
Simultaneous Pathoproteomic Evaluation of the Dystrophin-Glycoprotein Complex and Secondary Changes in the mdx-4cv Mouse Model of Duchenne Muscular Dystrophy

Abstract
:1. Introduction
2. Experimental Section
2.1. Chemicals and Materials
2.2. Animal Model of X-Linked Muscular Dystrophy
2.3. Subcellular Fractionation of Muscle Homogenates
2.4. Sample Preparation for Label-Free Liquid Chromatography Mass Spectrometric Analysis
2.5. Label-Free Liquid Chromatography Mass Spectrometric Analysis
2.6. Quantitative Proteomic Profiling by Label-Free LC-MS/MS Analysis
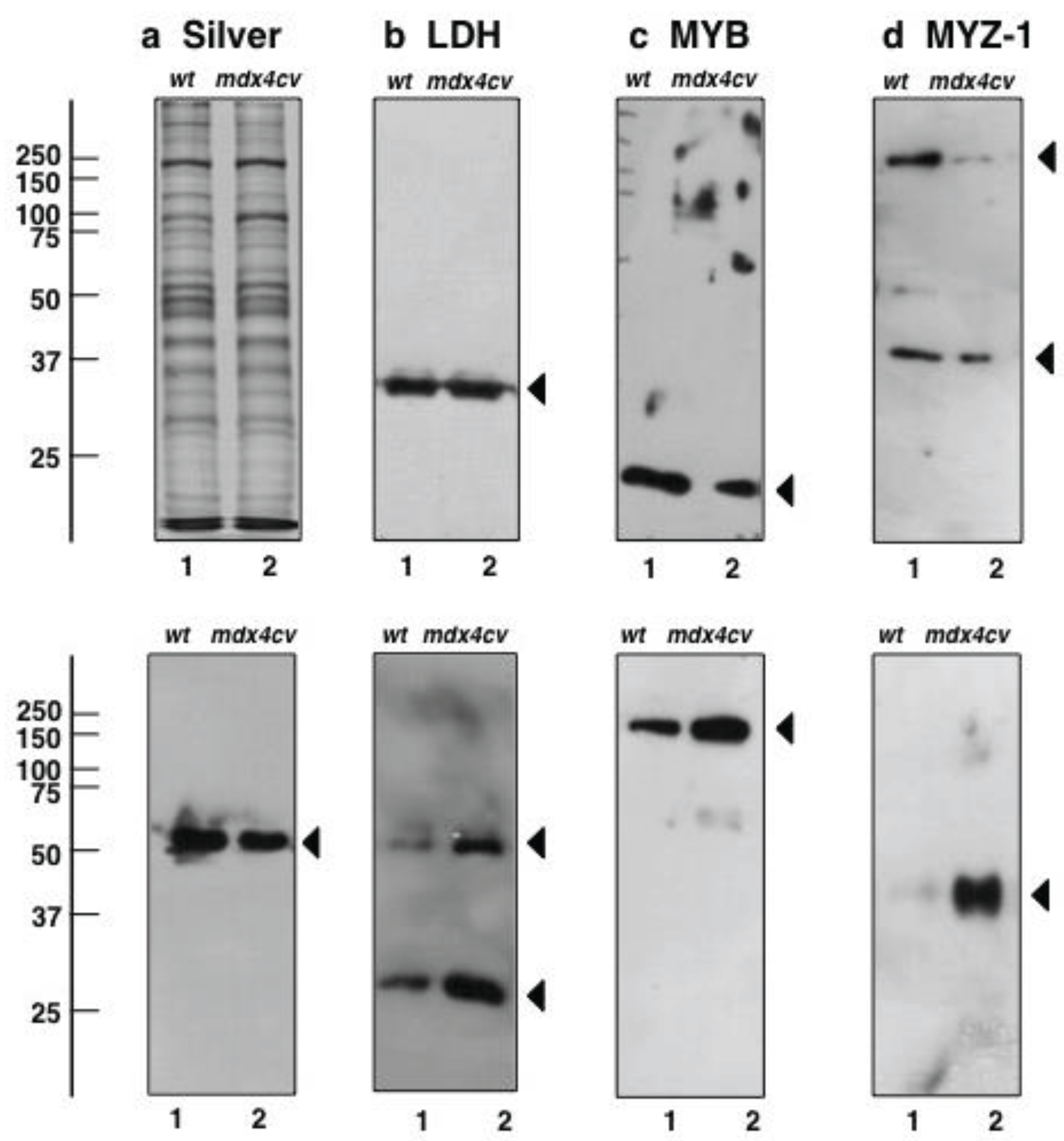
2.7. Verification of Key Proteomic Findings by Comparative Immunoblot Analysis
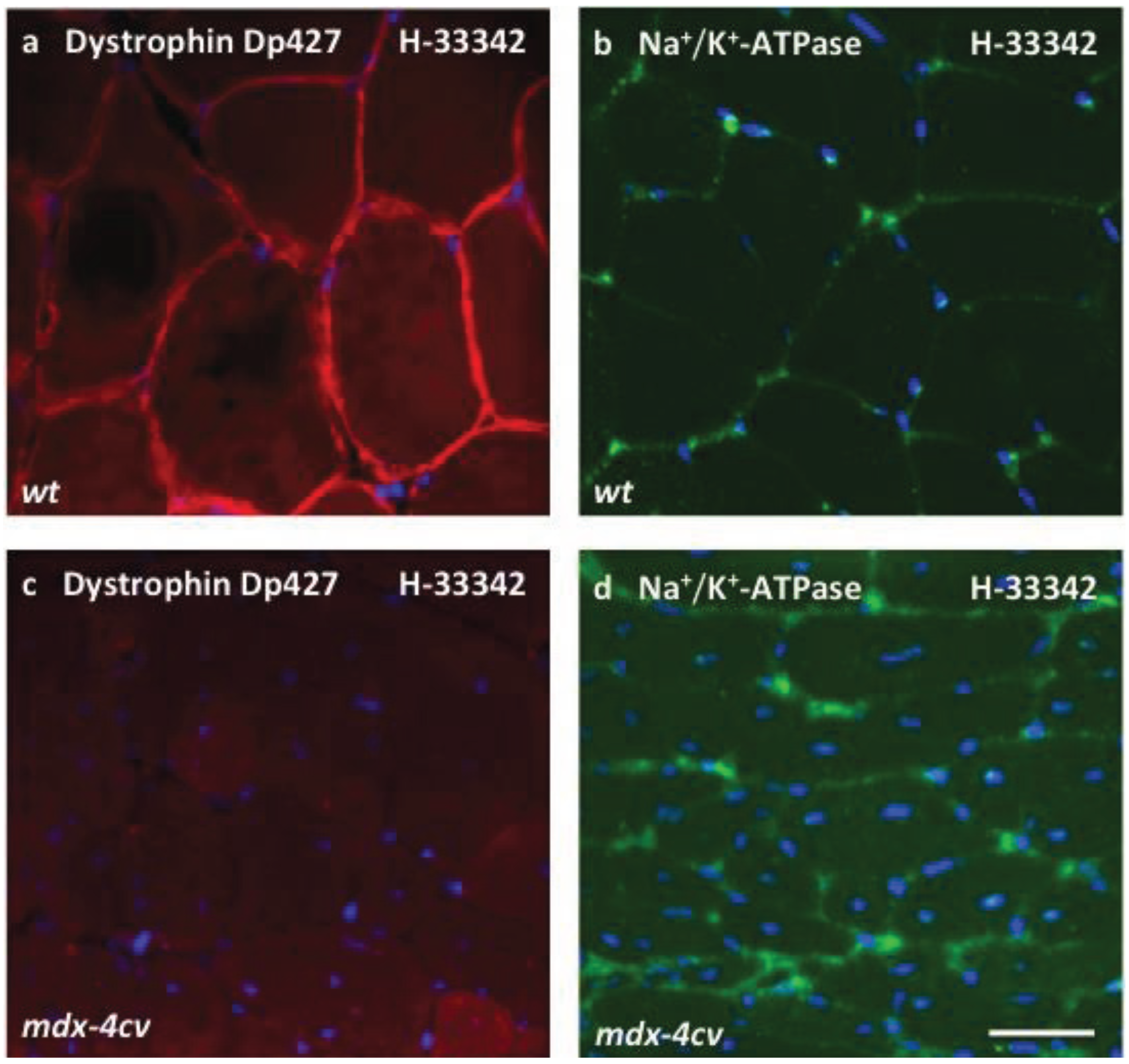
2.8. Immunofluorescence Microscopy
3. Results and Discussion
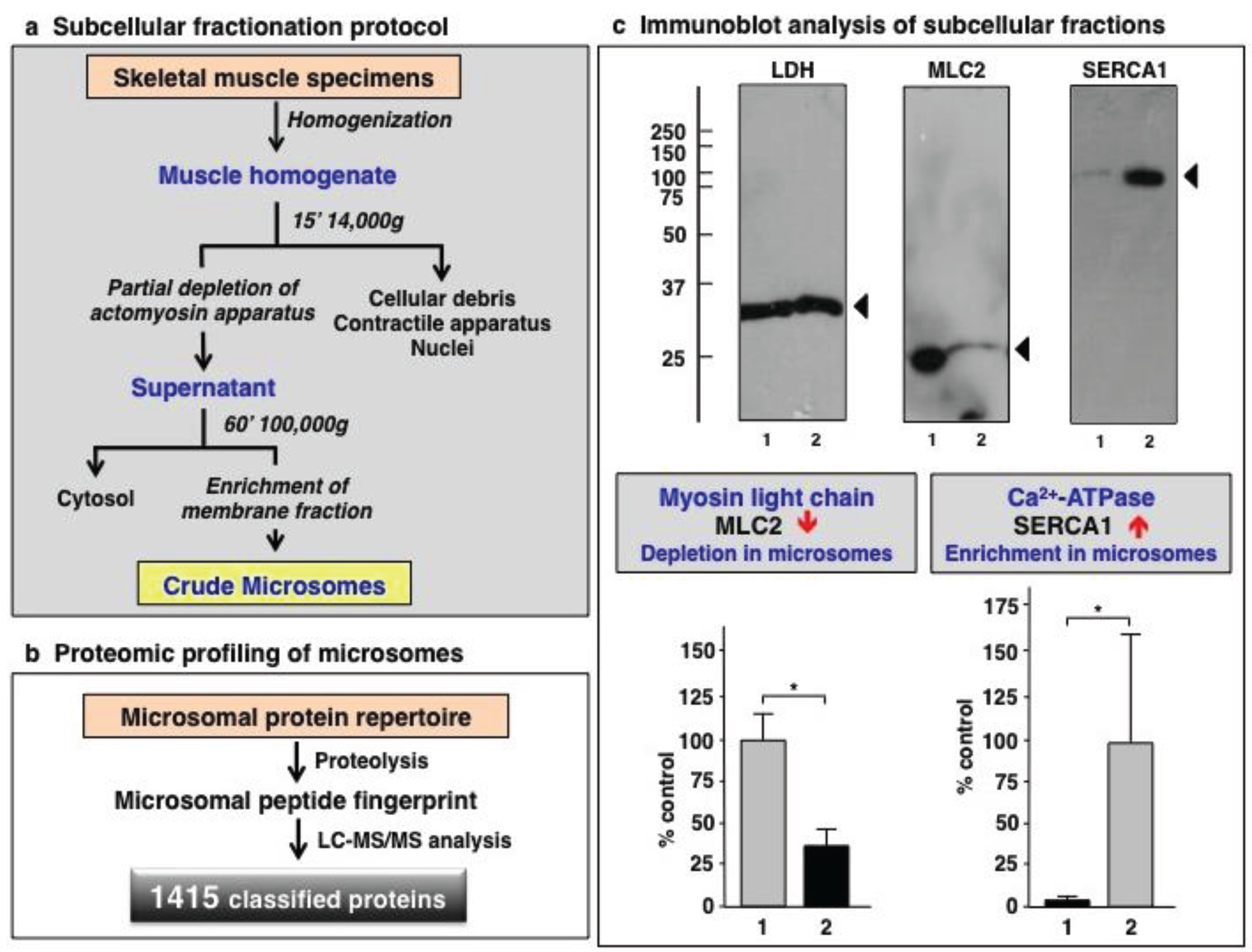
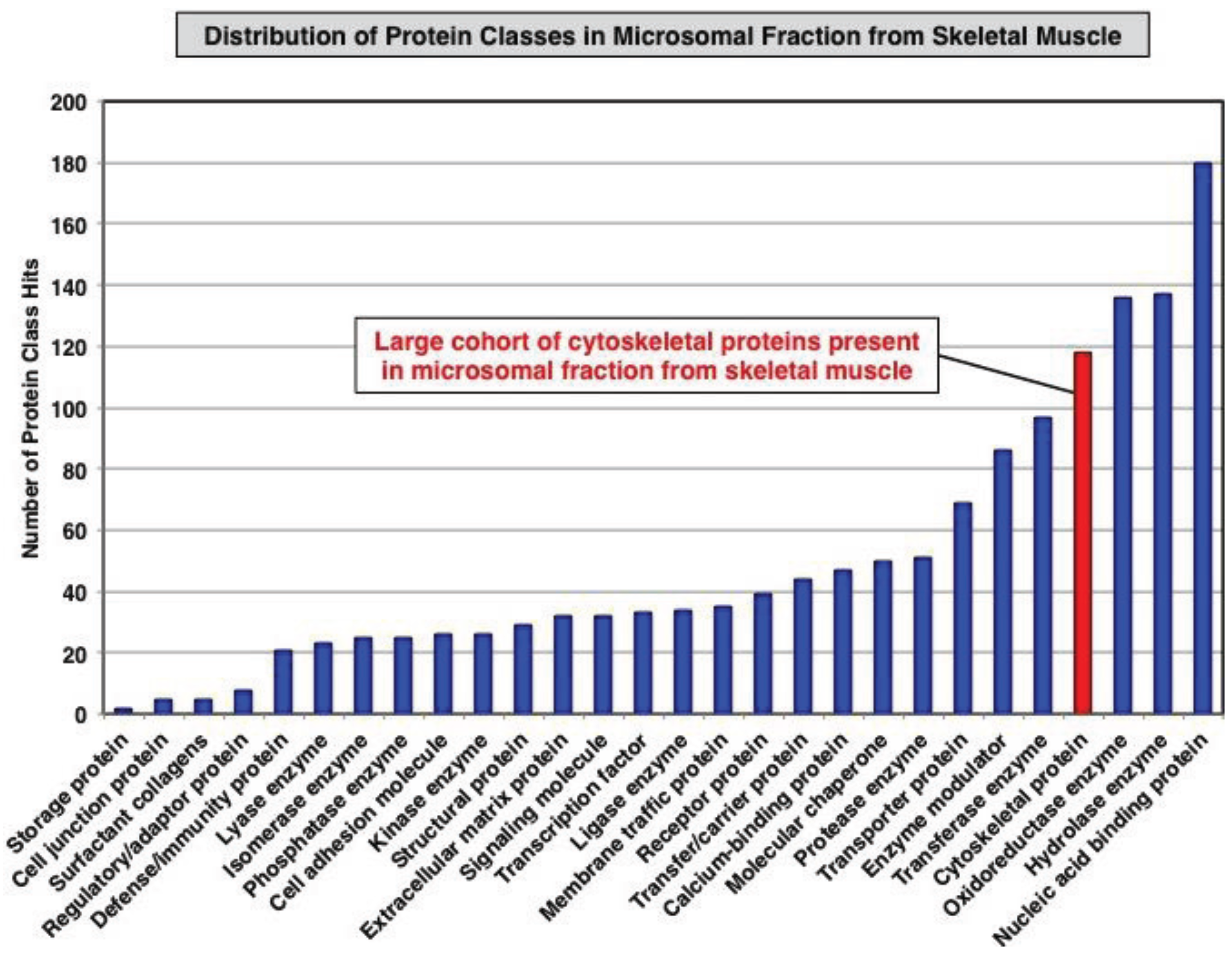
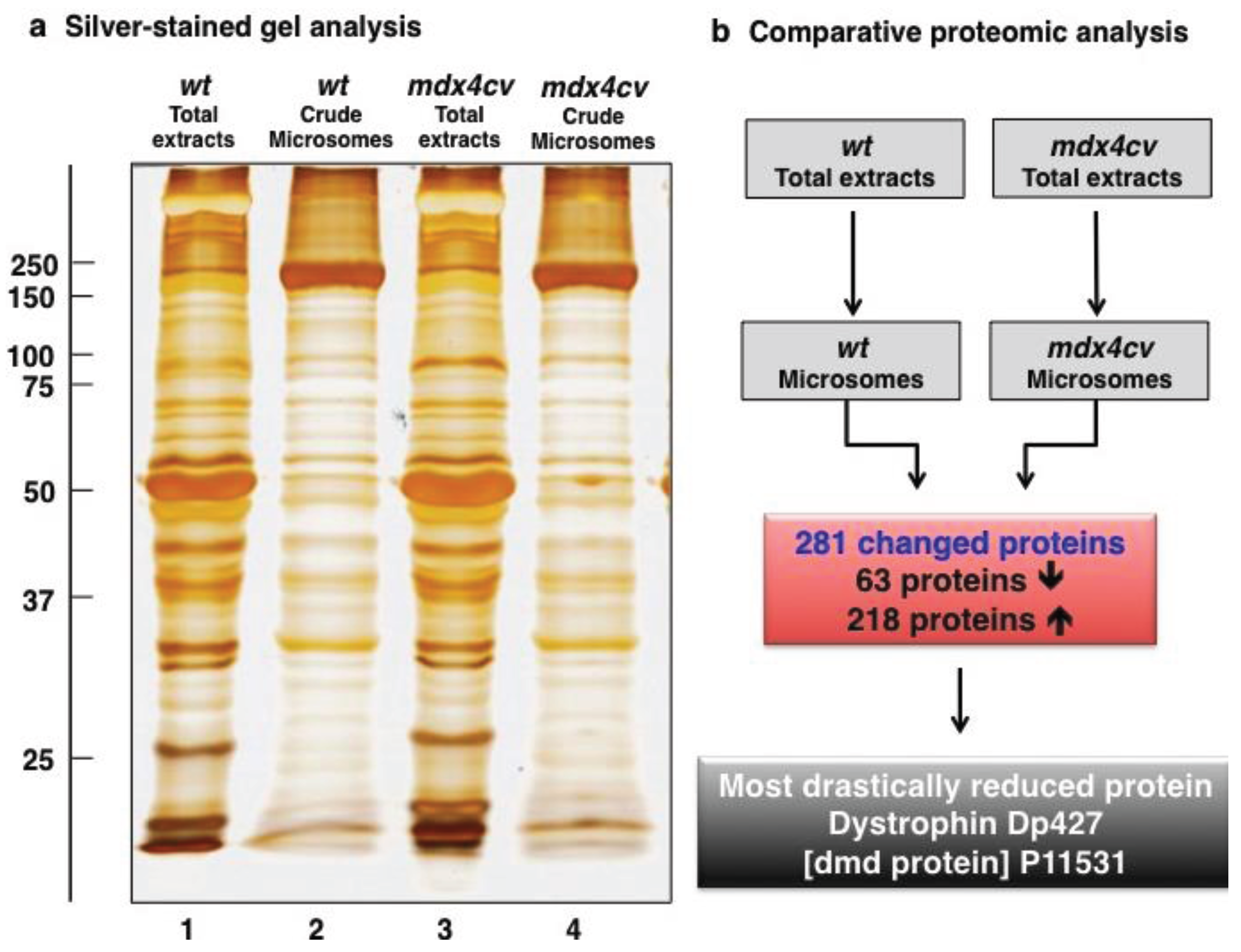
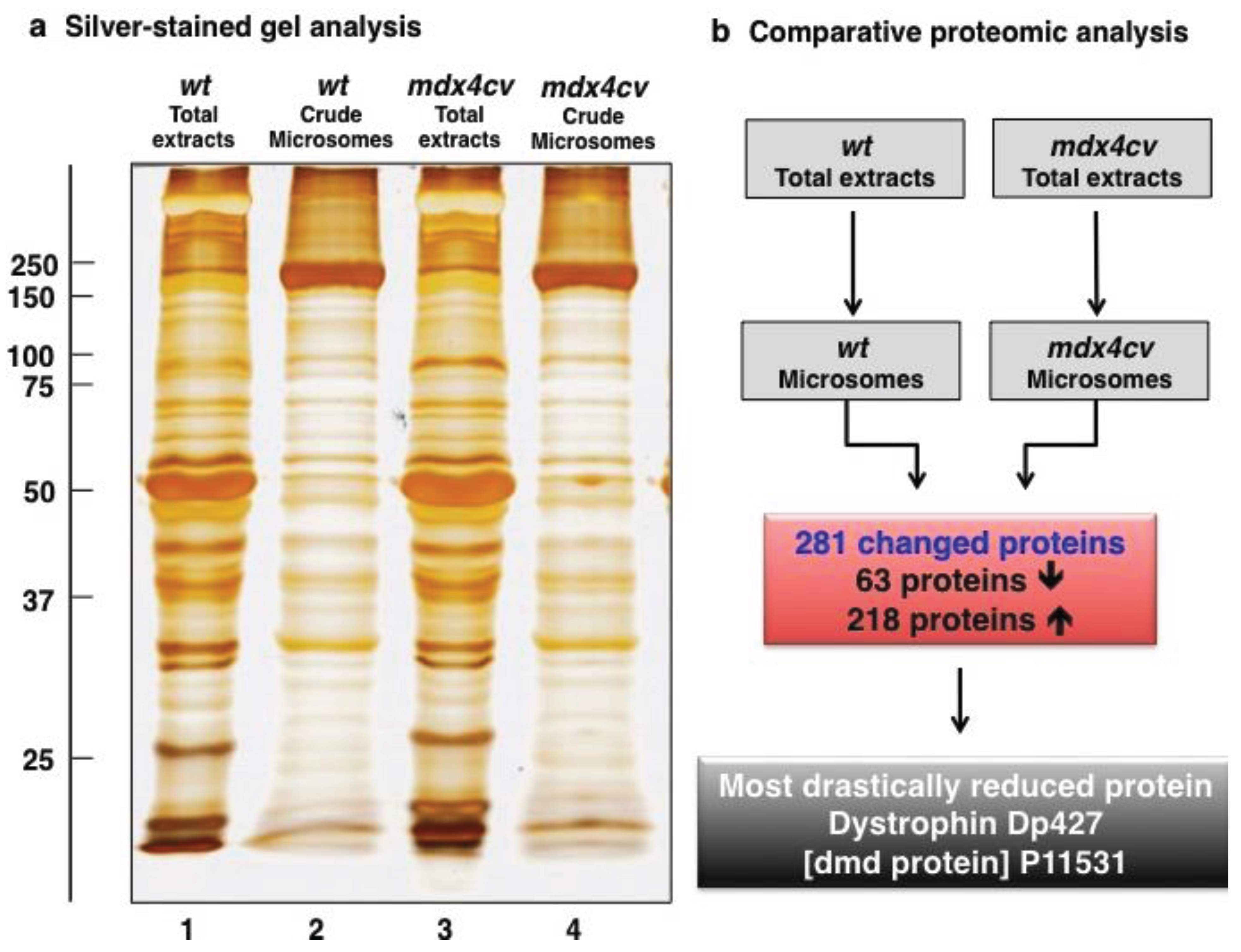
3.1. Organelle Proteomic Analysis of Skeletal Muscle
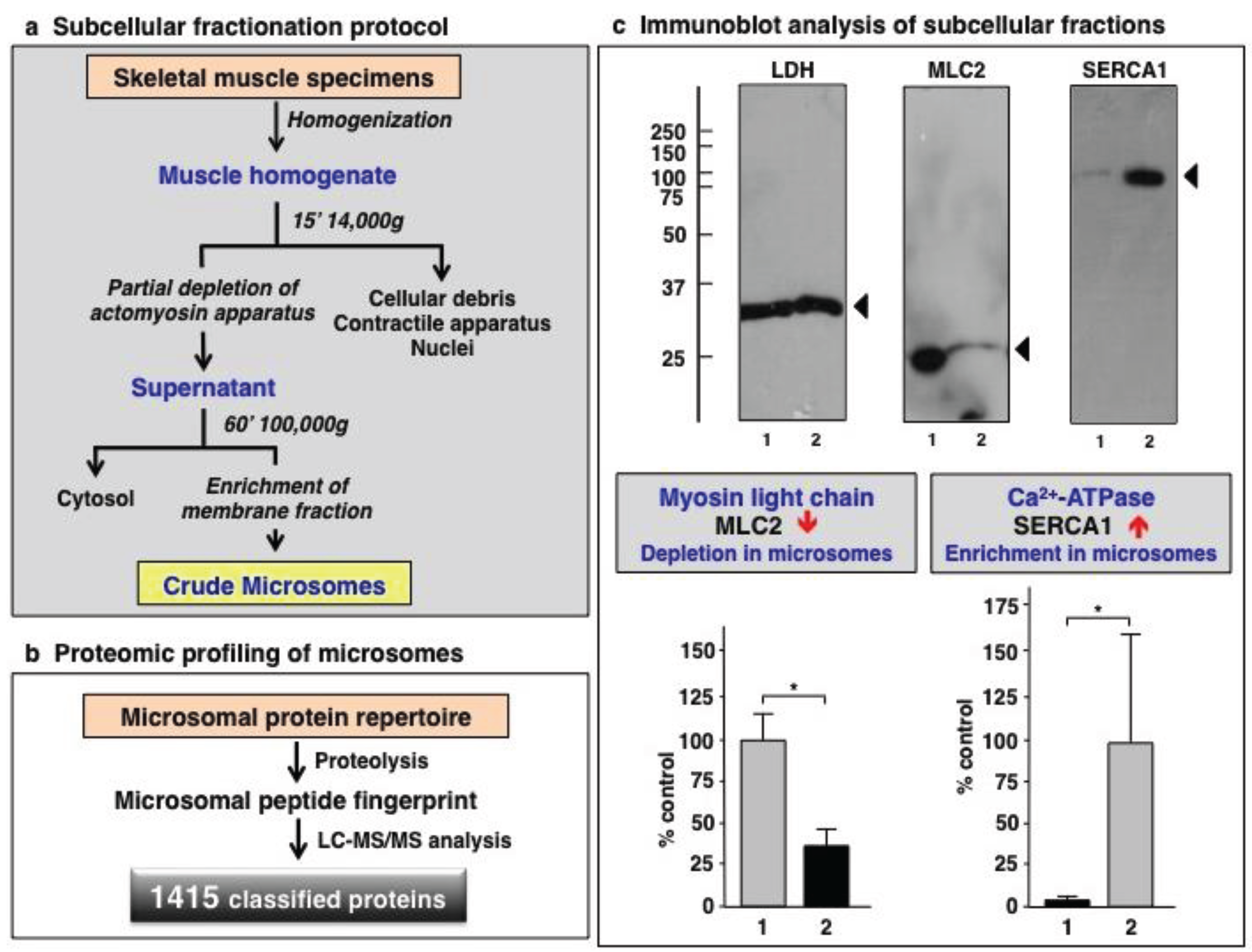
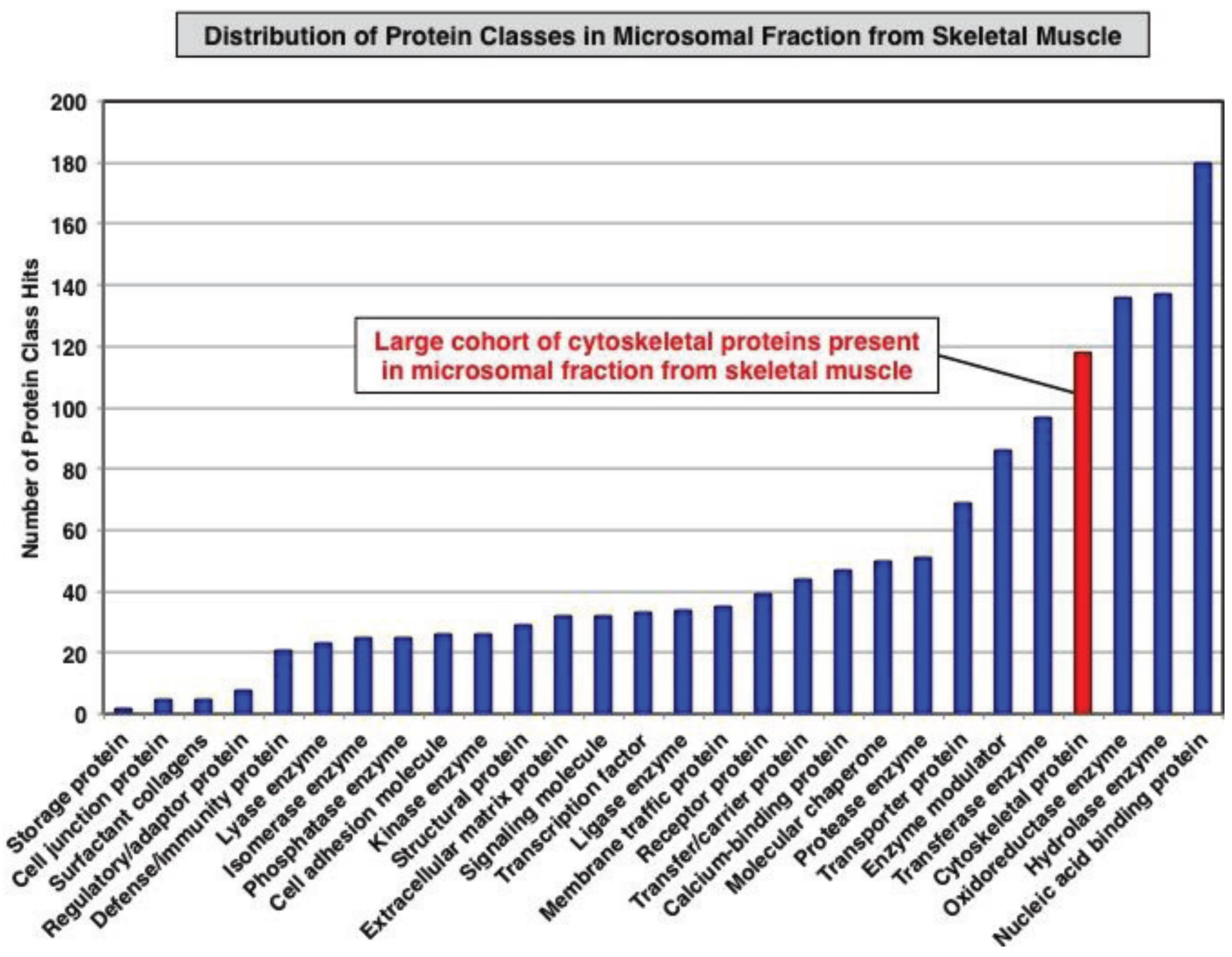
3.2. Label-Free LC-MS/MS Analysis of mdx-4cv versus Wild Type Skeletal Muscle Microsomes

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Accession No. | Protein Name | Peptide Count | Confidence Score | Anova (p) | Fold Change |
|---|---|---|---|---|---|
| P11531 | Dystrophin Dp427 | 15 | 744.3 | 5.95E-05 | –16.01 |
| Q62165 | Dystroglycan | 9 | 396.3 | 4.50E-06 | –9.17 |
| Q6P3A8 | 2-oxoisovalerate dehydrogenase subunit beta, mitochondrial | 2 | 172.9 | 0.007920 | –7.24 |
