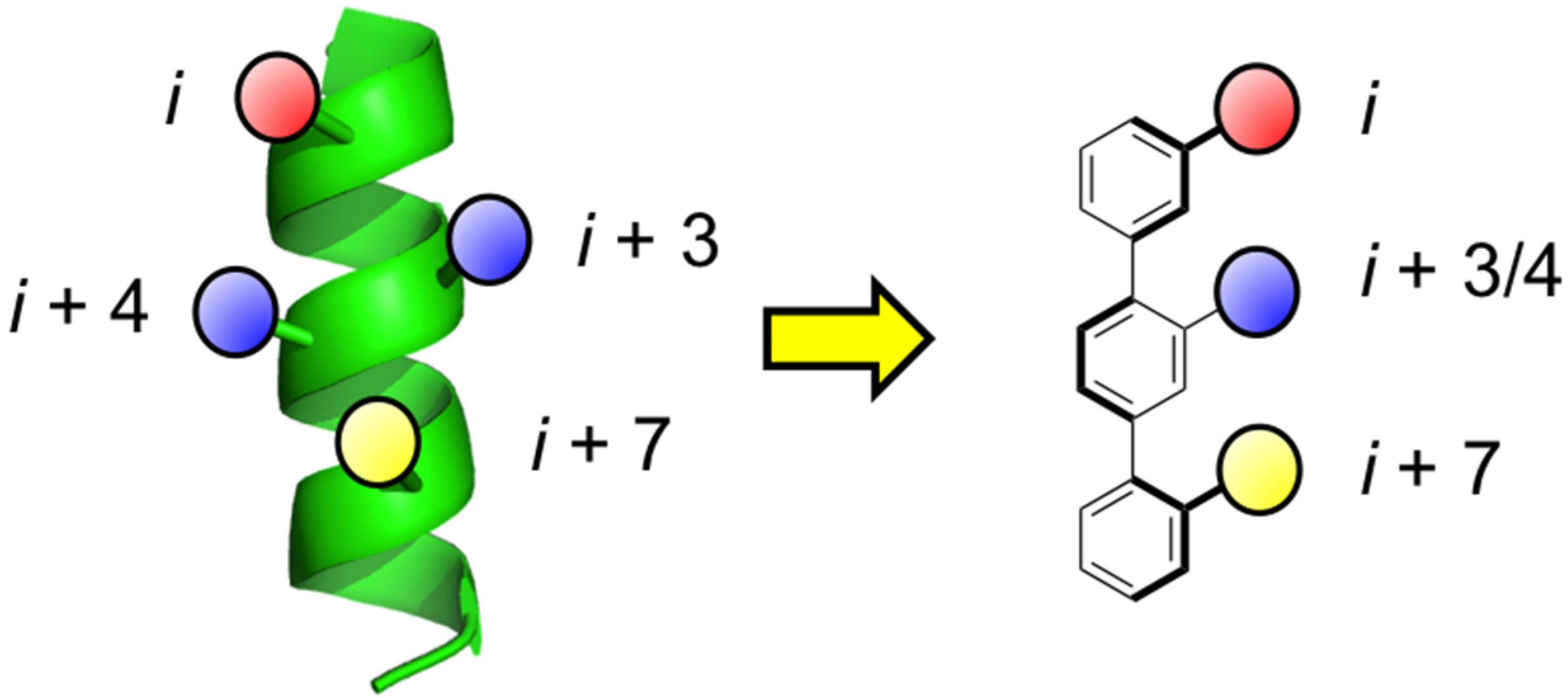
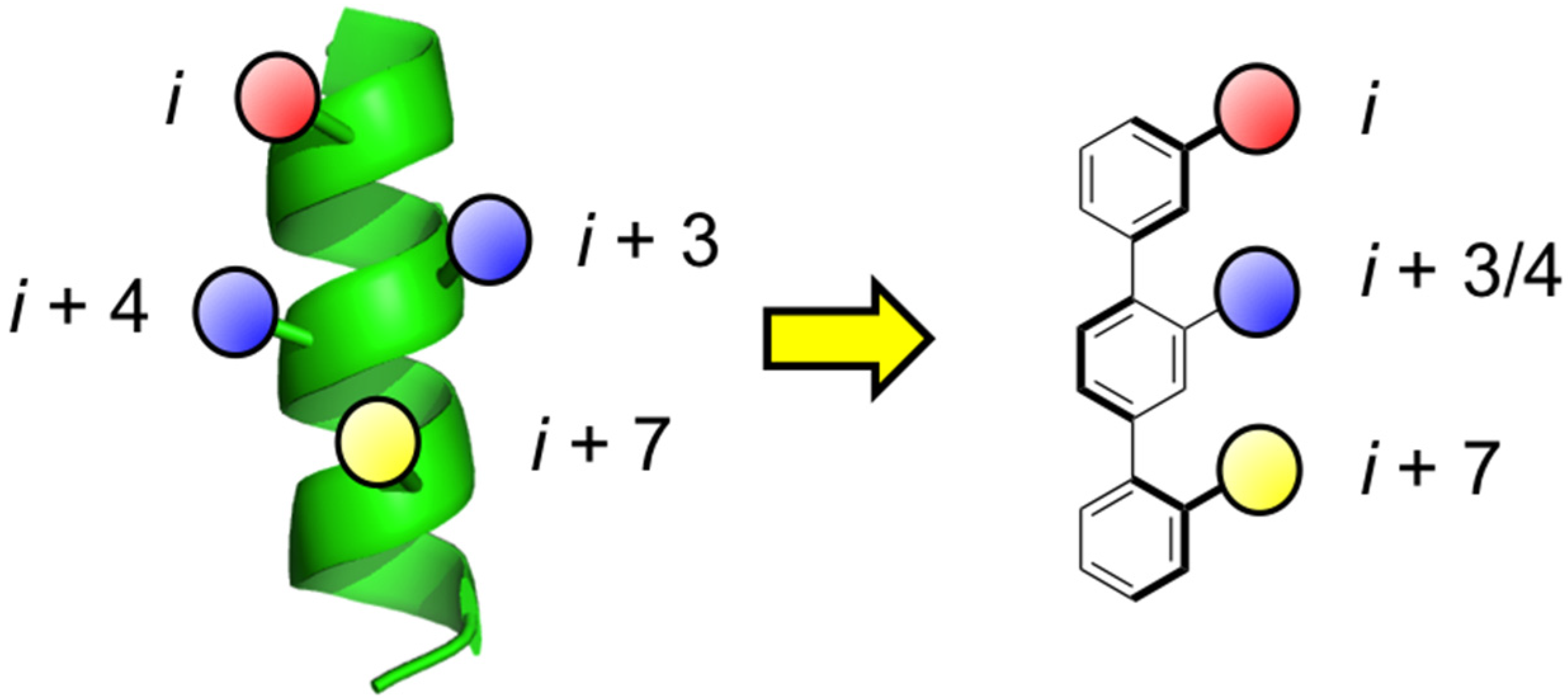
1. Introduction
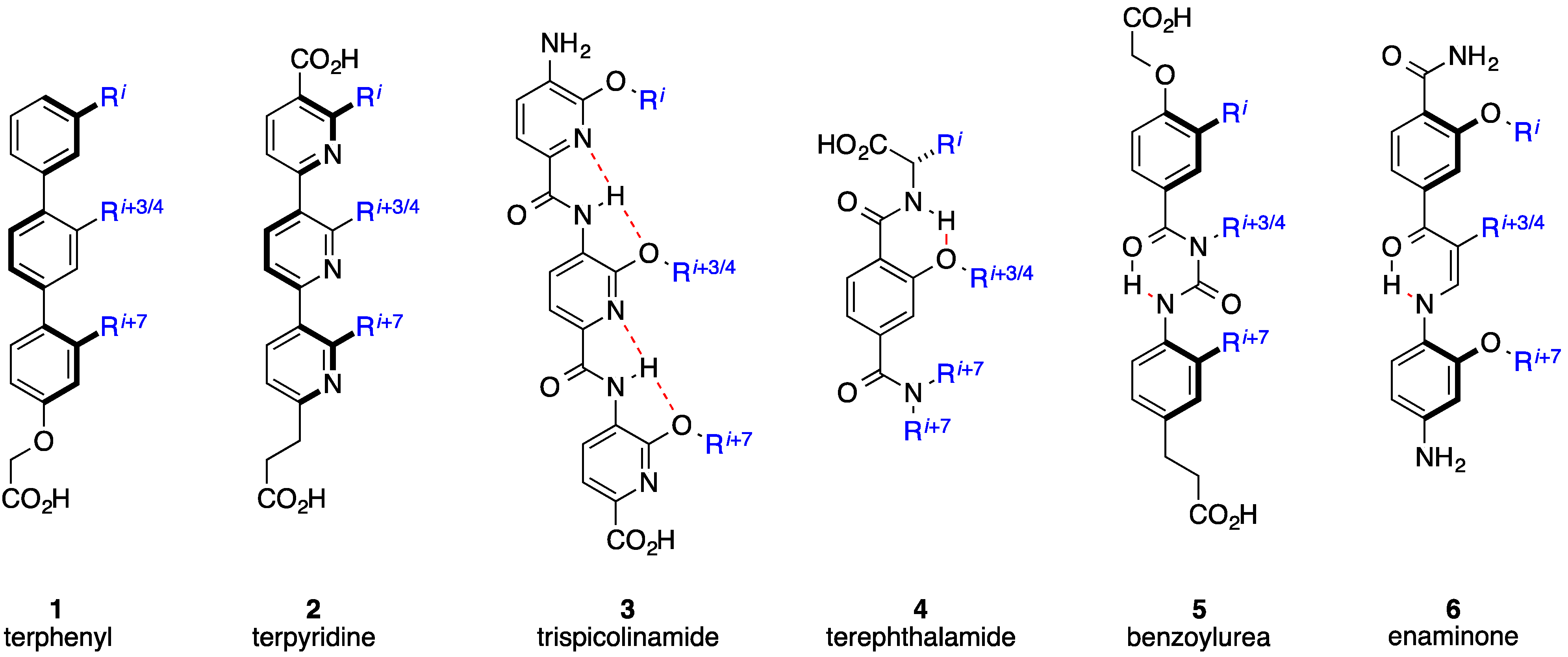
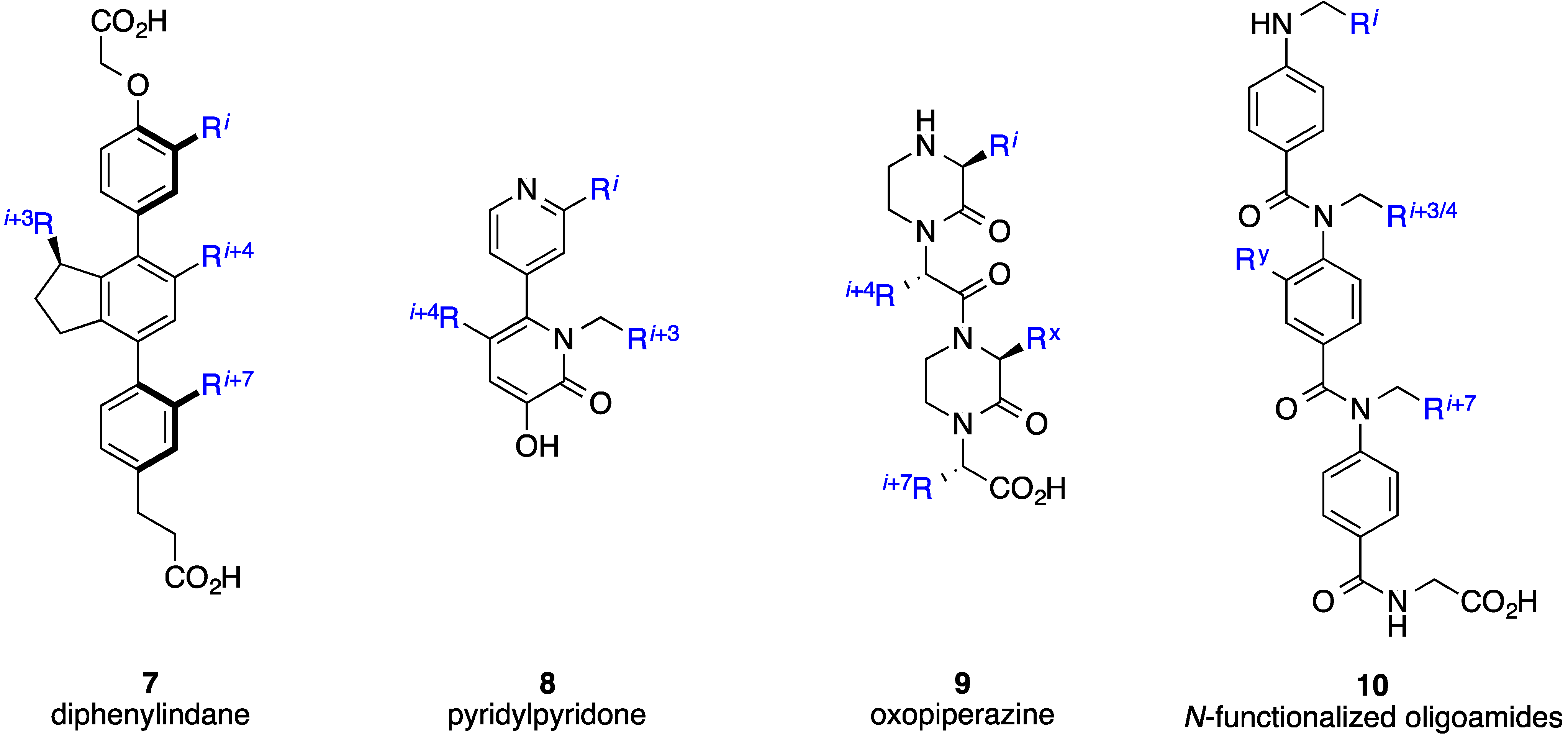
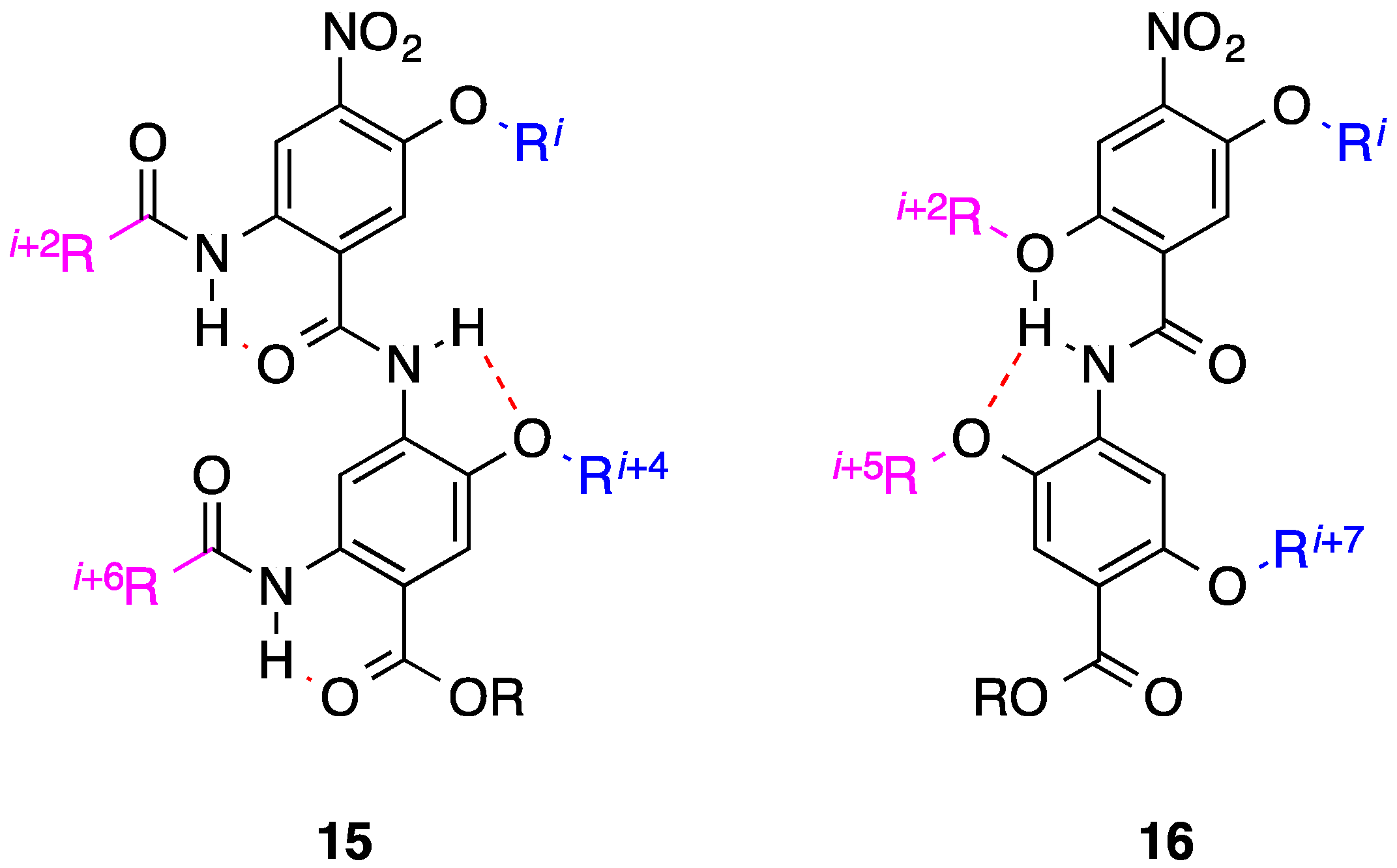
2. Bis-Benzamides
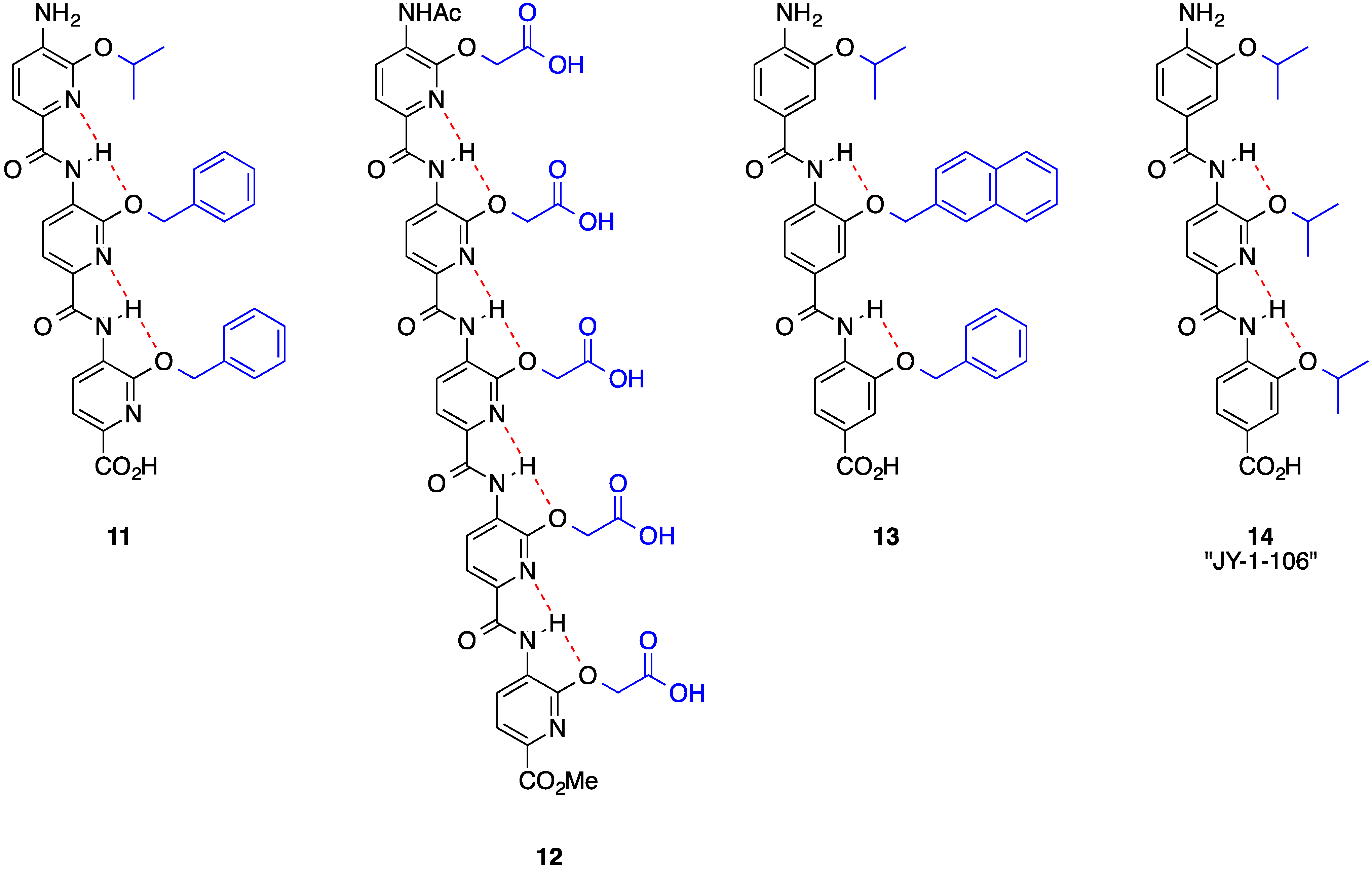
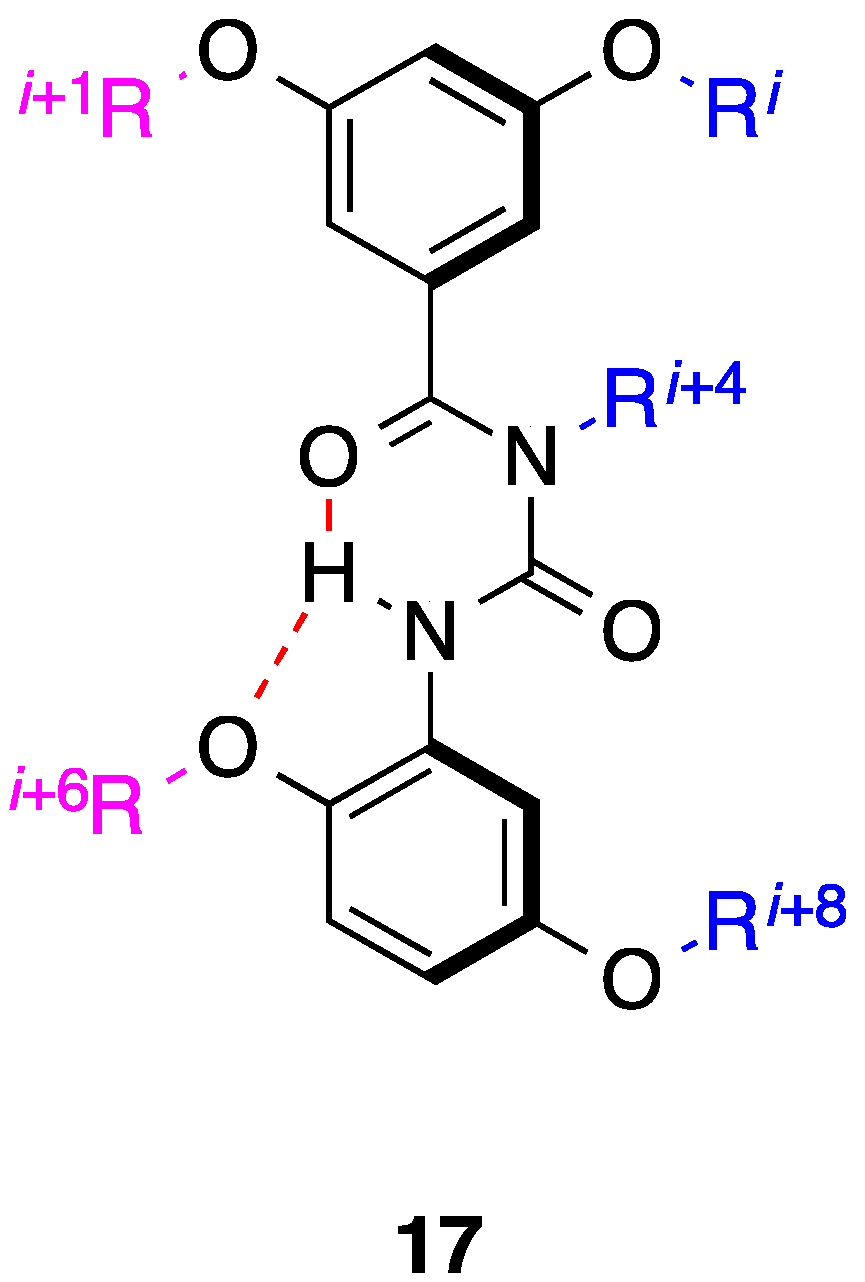
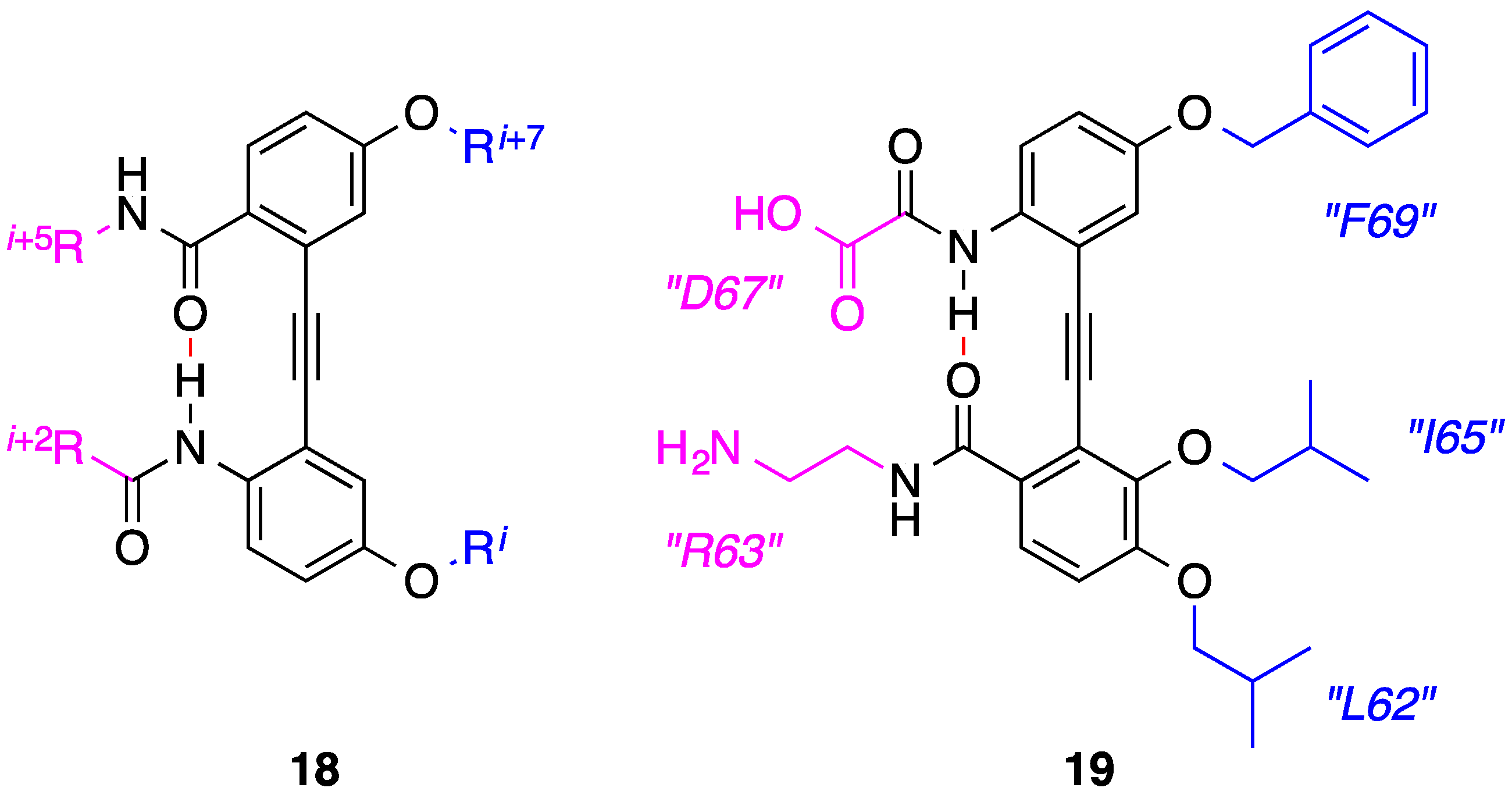
3. Benzoylureas
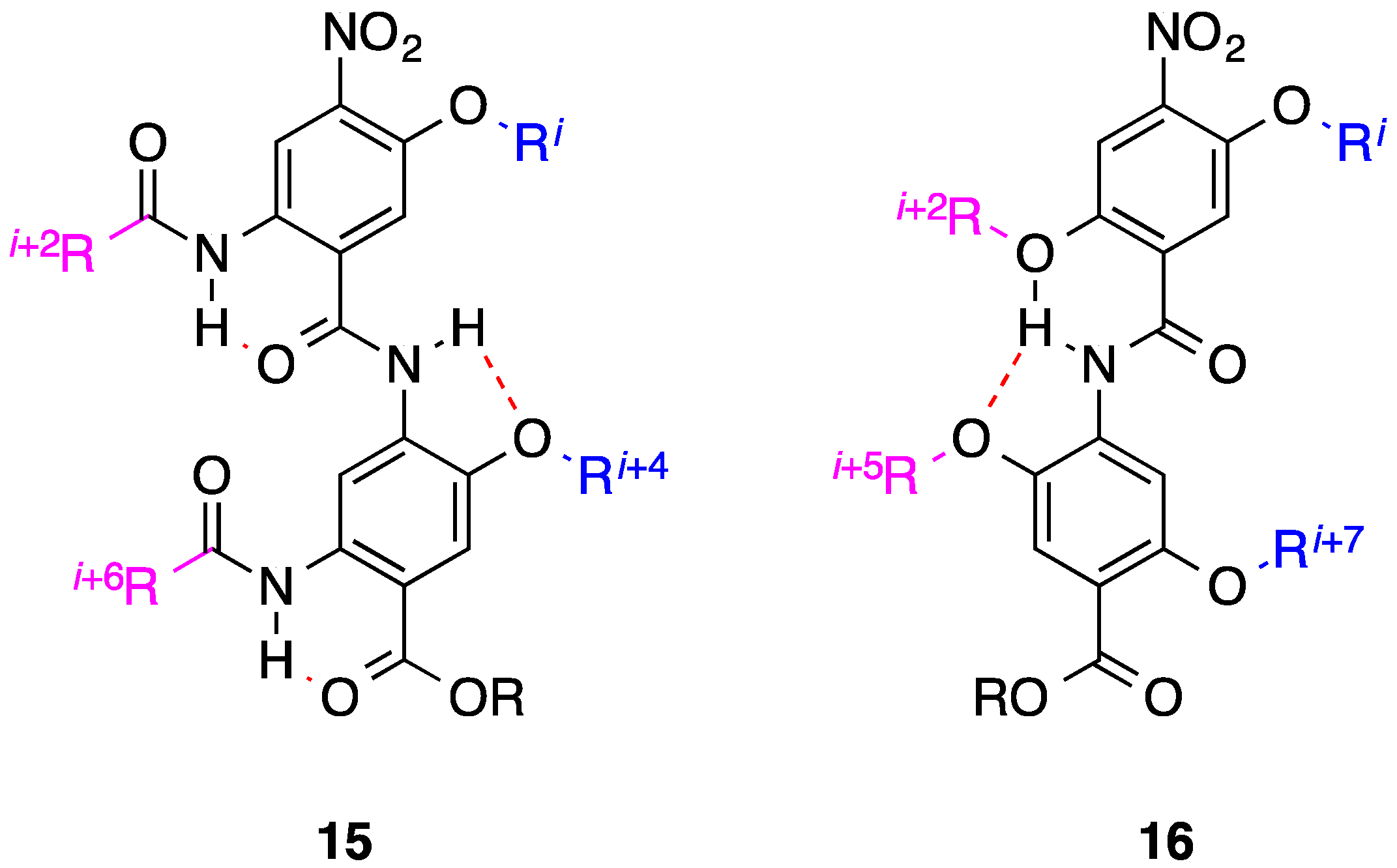
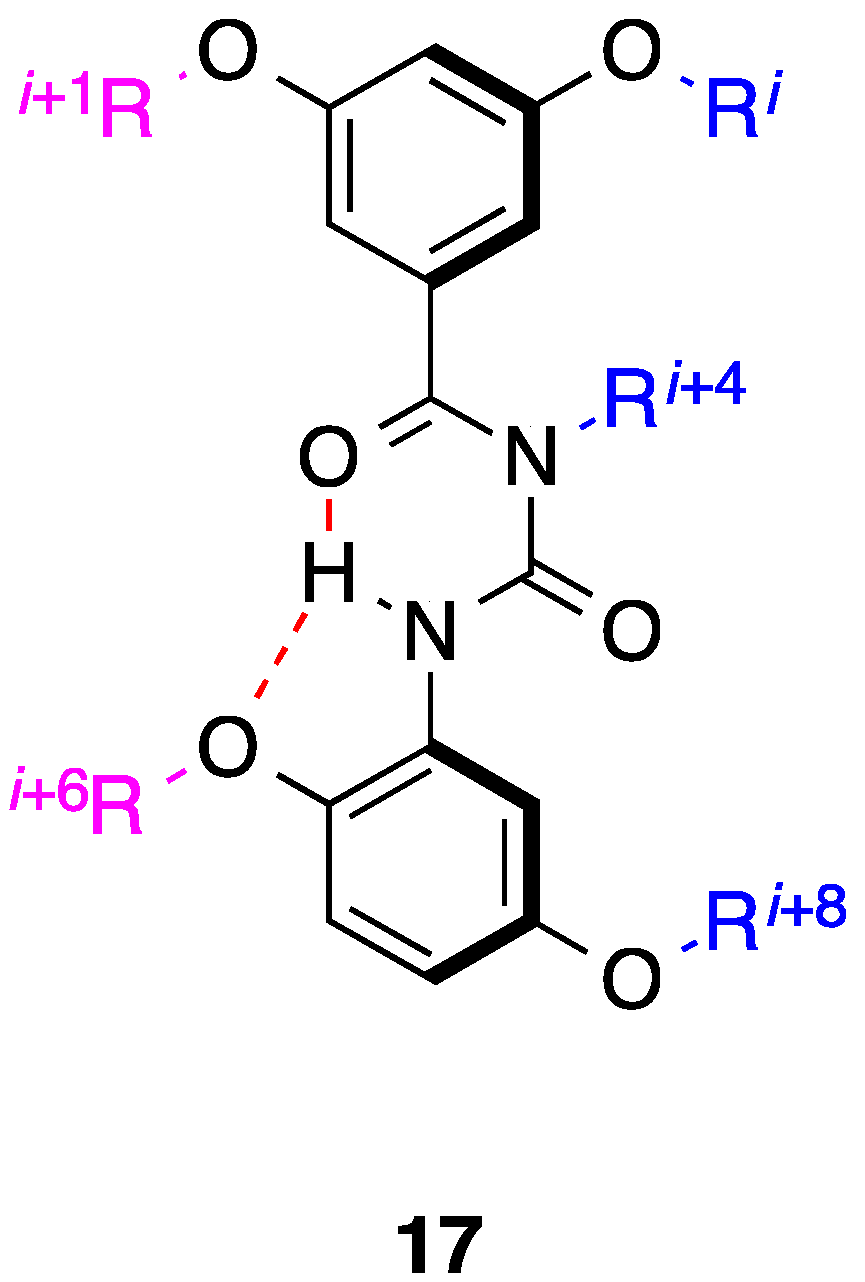
4. 1,2-Diphenylacetylenes
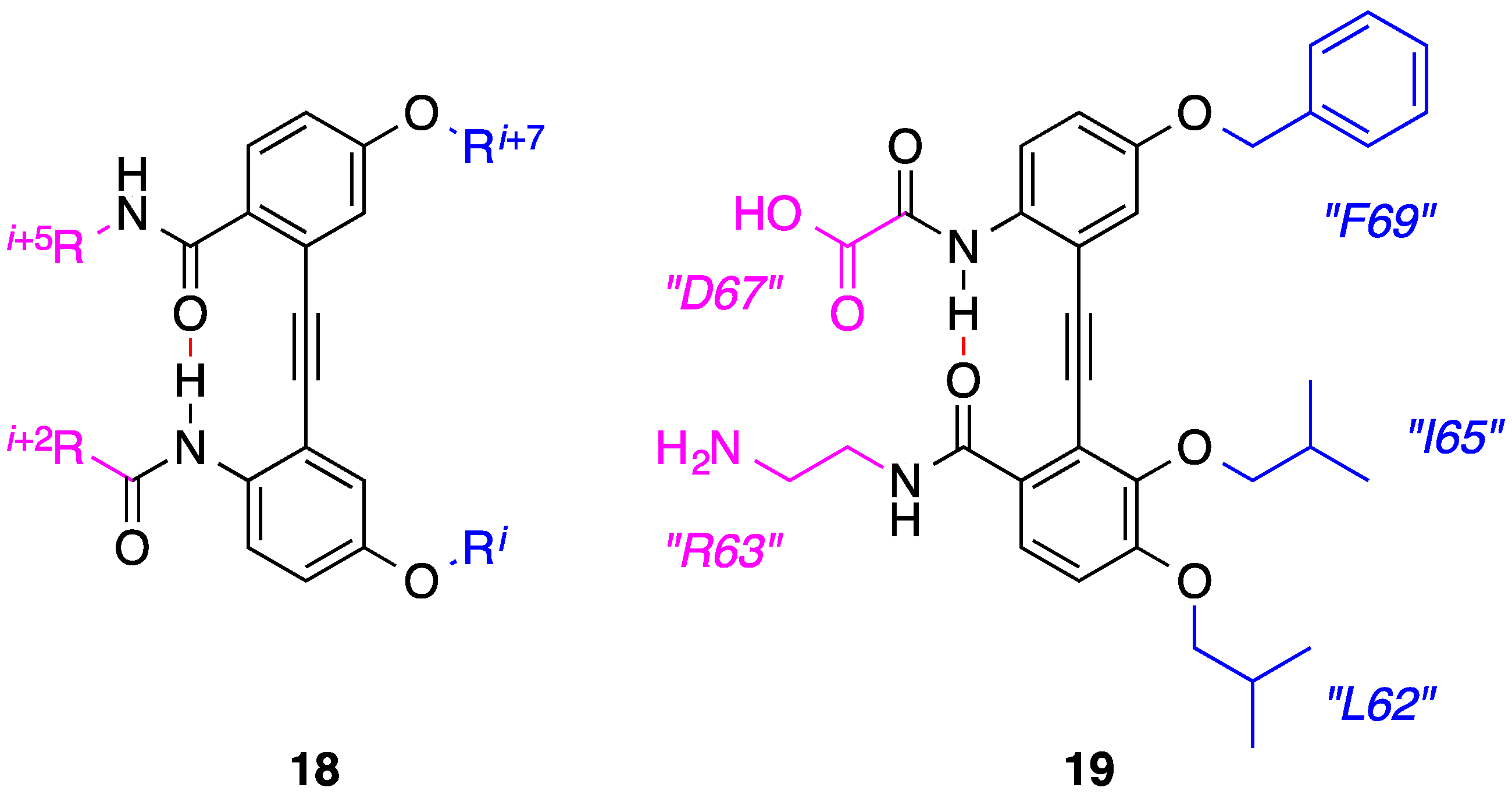
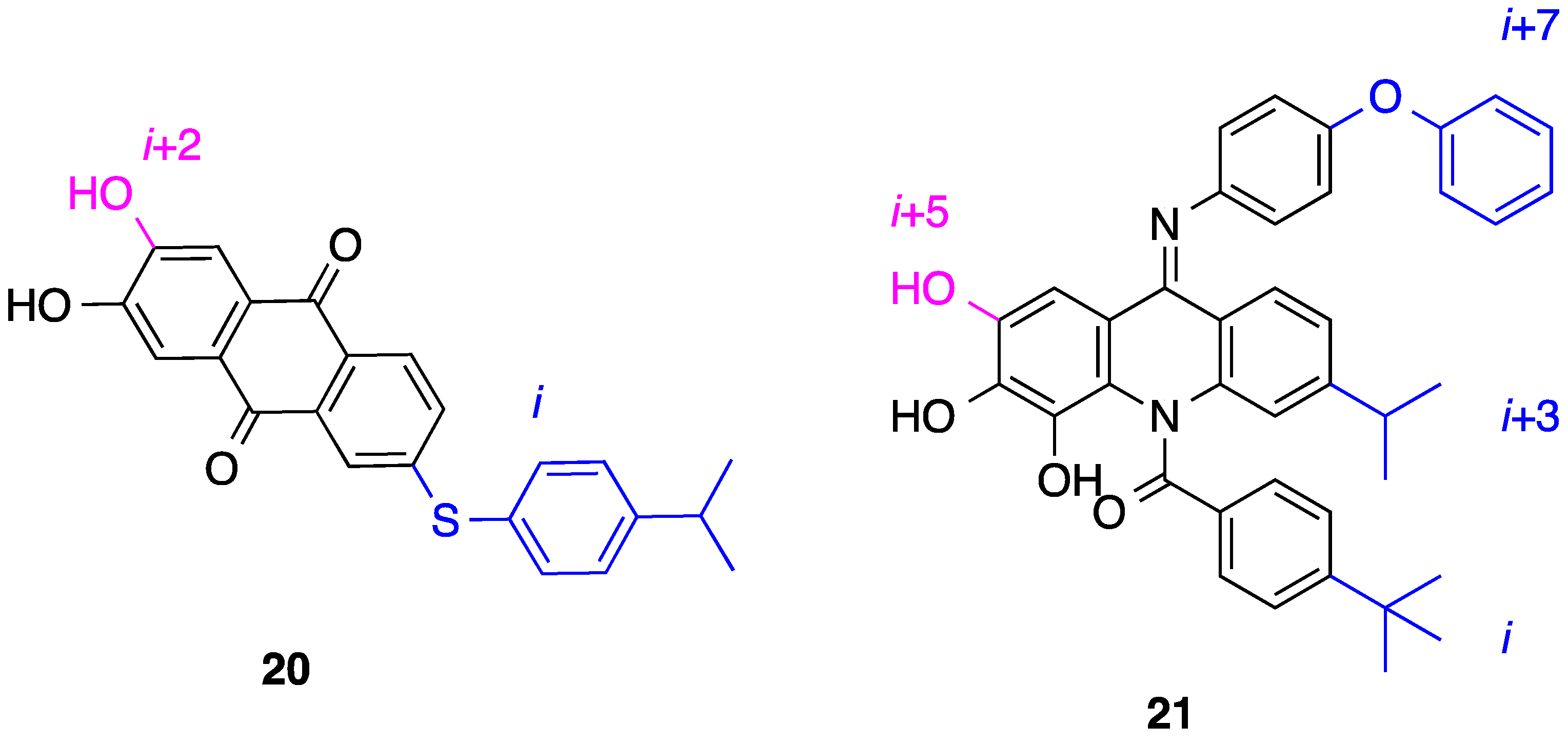
5. Anthraquinones and Acridines
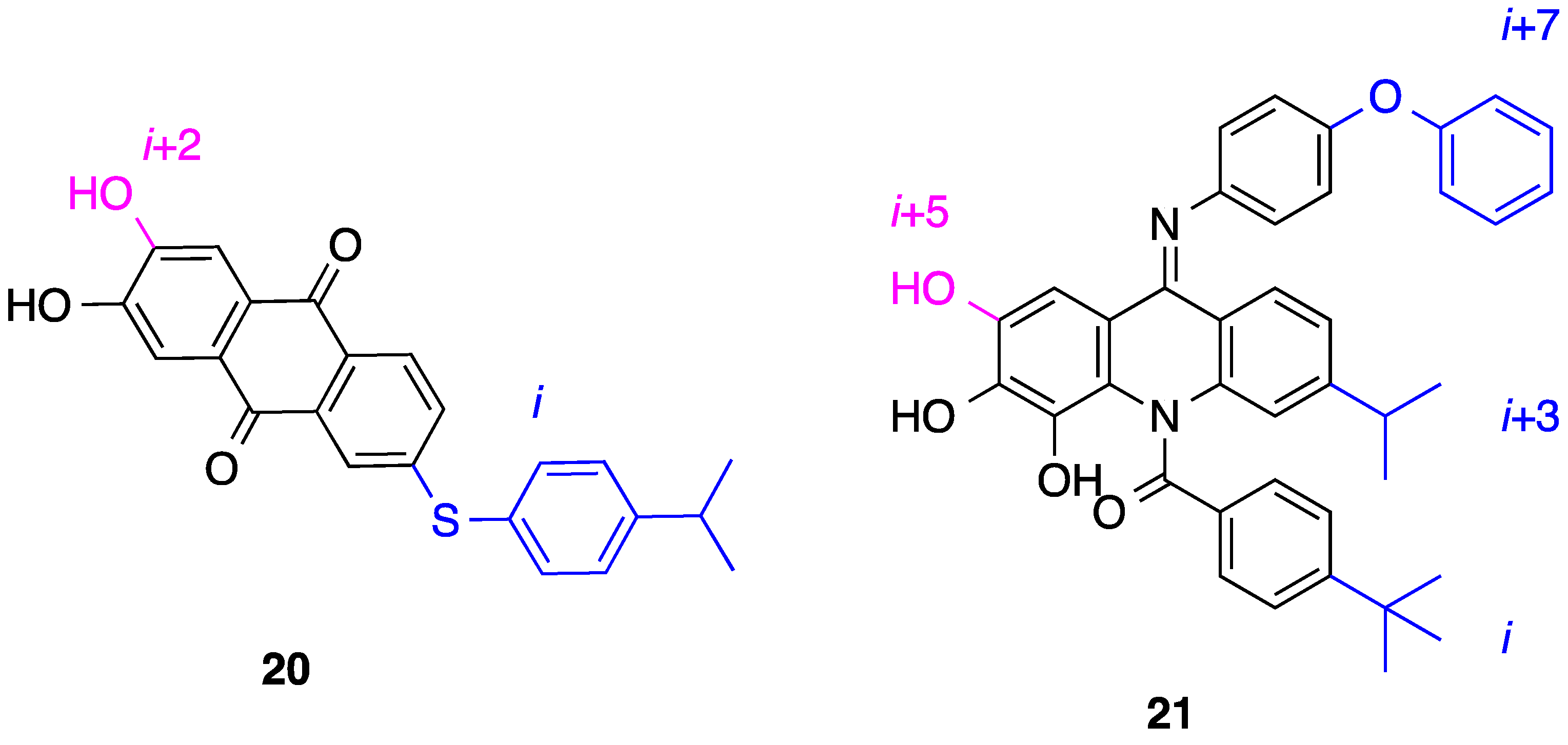
6. 2,6,9-Tri-Substituted Purines
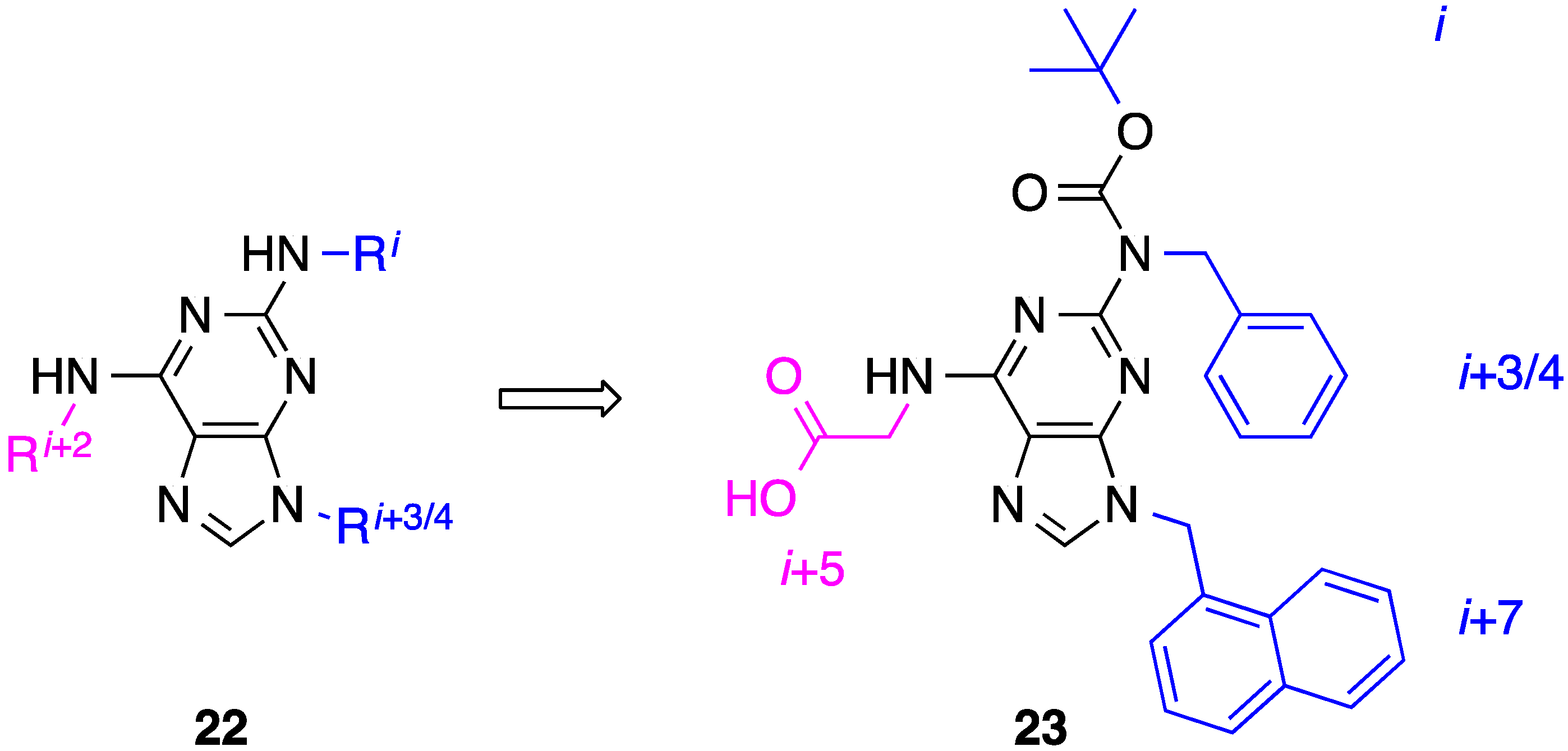
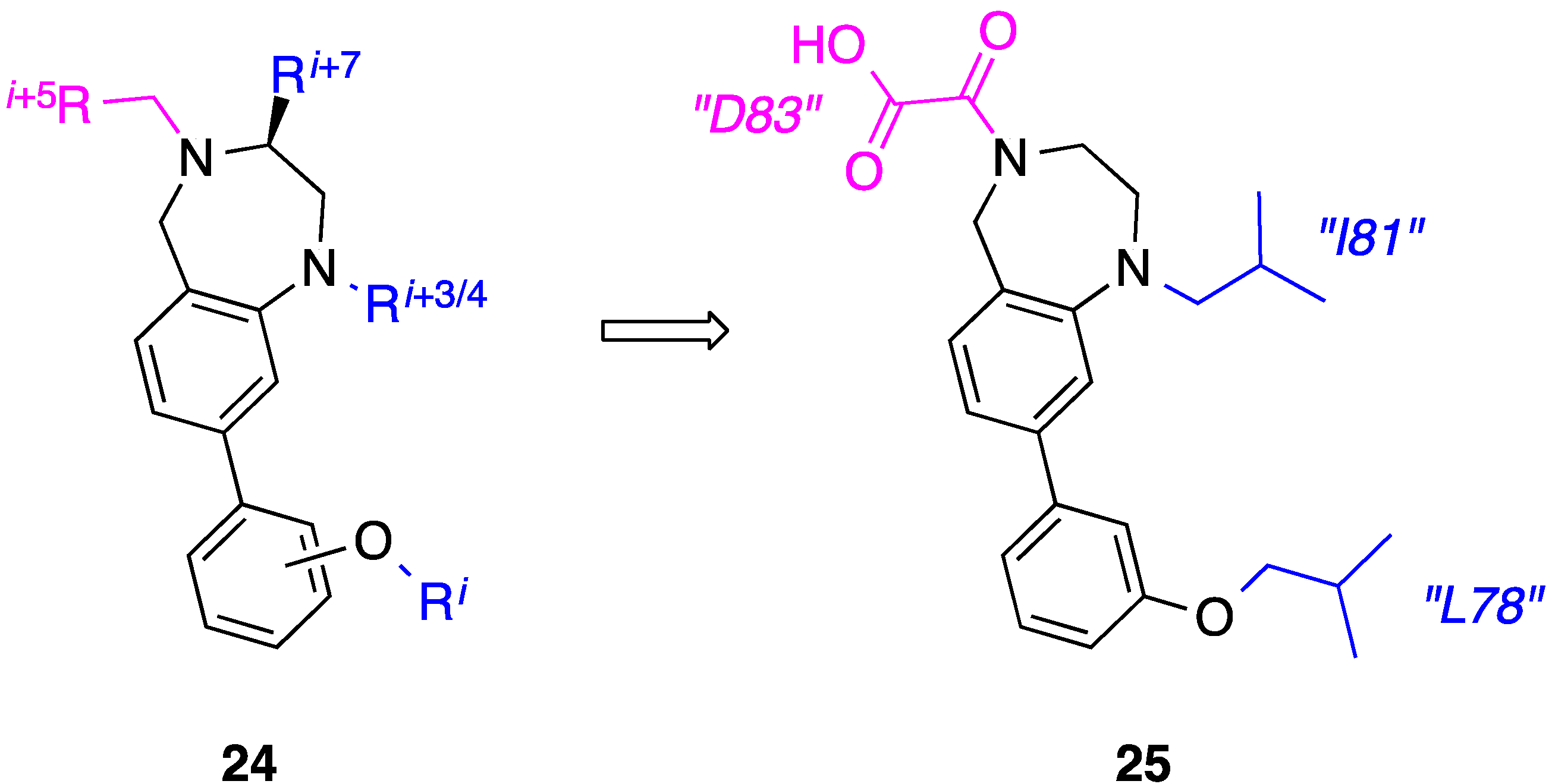
7. Tetrahydro-1H-benzo[e][1,4]diazepines
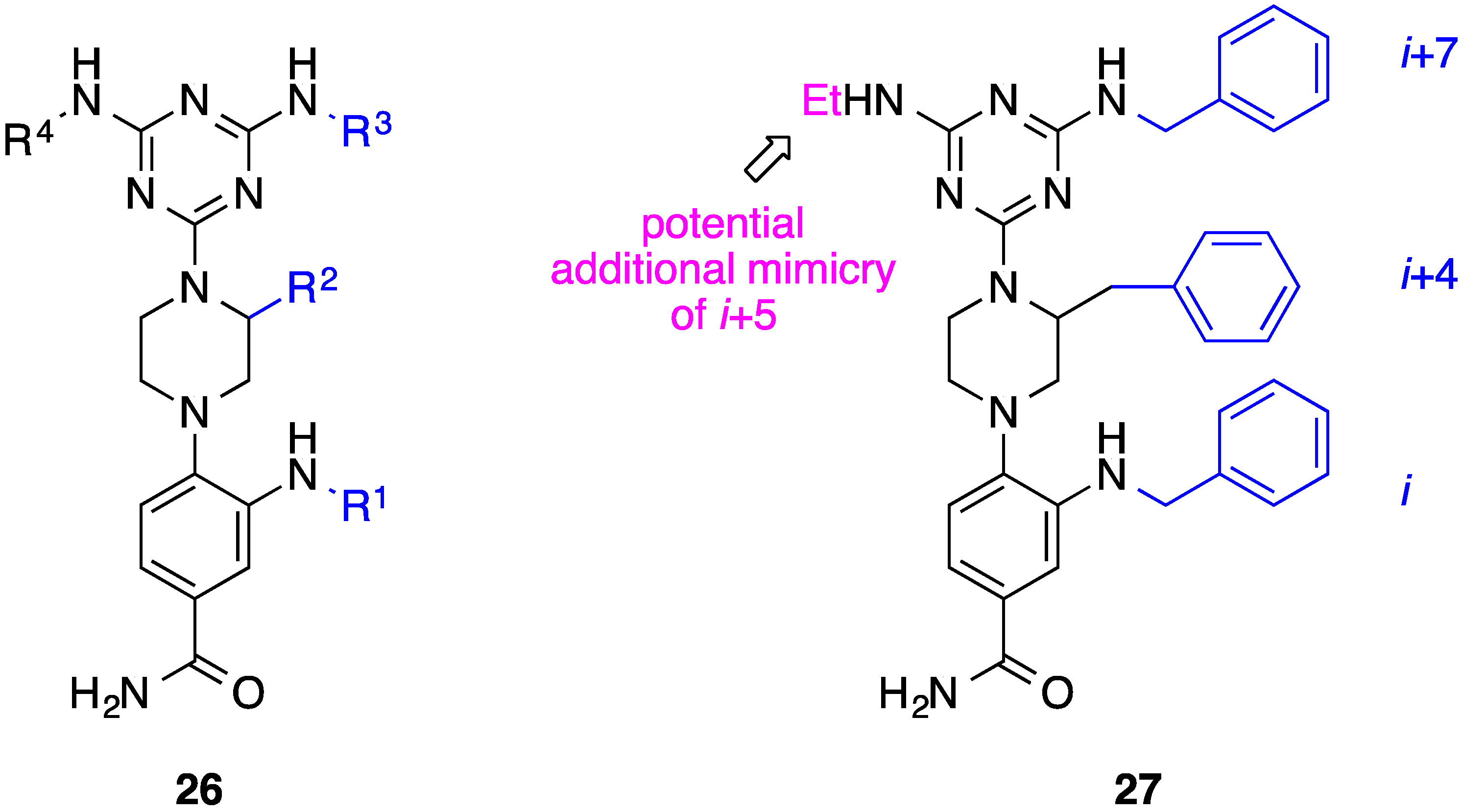
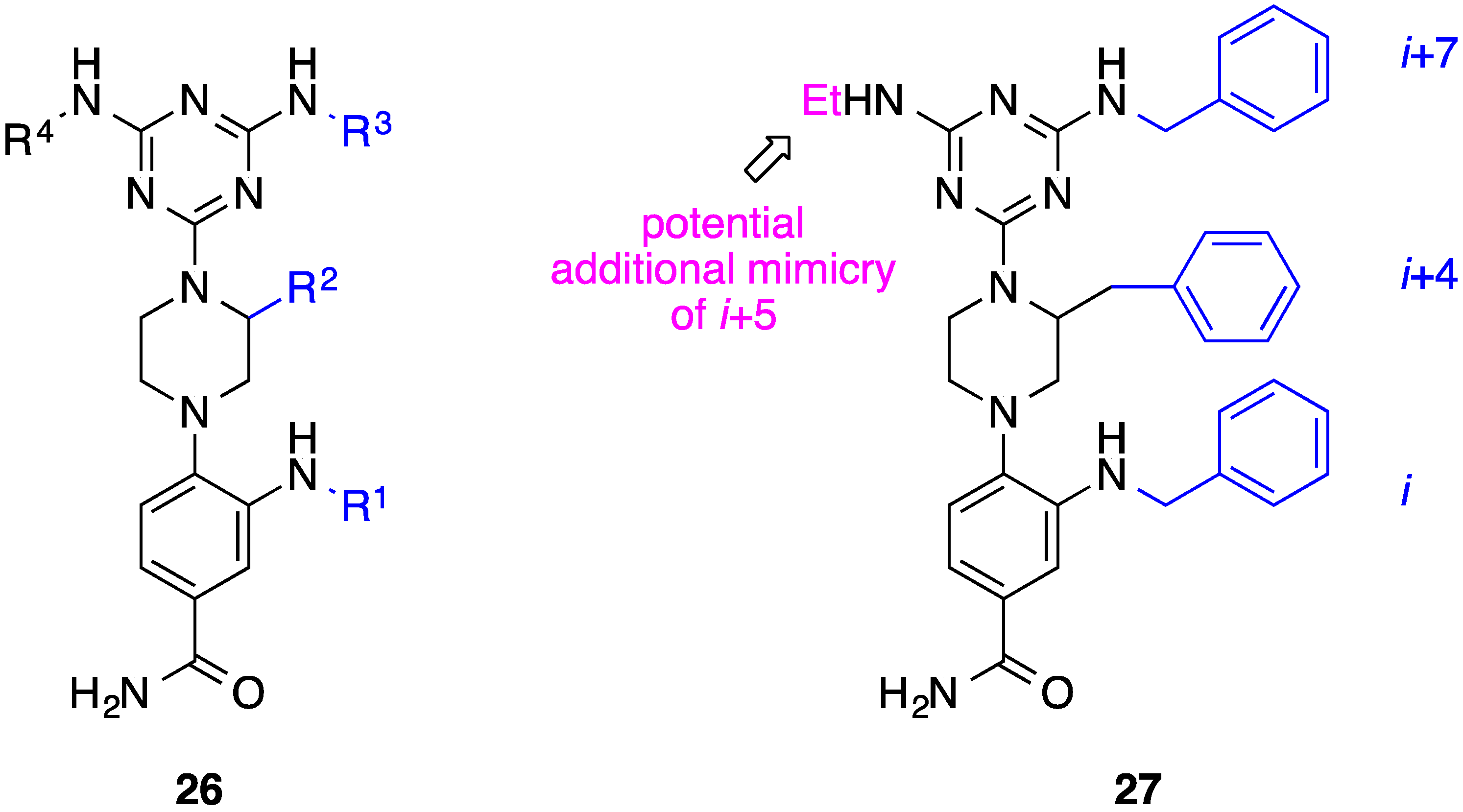
8. Phenyl-Piperazine-Triazine
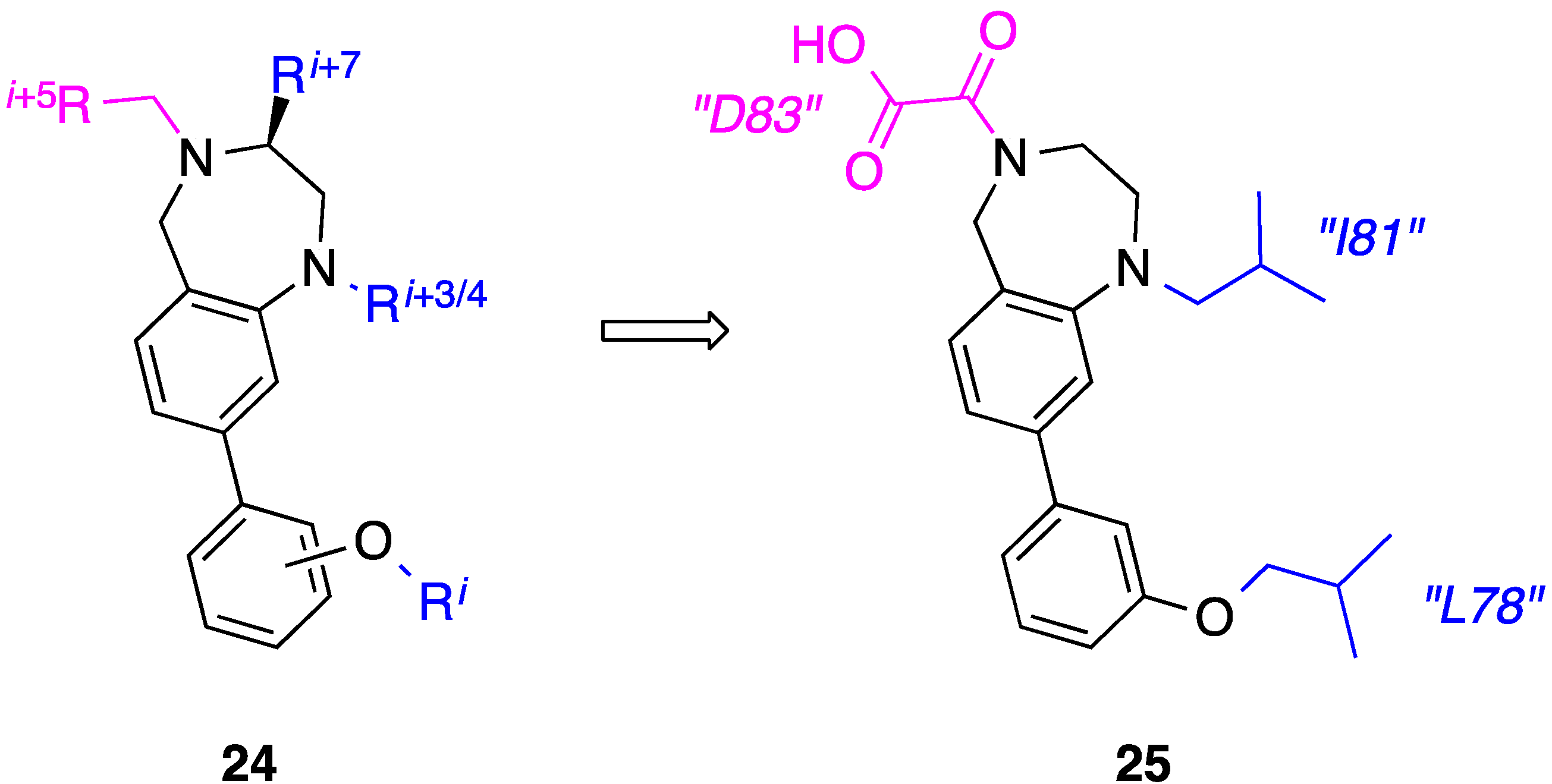
9. Azabicyclo[2.2.2]Octane Aryl Amide
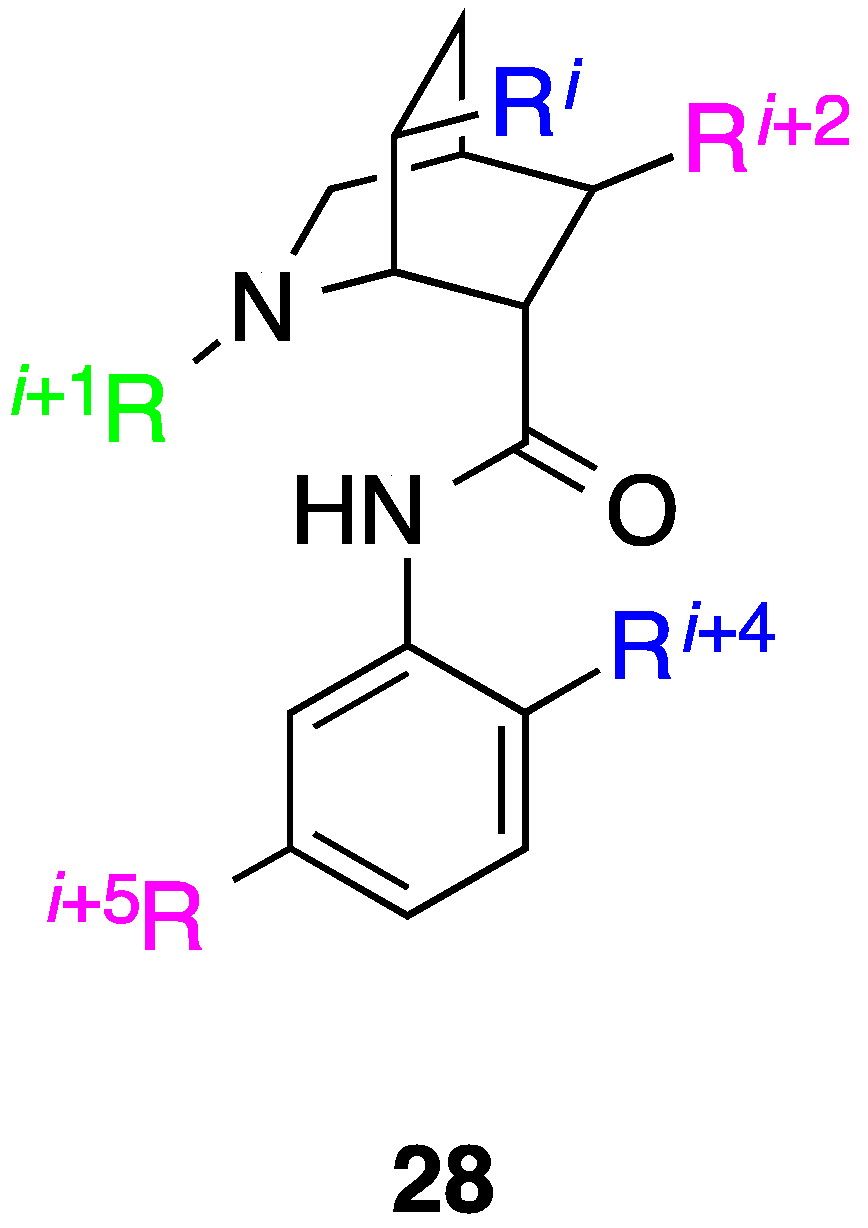
10. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Fletcher, S.; Hamilton, A.D. Protein surface recognition and proteomimetics: Mimics of protein surface structure and function. Curr. Opin. Chem. Biol. 2005, 9, 632–638. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.C.; Gestwicki, J.E. Features of protein-protein interactions that translate into potent inhibitors: Topology, surface area and affinity. Expert Rev. Mol. Med. 2012. [Google Scholar] [CrossRef] [PubMed]
- Nibbe, R.K.; Chowdhury, S.A.; Koyutürk, M.; Ewing, R.; Chance, M.R. Protein-protein interaction networks and subnetworks in the biology of disease. Wiley Interdiscip. Rev. Syst. Biol. Med. 2011, 3, 357–367. [Google Scholar] [CrossRef] [PubMed]
- Lessene, G.; Czabotar, P.E.; Colman, P.M. BCL-2 family antagonists for cancer therapy. Nat. Rev. Drug Discov. 2008, 7, 989–1000. [Google Scholar] [CrossRef] [PubMed]
- Knight, J.D.; Miranker, A.D. Phospholipid catalysis of diabetic amyloid assembly. J. Mol. Biol. 2004, 341, 1175–1187. [Google Scholar] [CrossRef] [PubMed]
- Al-Quobaili, F.; Montenarh, M. Pancreatic duodenal homeobox factor-1 and diabetes mellitus type 2. Int. J. Mol. Med. 2008, 21, 399–404. [Google Scholar] [PubMed]
- Mori, M.; Manetti, F.; Botta, M. Targeting protein-protein and protein-nucleic acid interactions for anti-HIV therapy. Curr. Pharm. Des. 2011, 17, 3713–3728. [Google Scholar] [CrossRef] [PubMed]
- Blazer, L.L.; Neubig, R.R. Small molecule protein-protein interaction inhibitors as CNS therapeutic agents: Current progress and future hurdles. Neuropsychopharmacology 2009, 34, 126–141. [Google Scholar] [CrossRef] [PubMed]
- Arkin, M.R.; Wells, J.A. Small-molecule inhibitors of protein-protein interactions: Progressing towards the dream. Nat. Rev. Drug Discov. 2004, 3, 301–317. [Google Scholar] [CrossRef] [PubMed]
- Arkin, M.R.; Tang, Y.; Wells, J.A. Small-molecule inhibitors of protein-protein interactions: Progressing toward the reality. Chem. Biol. 2014, 21, 1102–1114. [Google Scholar] [CrossRef] [PubMed]
- Bogan, A.A.; Thorn, K.S. Anatomy of hot spots in protein interfaces. J. Mol. Biol. 1998, 280, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Bullock, B.N.; Jochim, A.L.; Arora, P.S. Assessing helical protein interfaces for inhibitor design. J. Am. Chem. Soc. 2011, 133, 14220–14223. [Google Scholar] [CrossRef] [PubMed]
- Cabezas, E.; Satterthwait, A.C. The hydrogen bond mimic approach: Solid-phase synthesis of a peptide stabilized as an α-helix with a hydrazone link. J. Am. Chem. Soc. 1999, 121, 3862–3875. [Google Scholar] [CrossRef]
- Chapman, R.N.; Dimartino, G.; Arora, P.S. A highly stable short α-helix constrained by a main-chain hydrogen-bond surrogate. J. Am. Chem. Soc. 2004, 126, 12252–12253. [Google Scholar] [CrossRef] [PubMed]
- Henchey, L.K.; Porter, J.R.; Ghosh, I.; Arora, P.S. High specificity in protein recognition by hydrogen-bond-surrogate α-helices: Selective inhibition of the p53/MDM2 complex. ChemBioChem 2010, 11, 2104–2107. [Google Scholar] [CrossRef] [PubMed]
- Blackwell, H.E.; Grubbs, R.H. Highly efficient synthesis of covalently cross-linked peptide helices by ring-closing metathesis. Angew. Chem. Int. Ed. Engl. 1998, 37, 3281–3824. [Google Scholar] [CrossRef]
- Walensky, L.D.; Kung, A.L.; Escher, I.; Malia, T.J.; Barbuto, S.; Wright, R.D.; Wagner, G.; Verdine, G.L.; Korsmeyer, S.J. Activation of apoptosis in vivo by a hydrocarbon-stapled BH3 helix. Science 2004, 305, 1466–1470. [Google Scholar] [CrossRef] [PubMed]