| P68040 | Guanine nucleotide-binding protein subunit beta-2-like 1 | 2 | 113.4 | 0.007929 | –6.53 |
| P51667 | Myosin regulatory light chain 2, ventricular/cardiac muscle isoform | 3 | 167.4 | 0.037556 | –6.52 |
| P62737 | Actin, aortic smooth muscle | 7 | 407.6 | 0.008389 | –6.19 |
| P50136 | 2-oxoisovalerate dehydrogenase subunit alpha, mitochondrial | 7 | 360.6 | 8.15E-05 | –5.06 |
| Q9Z2I8 | Succinyl-CoA ligase (GDP-forming) subunit beta, mitochondrial | 2 | 65.4 | 0.005705 | –4.98 |
| P09542 | Myosin light chain 3 | 7 | 435.9 | 0.021966 | –4.68 |
| P82348 | Gamma-sarcoglycan | 3 | 139.3 | 0.013191 | –4.08 |
| P53395 | Lipoamide acyltransferase of branched alpha-keto acid dehydrogenase | 12 | 531.9 | 3.36E-05 | –3.97 |
| P68134 | Actin, alpha skeletal muscle | 2 | 67.6 | 0.019843 | –3.85 |
| Q9WUB3 | Glycogen phosphorylase, muscle form | 4 | 58.3 | 0.006224 | –3.85 |
| P70695 | Fructose-1,6-bisphosphatase isozyme 2 | 10 | 604.8 | 0.002383 | –3.83 |
| Q9CPP6 | NADH dehydrogenase [ubiquinone] 1 alpha subcomplex subunit 5 | 2 | 48.3 | 0.027033 | –3.74 |
| Q91YE8 | Synaptopodin-2 | 4 | 368.3 | 0.009175 | –3.70 |
| Q8K370 | Acyl-CoA dehydrogenase family member 10 | 11 | 414.0 | 0.000992 | –3.57 |
| Q9JI91 | Alpha-actinin-2 | 8 | 493.8 | 0.034239 | –3.43 |
| Q9JK37 | Myozenin-1 | 5 | 308.7 | 0.029472 | –3.41 |
| O88990 | Alpha-actinin-3 | 59 | 5419.6 | 0.008821 | –3.37 |
| P16015 | Carbonic anhydrase 3 | 22 | 1821.9 | 2.98E-05 | –3.31 |
| O88492 | Perilipin-4 | 2 | 130.3 | 1.58E-05 | –3.29 |
| Q8BVI4 | Dihydropteridine reductase | 3 | 206.2 | 0.000804 | –3.28 |
| P63323 | 40S ribosomal protein S12 | 2 | 116.9 | 0.027770 | –3.22 |