- Bouvier, M.; Taylor, J.W. Probing the functional conformation of neuropeptide Y through the design and study of cyclic analogues. J. Med. Chem. 1992, 35, 1145–1155. [Google Scholar] [CrossRef] [PubMed]
- Jackson, D.Y.; King, D.S.; Chmielewski, J.; Singh, S.; Schultz, P.G. General approach to the synthesis of short α-helical peptides. J. Am. Chem. Soc. 2002, 113, 9391–9392. [Google Scholar] [CrossRef]
- Marqusee, S.; Baldwin, R.L. Helix stabilization by Glu-...Lys+ salt bridges in short peptides of de novo design. Proc. Natl. Acad. Sci. USA 1987, 84, 8898–8902. [Google Scholar] [CrossRef] [PubMed]
- Kawamoto, S.A.; Coleska, A.; Ran, X.; Yi, H.; Yang, C.-Y.; Wang, S. Design of triazole-stapled BCL9 α-helical peptides to target the β-catenin/B-cell CLL/lymphoma 9 (BCL9) protein-protein interaction. J. Med. Chem. 2012, 55, 1137–1146. [Google Scholar] [CrossRef] [PubMed]
- Kritzer, J.A.; Lear, J.D.; Hodsdon, M.E.; Schepartz, A. Helical β-peptide inhibitors of the p53-hDM2 interaction. J. Am. Chem. Soc. 2004, 126, 9468–9469. [Google Scholar] [CrossRef] [PubMed]
- Johnson, L.M.; Mortenson, D.E.; Yun, H.G.; Horne, W.S.; Ketas, T.J.; Lu, M.; Moore, J.P.; Gellman, S.H. Enhancement of α-helix mimicry by an α/β-peptide foldamer via incorporation of a dense ionic side-chain array. J. Am. Chem. Soc. 2012, 134, 7317–7320. [Google Scholar] [CrossRef] [PubMed]
- Kazi, A.; Sun, J.; Doi, K.; Sung, S.-S.; Takahashi, Y.; Yin, H.; Rodriguez, J.M.; Becerril, J.; Berndt, N.; Hamilton, A.D.; et al. The BH3 α-helical mimic BH3-M6 disrupts Bcl-XL, Bcl-2, and MCL-1 protein-protein interactions with Bax, Bak, Bad, or Bim and induces apoptosis in a Bax- and Bim-dependent manner. J. Biol. Chem. 2011, 286, 9382–9392. [Google Scholar] [CrossRef] [PubMed]
- Cao, X.; Yap, J.L.; Newell-Rogers, M.K.; Peddaboina, C.; Jiang, W.; Papaconstantinou, H.T.; Jupitor, D.; Rai, A.; Jung, K.-Y.; Tubin, R.P.; et al. The novel BH3 α-helix mimetic JY-1-106 induces apoptosis in a subset of cancer cells (lung cancer, colon cancer and mesothelioma) by disrupting Bcl-xL and Mcl-1 protein-protein interactions with Bak. Mol. Cancer 2013. [Google Scholar] [CrossRef] [PubMed]
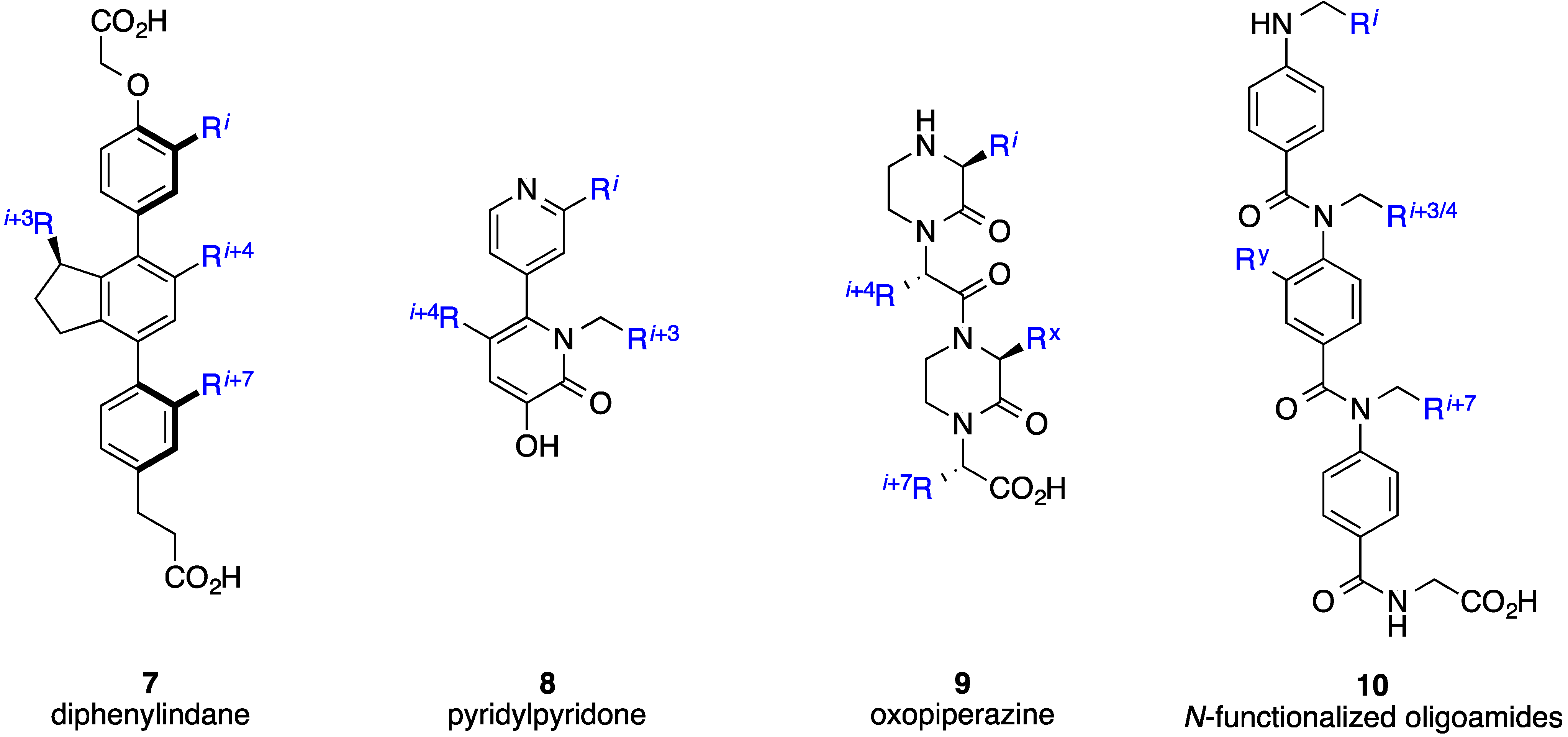