| Q91ZJ5 | UTP-glucose-1-phosphate uridylyltransferase | 20 | 1265.2 | 2.53E-05 | –3.16 |
| Q7TPR4 | Alpha-actinin-1 | 17 | 1205.8 | 0.011164 | –3.11 |
| Accession No. | Protein Name | Peptide Count | Confidence Score | Anova (p) | Fold Change |
|---|---|---|---|---|---|
| Q00898 | Alpha-1-antitrypsin 1–5 | 6 | 607.1 | 2.05E-05 | 158.43 |
| Q61941 | NAD(P) transhydrogenase, mitochondrial | 22 | 1020.3 | 0.001141 | 62.16 |
| Q8C013 | Trophoblast glycoprotein-like | 2 | 126.07 | 0.010285 | 52.42 |
| Q91WC3 | Long-chain-fatty-acid-CoA ligase 6 | 2 | 67.7 | 0.012257 | 32.60 |
| O70152 | Dolichol-phosphate mannosyltransferase | 2 | 47.5 | 0.000369 | 20.15 |
| Q61233 | Plastin-2 | 3 | 145.4 | 0.000393 | 18.30 |
| P41317 | Mannose-binding protein C | 6 | 527.3 | 1.80E-06 | 16.38 |
| Q62351 | Transferrin receptor protein 1 | 6 | 264.2 | 0.003053 | 16.02 |
| P16110 | Galectin-3 | 6 | 553.4 | 1.05E-06 | 15.93 |
| Q9D154 | Leukocyte elastase inhibitor A | 8 | 446.3 | 1.45E-06 | 15.48 |
| P09541 | Myosin light chain 4 | 2 | 121.1 | 0.000221 | 13.67 |
| Q61878 | Bone marrow proteoglycan | 2 | 80.3 | 0.000859 | 13.43 |
| Q69ZN7 | Myoferlin | 2 | 91.1 | 0.000864 | 12.72 |
| P14434 | H-2 class II histocompatibility antigen, A–B alpha chain | 3 | 109.3 | 0.000268 | 11.95 |
| Q924X2 | Carnitine O-palmitoyltransferase 1, muscle isoform | 2 | 66.4 | 0.032150 | 10.69 |
| Q62009 | Periostin | 4 | 156.5 | 0.000967 | 9.92 |
| Q8VCM7 | Fibrinogen gamma chain | 3 | 172.5 | 5.79E-05 | 9.15 |
| Q9DBS1 | Transmembrane protein 43 | 7 | 428.6 | 0.002721 | 8.61 |
| Q9CQW9 | Interferon-induced transmembrane protein 3 | 2 | 55.2 | 0.002680 | 8.44 |
| Q6PD26 | GPI transamidase component PIG-S | 4 | 158.2 | 0.013246 | 8.15 |
| Q9DC16 | Endoplasmic reticulum-Golgi intermediate compartment protein 1 E | 2 | 134.5 | 0.016933 | 7.76 |
| P28665 | Murinoglobulin-1 | 11 | 522.1 | 0.000139 | 7.51 |
| P05555 | Integrin alpha-M | 6 | 230.6 | 0.000560 | 7.39 |
| P09470 | Angiotensin-converting enzyme | 10 | 450.2 | 0.001196 | 7.28 |
| P14483 | H-2 class II histocompatibility antigen, A beta chain | 2 | 9.3 | 0.001178 | 7.13 |
| Q68FD5 | Clathrin heavy chain 1 | 4 | 158.9 | 0.012682 | 7.01 |
| Q9R069 | Basal cell adhesion molecule | 4 | 131.2 | 0.000134 | 6.98 |
| Q06890 | Clusterin | 6 | 271.7 | 0.003051 | 6.97 |
| P61620 | Protein transport protein Sec61 subunit alpha isoform 1 | 2 | 115.9 | 0.032657 | 6.80 |
| P01898 | H-2 class I histocompatibility antigen, Q10 alpha chain | 2 | 45.0 | 0.000994 | 6.47 |
| Q6IRU2 | Tropomyosin alpha-4 chain | 2 | 59.5 | 0.001981 | 6.39 |
| Q8BMD8 | Calcium-binding mitochondrial carrier protein SCaMC-1 | 2 | 94.8 | 0.018072 | 6.30 |
| Q8K0E8 | Fibrinogen beta chain | 4 | 221.8 | 9.94E-05 | 6.28 |
| Q8BMK4 | Cytoskeleton-associated protein 4 | 8 | 529.1 | 0.002108 | 6.27 |
| Q9WUQ2 | Prolactin regulatory element-binding protein | 2 | 45.5 | 0.024108 | 6.23 |
| P11835 | Integrin beta-2 | 4 | 327.7 | 0.002931 | 6.22 |
| Q9QZF2 | Glypican-1 | 6 | 405.6 | 0.001895 | 5.96 |
| Q8C129 | Leucyl-cystinyl aminopeptidase | 5 | 240.9 | 0.009879 | 5.95 |
| P49290 | Eosinophil peroxidase | 11 | 581.4 | 0.010892 | 5.67 |
| Q9D7J6 | Deoxyribonuclease-1-like 1 | 2 | 145.7 | 0.029576 | 5.65 |