- Horwell, D.C.; Howson, W.; Nolan, W.P.; Ratcliffe, G.S.; Rees, D.C.; Willems, H.M.G. The design of dipeptide helical mimetics, Part I: The synthesis of 1,6-Disubstituted indanes. Tetrahedron 1995, 51, 203–216. [Google Scholar] [CrossRef]
- Horwell, D.C.; Howson, W.; Ratcliffe, G.S.; Willems, H.M.G. The design of dipeptide helical mimetics: The synthesis, tachykinin receptor affinity and conformational analysis of 1,1,6-trisubstituted indanes. Bioorg. Med. Chem. 1996, 4, 33–42. [Google Scholar] [CrossRef]
- Orner, B.P.; Ernst, J.T.; Hamilton, A.D. Toward proteomimetics: Terphenyl derivatives as structural and functional mimics of extended regions of an α-helix. J. Am. Chem. Soc. 2001, 123, 5382–5383. [Google Scholar] [CrossRef] [PubMed]
- Kutzki, O.; Park, H.-S.; Ernst, J.T.; Orner, B.P.; Yin, H.; Hamilton, A.D. Development of a potent Bcl-xL antagonist based on α-helix mimicry. J. Am. Chem. Soc. 2002, 124, 11838–11839. [Google Scholar] [CrossRef] [PubMed]
- Davis, J.M.; Truong, A.; Hamilton, A.D. Synthesis of a 2,3';6',3''-Terpyridine scaffold as an α-helix mimetic. Org. Lett. 2005, 7, 5405–5408. [Google Scholar] [CrossRef] [PubMed]
- Ernst, J.T.; Becerril, J.; Park, H.S.; Yin, H.; Hamilton, A.D. Design and application of an α-helix-mimetic scaffold based on an oligoamide-foldamer strategy: Antagonism of the Bak BH3/Bcl‐xL complex. Angew. Chem. Int. Ed. Engl. 2003, 115, 553–557. [Google Scholar] [CrossRef]
- Yin, H.; Hamilton, A.D. Terephthalamide derivatives as mimetics of the helical region of Bak peptide target Bcl-xL protein. Bioorg. Med. Chem. Lett. 2004, 14, 1375–1379. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.; Lee, G.-I.; Sedey, K.A.; Kutzki, O.; Park, H.-S.; Orner, B.P.; Ernst, J.T.; Wang, H.-G.; Sebti, S.M.; Hamilton, A.D. Terphenyl-based Bak BH3 α-helical proteomimetics as low-molecular-weight antagonists of Bcl-xL. J. Am. Chem. Soc. 2005, 127, 10191–10196. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, J.M.; Ross, N.T.; Katt, W.P.; Dhar, D.; Lee, G.I.; Hamilton, A.D. Structure and function of benzoylurea-derived α-helix mimetics targeting the Bcl‐xL/Bak binding interface. ChemMedChem 2009, 4, 649–656. [Google Scholar] [CrossRef] [PubMed]
- Adler, M.J.; Hamilton, A.D. Oligophenylenaminones as scaffolds for α-helix mimicry. J. Org. Chem. 2011, 76, 7040–7047. [Google Scholar] [CrossRef] [PubMed]
- Adler, M.J.; Scott, R.T.W.; Hamilton, A.D. Enaminone-based mimics of extended and hydrophilic α-helices. Chem. Eur. J. 2012, 18, 12974–12977. [Google Scholar] [CrossRef] [PubMed]
- Davis, J.M.; Tsou, L.K.; Hamilton, A.D. Synthetic non-peptide mimetics of α-helices. Chem. Soc. Rev. 2007, 36, 326–334. [Google Scholar] [CrossRef] [PubMed]
- Cummings, C.G.; Hamilton, A.D. Disrupting protein-protein interactions with non-peptidic, small molecule α-helix mimetics. Curr. Opin. Chem. Biol. 2010, 14, 341–346. [Google Scholar] [CrossRef] [PubMed]
- Azzarito, V.; Long, K.; Murphy, N.S.; Wilson, A.J. Inhibition of α-helix-mediated protein-protein interactions using designed molecules. Nature Chem. 2013, 5, 161–173. [Google Scholar] [CrossRef] [PubMed]
- Lanning, M.; Fletcher, S. Recapitulating the α-helix: Nonpeptidic, low-molecular-weight ligands for the modulation of helix-mediated protein-protein interactions. Future Med. Chem. 2013, 5, 2157–2174. [Google Scholar] [CrossRef] [PubMed]
- Moon, H.; Lim, H.-S. Synthesis and screening of small-molecule α-helix mimetic libraries targeting protein-protein interactions. Curr. Opin. Chem. Biol. 2015, 24, 38–47. [Google Scholar] [CrossRef] [PubMed]
- Pelay-Gimeno, M.; Glas, A.; Koch, O.; Grossmann, T.N. Structure-based design of inhibitors of protein-protein interactions: Mimicking peptide binding epitopes. Angew. Chem. Int. Ed. Engl. 2015, 54, 8896–8927. [Google Scholar] [CrossRef] [PubMed]
- Heindel, D.W.; Bonneau, R.; Arora, P.S. Rational design of topographical helix mimics as potent inhibitors of protein-protein interactions. J. Am. Chem. Soc. 2014, 136, 7877–7888. [Google Scholar]
- Barnard, A.; Long, K.; Yeo, D.J.; Miles, J.A.; Azzarito, V.; Burslem, G.M.; Prabhakaran, P.; Edwards, T.A.; Wilson, A.J. Orthogonal functionalisation of α-helix mimetics. Org. Biomol. Chem. 2014, 12, 6794–6799. [Google Scholar] [CrossRef] [PubMed]
- Becerril, J.; Hamilton, A.D. Helix mimetics as inhibitors of the interaction of the estrogen receptor with coactivator peptides. Angew. Chem. Int. Ed. Engl. 2007, 46, 4471–4473. [Google Scholar] [CrossRef] [PubMed]
- Kim, I.; Hamilton, A.D. Diphenylindane-based proteomimetics reproduce the projection of the i, i + 3, i + 4, and i + 7 residues on an α-helix. Org. Lett. 2006, 8, 1751–1754. [Google Scholar] [CrossRef] [PubMed]