| Q9ERD7 | Tubulin beta-3 chain | 2 | 96.3 | 0.002934 | 5.57 |
| P14426 | H-2 class I histocompatibility antigen, D–K alpha chain | 2 | 156.2 | 0.002441 | 5.47 |
| P57716 | Nicastrin | 2 | 114.6 | 0.006051 | 5.43 |
| Q61704 | Inter-alpha-trypsin inhibitor heavy chain H3 | 2 | 123.4 | 0.011644 | 5.37 |
| Q60854 | Serpin B6 | 11 | 601.5 | 1.68E-06 | 5.07 |
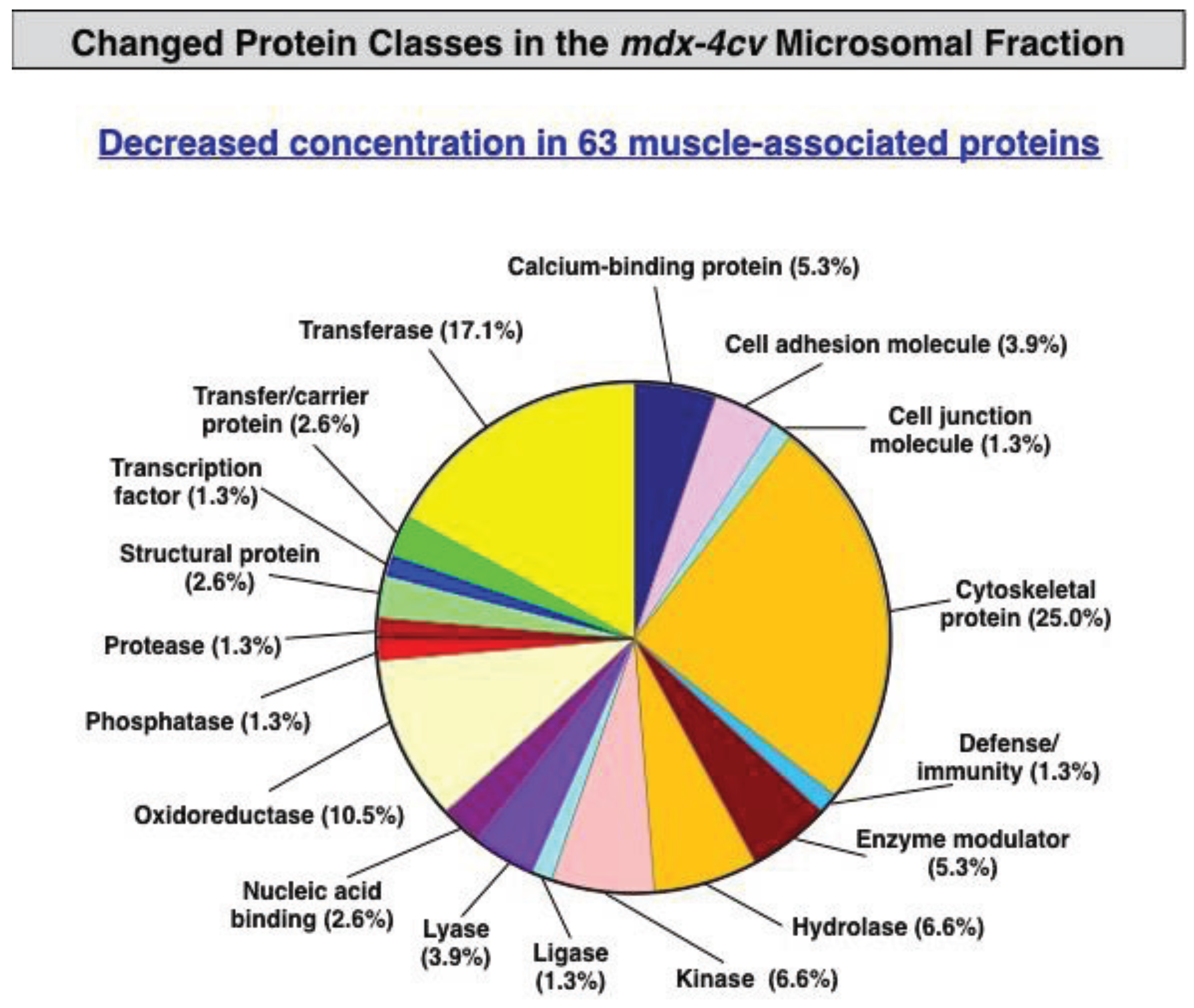
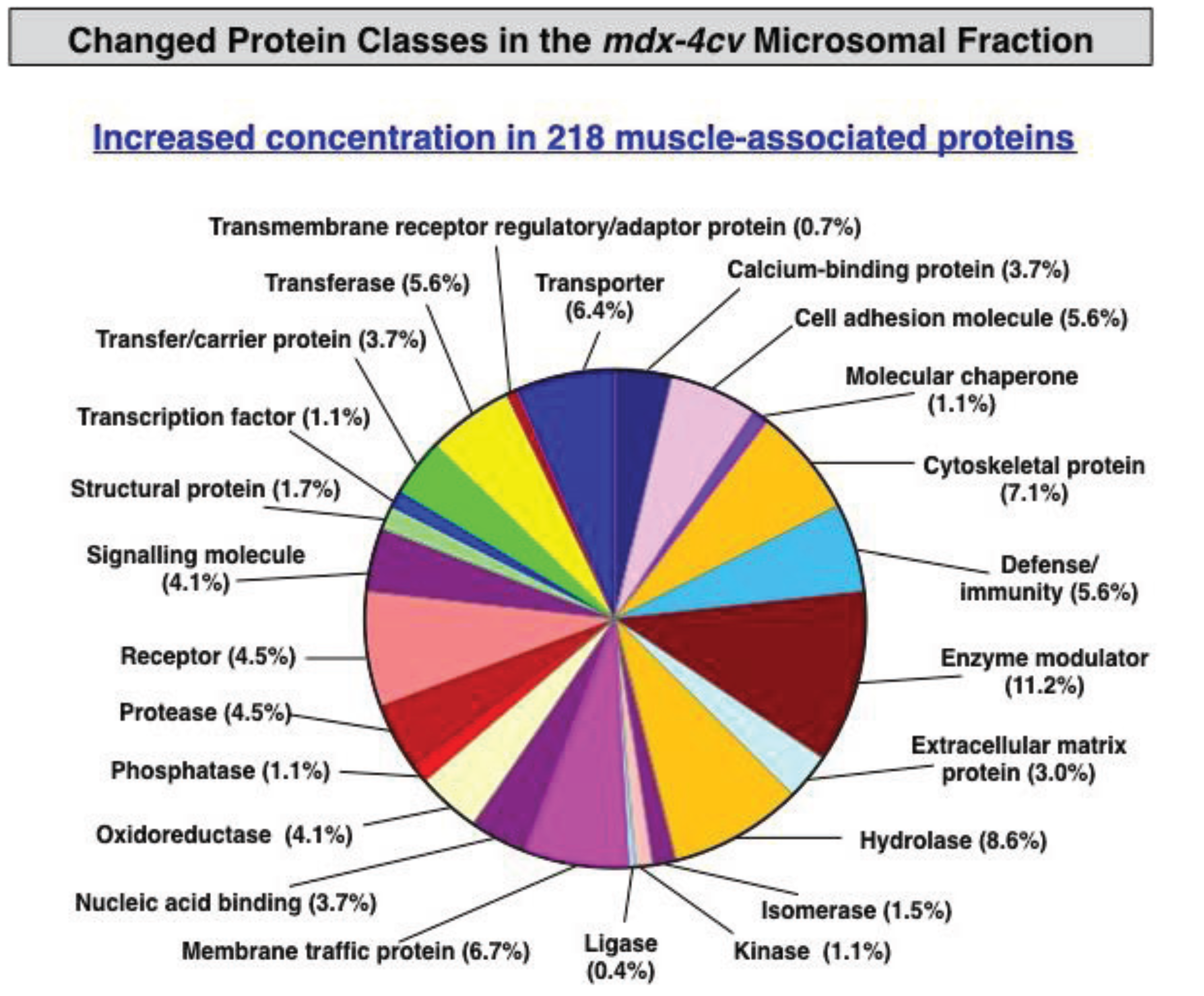
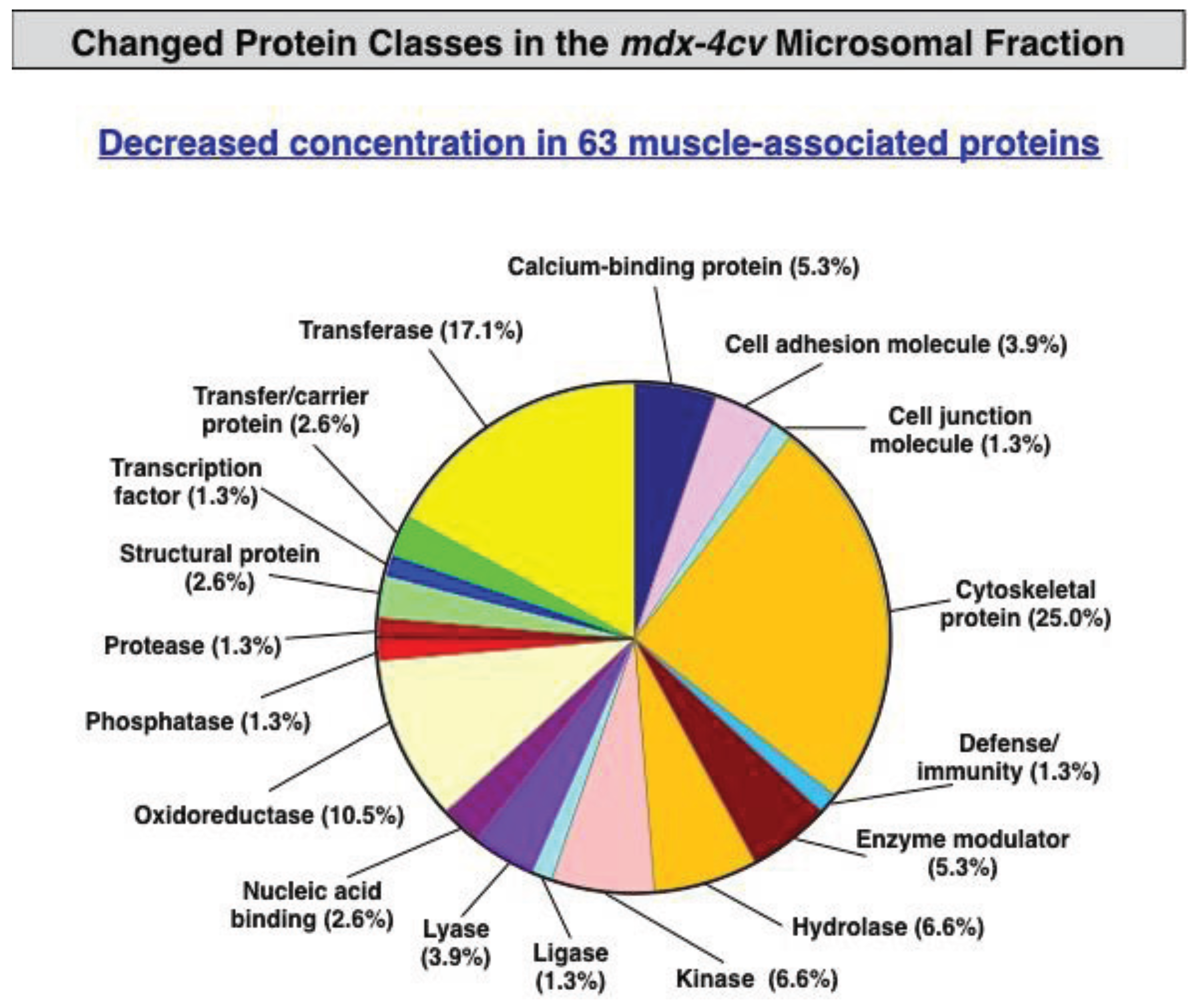
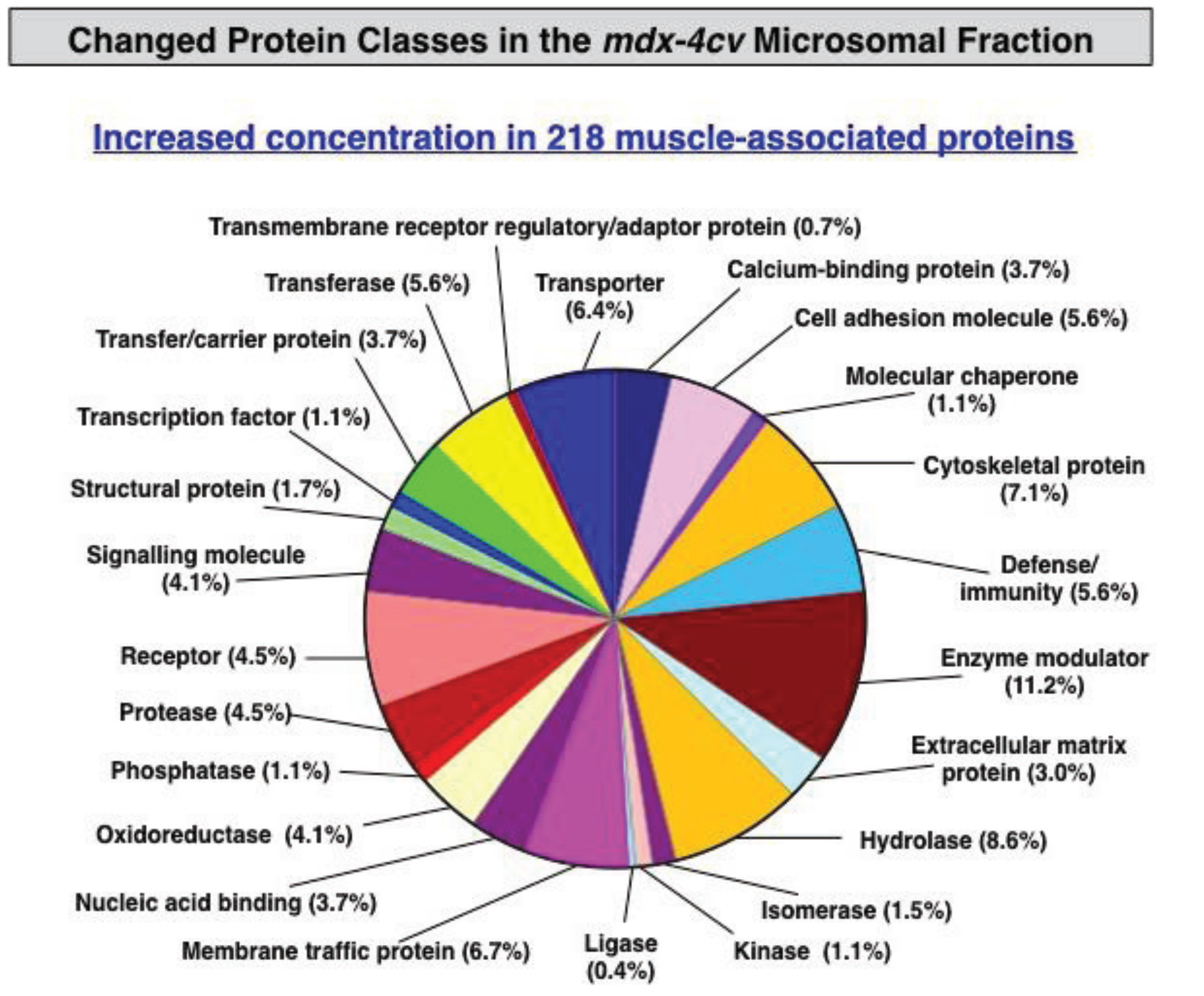
3.3. Bioinformatics Evaluation of the Label-Free LC-MS/MS Analysis of mdx-4cv Muscle Microsomes
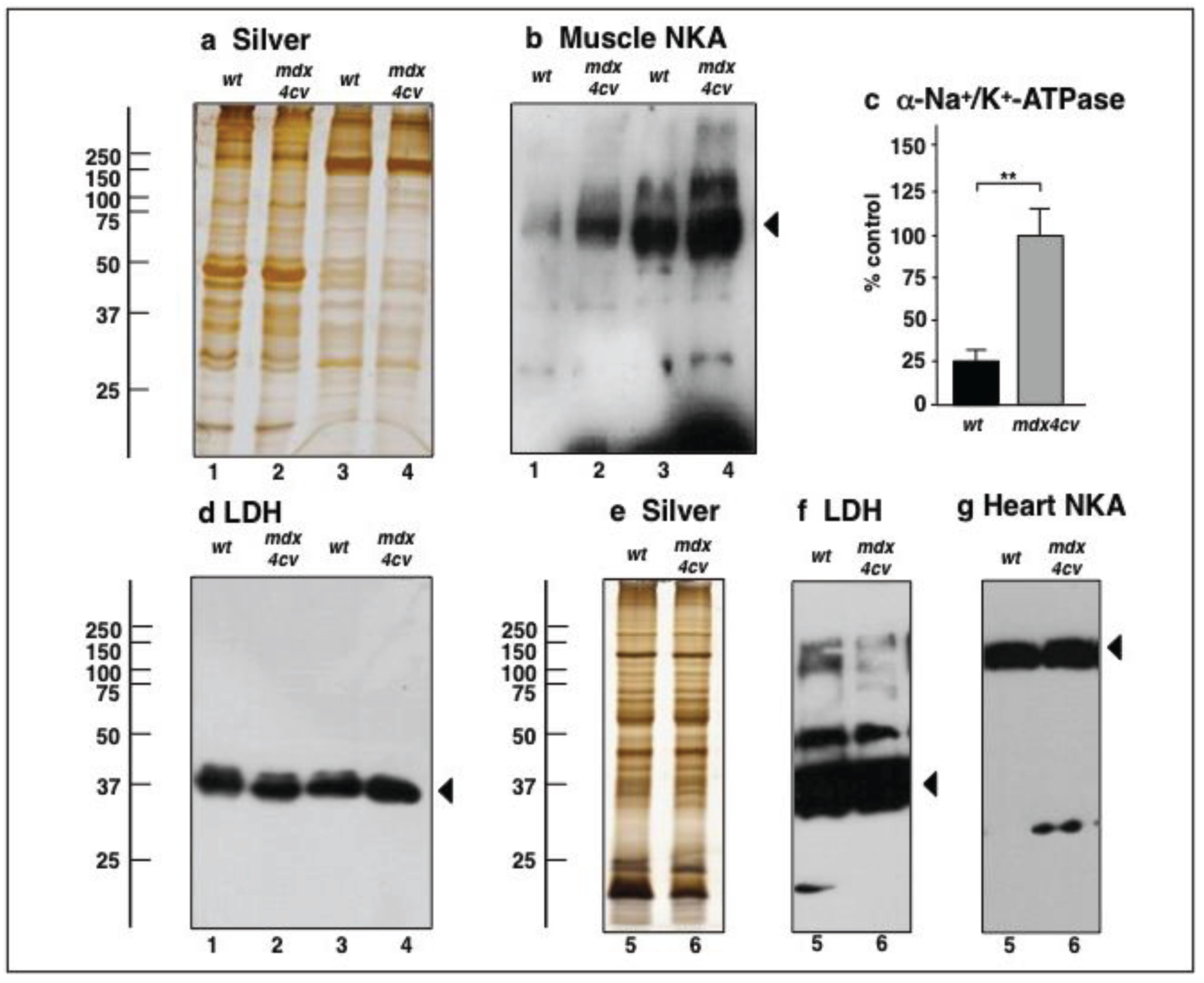
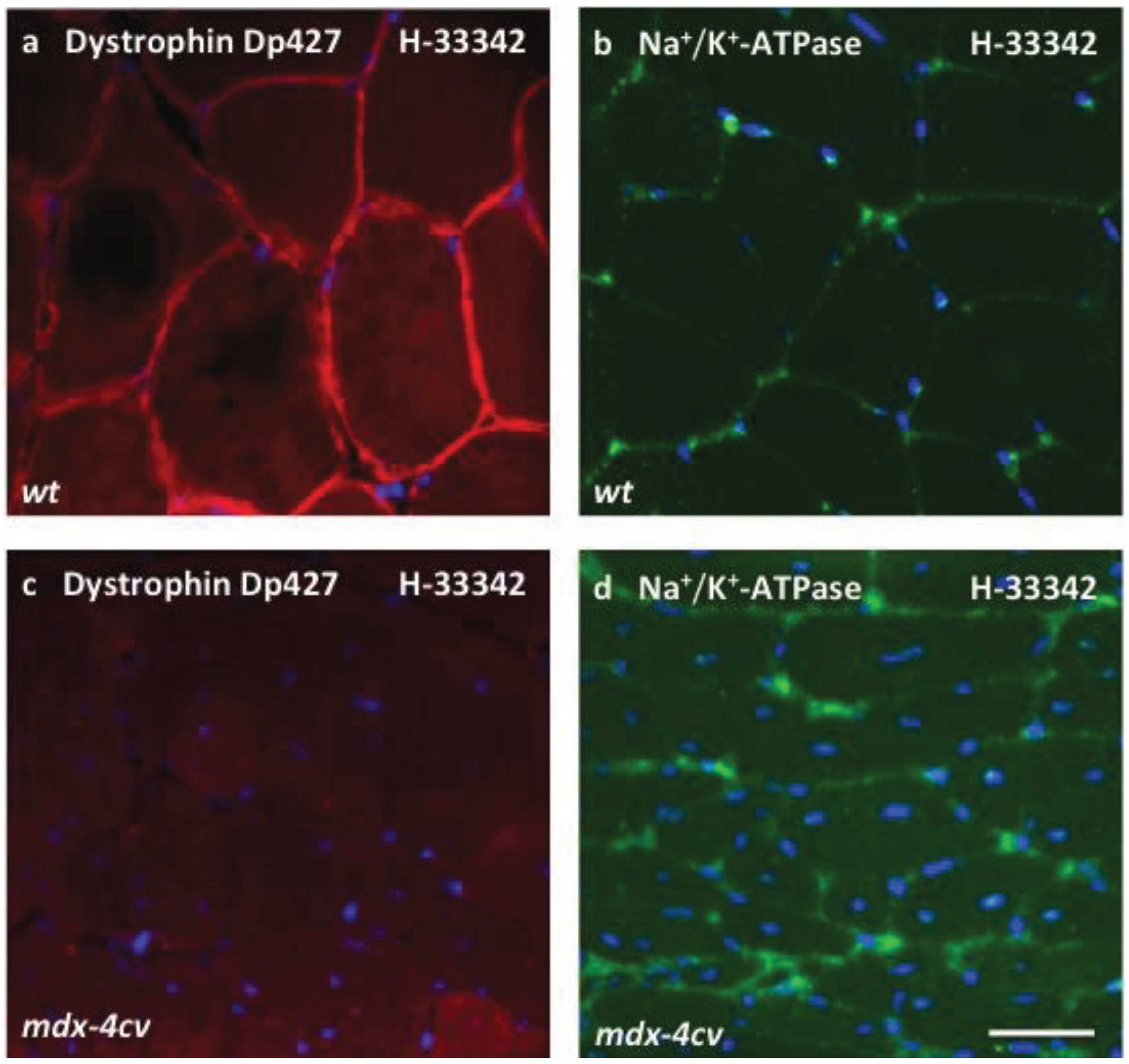
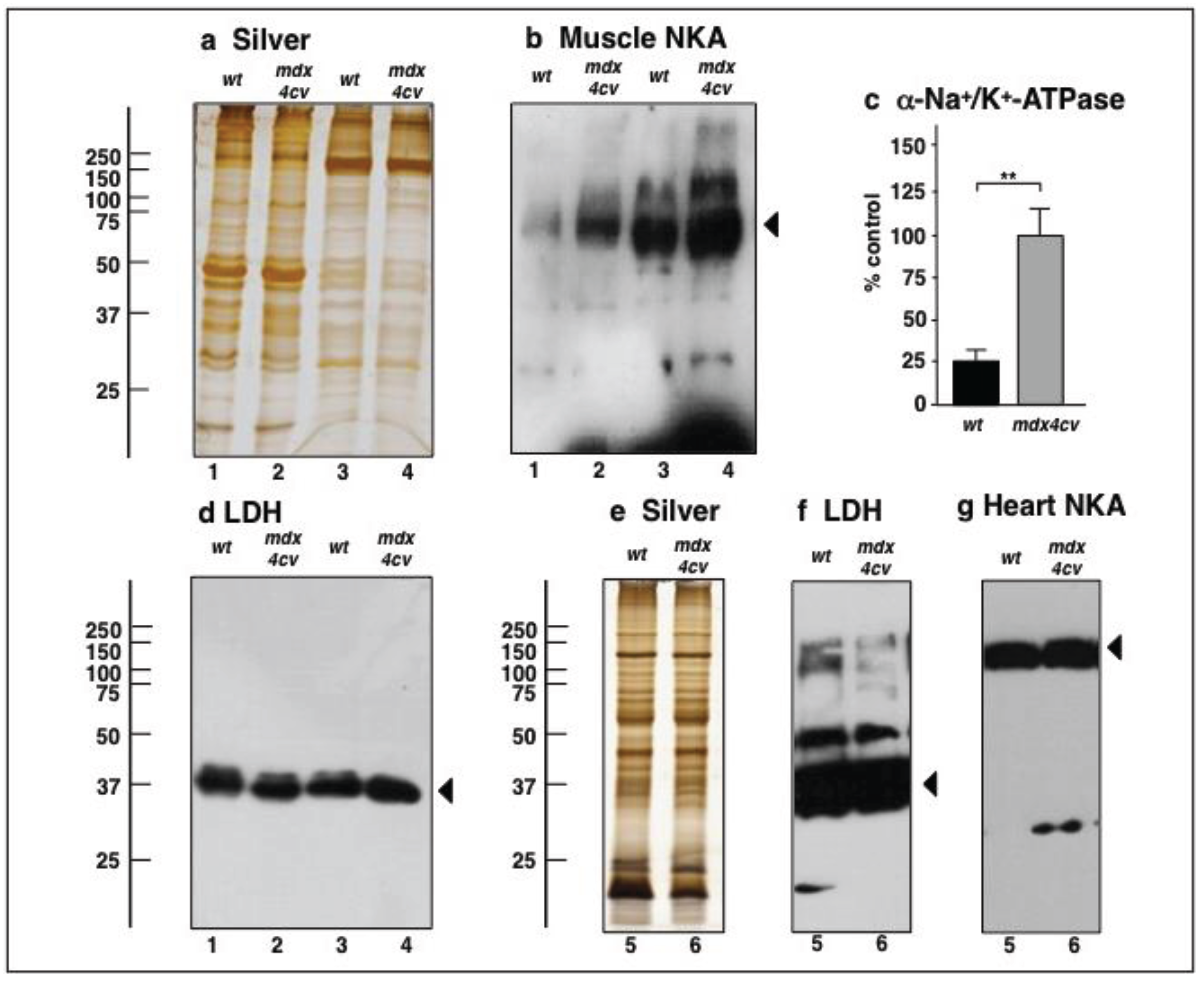
3.4. Verification Analysis of Increased Levels of the Na+/K+-ATPase in mdx-4cv Skeletal Muscle
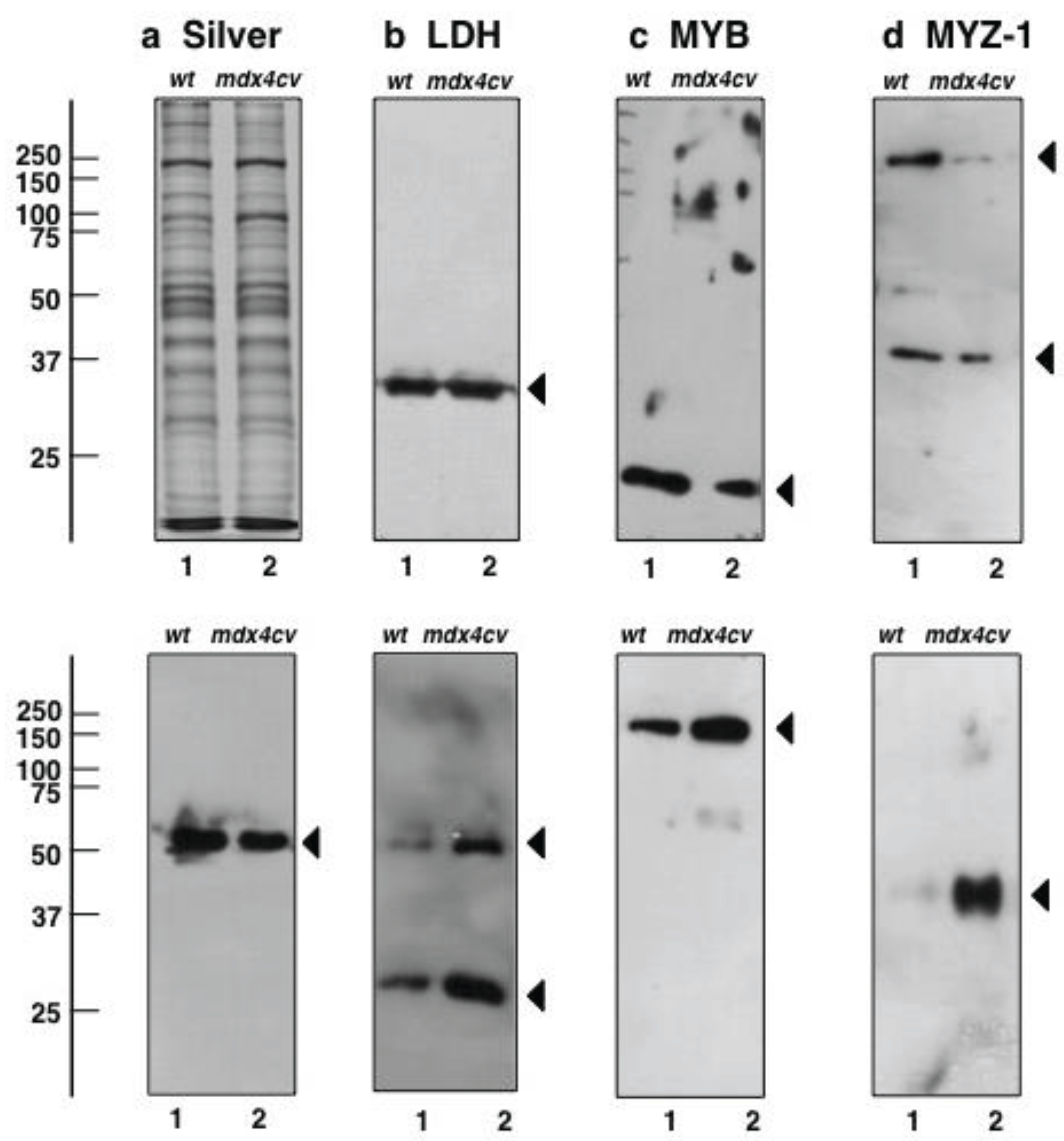
3.5. Immunoblotting Survey of mdx-4cv versus Wild Type Skeletal Muscle
4. Conclusions
Supplementary Files
Supplementary File 1Acknowledgments
Author Contributions
Conflicts of Interest
References
- Geiger, T.; Velic, A.; Macek, B.; Lundberg, E.; Kampf, C.; Nagaraj, N.; Uhlen, M.; Cox, J.; Mann, M. Initial quantitative proteomic map of 28 mouse tissues using the SILAC mouse. Mol. Cell. Proteomics 2013, 12, 1709–1722. [Google Scholar] [CrossRef] [PubMed]
- Højlund, K.; Yi, Z.; Hwang, H.; Bowen, B.; Lefort, N.; Flynn, C.R.; Langlais, P.; Weintraub, S.T.; Mandarino, L.J. Characterization of the human skeletal muscle proteome by one-dimensional gel electrophoresis and HPLC-ESI-MS/MS. Mol. Cell. Proteomics 2008, 7, 257–267. [Google Scholar] [CrossRef] [PubMed]