- Jayatunga, M.K.P.; Thompson, S.; Hamilton, A.D. α-Helix mimetics: Outwards and upwards. Bioorg. Med. Chem. Lett. 2014, 24, 717–724. [Google Scholar] [CrossRef] [PubMed]
- Schäfer, G.; Milić, J.; Eldahshan, A.; Götz, F.; Zühlke, K.; Schillinger, C.; Kreuchwig, A.; Elkins, J.M.; Abdul Azeez, K.R.; Oder, A.; et al. Highly functionalized terpyridines as competitive inhibitors of AKAP–PKA interactions. Angew. Chem. Int. Ed. Engl. 2013, 52, 12187–12191. [Google Scholar] [CrossRef] [PubMed]
- Saraogi, I.; Hebda, J.A.; Becerril, J.; Estroff, L.A.; Miranker, A.D.; Hamilton, A.D. Synthetic α-helix mimetics as agonists and antagonists of islet amyloid polypeptide aggregation. Angew. Chem. Int. Ed. Engl. 2010, 122, 748–751. [Google Scholar] [CrossRef]
- Kulikov, O.V.; Kumar, S.; Magzoub, M.; Knipe, P.C.; Saraogi, I.; Thompson, S.; Miranker, A.D.; Hamilton, A.D. Amphiphilic oligoamide α-helix peptidomimetics inhibit islet amyloid polypeptide aggregation. Tetrahedron Lett. 2015, 56, 3670–3673. [Google Scholar] [CrossRef]
- Plante, J.P.; Burnley, T.; Malkova, B.; Webb, M.E.; Warriner, S.L.; Edwards, T.A.; Wilson, A.J. Oligobenzamide proteomimetic inhibitors of the p53–HDM2 protein-protein interaction. Chem. Commun. 2009, 5091–5093. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Aguilar, A.; Bernard, D.; Wang, S. Small-molecule inhibitors of the MDM2-p53 protein-protein interaction (MDM2 inhibitors) in clinical trials for cancer treatment. J. Med. Chem. 2015, 58, 1038–1052. [Google Scholar] [CrossRef] [PubMed]
- Yap, J.L.; Cao, X.; Vanommeslaeghe, K.; Jung, K.-Y.; Peddaboina, C.; Wilder, P.T.; Nan, A.; MacKerell, A.D.; Smythe, W.R.; Fletcher, S. Relaxation of the rigid backbone of an oligoamide-foldamer-based α-helix mimetic: Identification of potent Bcl-xL inhibitors. Org. Biomol. Chem. 2012, 10, 2928–2933. [Google Scholar] [CrossRef] [PubMed]
- Thompson, S.; Vallinayagam, R.; Adler, M.J.; Scott, R.T.W.; Hamilton, A.D. Double-sided α-helix mimetics. Tetrahedron 2012, 68, 4501–4505. [Google Scholar] [CrossRef]
- Marimganti, S.; Cheemala, M.N.; Ahn, J.-M. Novel amphiphilic α-helix mimetics based on a bis-benzamide scaffold. Org. Lett. 2009, 11, 4418–4421. [Google Scholar] [CrossRef] [PubMed]