- Raddatz, K.; Albrecht, D.; Hochgräfe, F.; Hecker, M.; Gotthardt, M. A proteome map of murine heart and skeletal muscle. Proteomics 2008, 8, 1885–1897. [Google Scholar] [CrossRef] [PubMed]
- Parker, K.C.; Walsh, R.J.; Salajegheh, M.; Amato, A.A.; Krastins, B.; Sarracino, D.A.; Greenberg, S.A. Characterization of human skeletal muscle biopsy samples using shotgun proteomics. J. Proteome Res. 2009, 8, 3265–3277. [Google Scholar] [CrossRef] [PubMed]
- Burniston, J.G.; Connolly, J.; Kainulainen, H.; Britton, S.L.; Koch, L.G. Label-free profiling of skeletal muscle using high-definition mass spectrometry. Proteomics 2014, 14, 2339–2344. [Google Scholar] [CrossRef] [PubMed]
- Deshmukh, A.S.; Murgia, M.; Nagaraja, N.; Treebak, J.T.; Cox, J.; Mann, M. Deep proteomics of mouse skeletal muscle enables quantitation of protein isoforms, metabolic pathways and transcription factors. Mol. Cell. Proteomics 2015, 14, 841–853. [Google Scholar] [CrossRef] [PubMed]
- Schiaffino, S.; Reggiani, C. Fiber types in mammalian skeletal muscles. Physiol. Rev. 2011, 91, 1447–1531. [Google Scholar] [CrossRef] [PubMed]
- Pette, D.; Staron, R.S. Myosin isoforms, muscle fiber types, and transitions. Microsc. Res. Tech. 2000, 50, 500–509. [Google Scholar] [CrossRef]
- Froemming, G.R.; Murray, B.E.; Harmon, S.; Pette, D.; Ohlendieck, K. Comparative analysis of the isoform expression pattern of Ca2+-regulatory membrane proteins in fast-twitch, slow-twitch, cardiac, neonatal and chronic low-frequency stimulated muscle fibers. Biochim. Biophys. Acta 2000, 1466, 151–168. [Google Scholar] [CrossRef]