- Thompson, S.; Hamilton, A.D. Amphiphilic α-helix mimetics based on a benzoylurea scaffold. Org. Biomol. Chem. 2012, 10, 5780–5782. [Google Scholar] [CrossRef] [PubMed]
- Kemp, D.S.; Li, Z.Q. 2-Amino-2′-carboxydiphenylacetylenes as β-turn mimetics. Synthesis and conformational properties. Tetrahedron Lett. 1995, 36, 4175–4178. [Google Scholar] [CrossRef]
- Kemp, D.S.; Li, Z.Q. A short β-sheet containing proline nucleated by a 2,2′-substituted tolan β-turn mimetic. Tetrahedron Lett. 1995, 36, 4179–4180. [Google Scholar] [CrossRef]
- Spivey, A.C.; McKendrick, J.; Srikaran, R.; Helm, B.A. Solid-phase synthesis of an AB loop mimetic of the Cε3 domain of human IgE: Macrocyclization by sonogashira coupling. J. Org. Chem. 2003, 68, 1843–1851. [Google Scholar] [CrossRef] [PubMed]
- Lingard, H.; Han, J.T.; Thompson, A.L.; Leung, I.K.H.; Scott, R.T.W.; Thompson, S.; Hamilton, A.D. Diphenylacetylene-linked peptide strands induce bidirectional β‐sheet formation. Angew. Chem. Int. Ed. Engl. 2014, 126, 3724–3727. [Google Scholar] [CrossRef]
- Jung, K.-Y.; Vanommeslaeghe, K.; Lanning, M.E.; Yap, J.L.; Gordon, C.; Wilder, P.T.; MacKerell, A.D.; Fletcher, S. Amphipathic α-helix mimetics based on a 1,2-diphenylacetylene scaffold. Org. Lett. 2013, 15, 3234–3237. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Li, X.; Song, T.; Zhao, Y.; Feng, Y. An anthraquinone scaffold for putative, two-face Bim BH3 α-helix mimic. J. Med. Chem. 2012, 55, 10735–10741. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wang, Z.; Feng, Y.; Song, T.; Su, P.; Chen, C.; Chai, G.; Yang, Y.; Zhang, Z. Two-face, two-turn α-helix mimetics based on a cross-acridine scaffold: Analogues of the Bim BH3 domain. ChemBioChem 2014, 15, 1280–1285. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Zhang, Q.; Jo, S.; Chai, S.C.; Oh, M.; Im, W.; Lu, H.; Lim, H.-S. Novel pyrrolopyrimidine-based α-helix mimetics: Cell-permeable inhibitors of protein-protein interactions. J. Am. Chem. Soc. 2010, 133, 676–679. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, S.; Shahani, V.M.; Gunning, P.T. Facile and efficient access to 2,6,9-tri-substituted purines through sequential N9, N2 Mitsunobu reactions. Tetrahedron Lett. 2009, 50, 4258–4261. [Google Scholar] [CrossRef]