- Quiroz-Rothe, E.; Rivero, J.L. Coordinated expression of myosin heavy chains, metabolic enzymes, and morphological features of porcine skeletal muscle fiber types. Microsc. Res. Tech. 2004, 65, 43–61. [Google Scholar] [CrossRef] [PubMed]
- Blaauw, B.; Schiaffino, S.; Reggiani, C. Mechanisms modulating skeletal muscle phenotype. Compr. Physiol. 2013, 3, 1645–1687. [Google Scholar] [PubMed]
- Okumura, N.; Hashida-Okumura, A.; Kita, K.; Matsubae, M.; Matsubara, T.; Takao, T.; Nagai, K. Proteomic analysis of slow- and fast-twitch skeletal muscles. Proteomics 2005, 5, 2896–2906. [Google Scholar] [CrossRef] [PubMed]
- Gelfi, C.; Viganò, A.; de Palma, S.; Ripamonti, M.; Begum, S.; Cerretelli, P.; Wait, R. 2-D protein maps of rat gastrocnemius and soleus muscles: A tool for muscle plasticity assessment. Proteomics 2006, 6, 321–340. [Google Scholar] [CrossRef] [PubMed]
- Drexler, H.C.; Ruhs, A.; Konzer, A.; Mendler, L.; Bruckskotten, M.; Looso, M.; Günther, S.; Boettger, T.; Krüger, M.; Braun, T. On marathons and Sprints: An integrated quantitative proteomics and transcriptomics analysis of differences between slow and fast muscle fibers. Mol. Cell. Proteomics 2012. [Google Scholar] [CrossRef] [PubMed]
- Murgia, M.; Nagaraj, N.; Deshmukh, A.S.; Zeiler, M.; Cancellara, P.; Moretti, I.; Reggiani, C.; Schiaffino, S.; Mann, M. Single muscle fiber proteomics reveals unexpected mitochondrial specialization. EMBO Rep. 2015, 16, 387–395. [Google Scholar] [CrossRef] [PubMed]
- Ohlendieck, K. Proteomics of skeletal muscle differentiation, neuromuscular disorders and fiber aging. Expert Rev. Proteomics 2010, 7, 283–296. [Google Scholar] [CrossRef] [PubMed]