- Lanning, M.E.; Wilder, P.T.; Drennen, B.; Cavalier, M.; Bailey, H.; Chen, L.; Yap, J.; Raje, M.; Fletcher, S. Towards more drug-like proteomimetics: Two-faced, synthetic α-helix mimetics based on a purine scaffold. Org. Biomol. Chem. 2015, 13, 8642–8646. [Google Scholar] [CrossRef] [PubMed]
- Ripka, W.C.; de Lucca, G.V.; Bach, A.C., II; Pottorf, R.S.; Blaney, J.M. Protein β-turn mimetics I. Design, synthesis, and evaluation in model cyclic peptides. Tetrahedron 1993, 49, 3593–3608. [Google Scholar] [CrossRef]
- Cummings, M.D.; Schubert, C.; Parks, D.J.; Calvo, R.R.; LaFrance, L.V.; Lattanze, J.; Milkiewicz, K.L.; Lu, T. Substituted 1,4‐benzodiazepine-2,5-diones as α-helix mimetic antagonists of the HDM2‐p53 protein-protein interaction. Chem. Biol. Drug Des. 2006, 67, 201–205. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Jung, K.-Y.; Fletcher, S. MEDI-134: Amphipathic α-Helix Mimetics based on a 1,4-Disubstituted 2,3,4,5-Tetrahydro-1H-Benzo[e][1,4]Diazepine: Inhibition of the Mcl-1 Oncoprotein. In Proceedings of the 249th ACS National Meeting & Exposition, Denver, CO, USA, 22–26 March 2015.
- Moon, H.; Lee, W.S.; Oh, M.; Lee, H.; Lee, J.H.; Im, W.; Lim, H.-S. Design, solid-phase synthesis, and evaluation of a phenyl-piperazine-triazine scaffold as α-helix mimetics. ACS Comb. Sci. 2014, 16, 695–701. [Google Scholar] [CrossRef]
- Bayly, A.R.; White, A.J.P.; Spivey, A.C. Design and synthesis of a prototype scaffold for five-residue α-helix mimetics. Eur. J. Org. Chem. 2013, 2013, 5566–5569. [Google Scholar] [CrossRef]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lanning, M.E.; Fletcher, S. Multi-Facial, Non-Peptidic α-Helix Mimetics. Biology 2015, 4, 540-555. https://doi.org/10.3390/biology4030540
Lanning ME, Fletcher S. Multi-Facial, Non-Peptidic α-Helix Mimetics. Biology. 2015; 4(3):540-555. https://doi.org/10.3390/biology4030540
Chicago/Turabian StyleLanning, Maryanna E., and Steven Fletcher. 2015. "Multi-Facial, Non-Peptidic α-Helix Mimetics" Biology 4, no. 3: 540-555. https://doi.org/10.3390/biology4030540
APA StyleLanning, M. E., & Fletcher, S. (2015). Multi-Facial, Non-Peptidic α-Helix Mimetics. Biology, 4(3), 540-555. https://doi.org/10.3390/biology4030540