- Gelfi, C.; Vasso, M.; Cerretelli, P. Diversity of human skeletal muscle in health and disease: Contribution of proteomics. J. Proteomics 2011, 74, 774–795. [Google Scholar] [CrossRef] [PubMed]
- Holland, A.; Ohlendieck, K. Proteomic profiling of the contractile apparatus from skeletal muscle. Expert Rev. Proteomics 2013, 10, 239–257. [Google Scholar] [CrossRef] [PubMed]
- Holland, A.; Ohlendieck, K. Comparative proteomics for studying muscular dystrophy: Intrinsic biological and analytical issues associated with the systematic utilization of tissue specimens. J. Proteomics Bioinform. 2014. [Google Scholar] [CrossRef]
- Gauthier, D.J.; Lazure, C. Complementary methods to assist subcellular fractionation in organellar proteomics. Expert Rev. Proteomics 2008, 5, 603–617. [Google Scholar] [CrossRef] [PubMed]
- Altelaar, A.F.; Heck, A.J. Trends in ultrasensitive proteomics. Curr. Opin. Chem. Biol. 2012, 16, 206–213. [Google Scholar] [CrossRef] [PubMed]
- Drissi, R.; Dubois, M.L.; Boisvert, F.M. Proteomics methods for subcellular proteome analysis. FEBS J. 2013, 280, 5626–5634. [Google Scholar] [CrossRef] [PubMed]
- Ohlendieck, K. Organelle proteomics in skeletal muscle biology. J. Integr. Omics 2012, 2, 27–38. [Google Scholar] [CrossRef]
- Vitorino, R.; Ferreira, R.; Neuparth, M.; Guedes, S.; Williams, J.; Tomer, K.B.; Domingues, P.M.; Appell, H.J.; Duarte, J.A.; Amado, F.M. Subcellular proteomics of mice gastrocnemius and soleus muscles. Anal. Biochem. 2007, 366, 156–169. [Google Scholar] [CrossRef] [PubMed]
- Maughan, D.W.; Henkin, J.A.; Vigoreaux, J.O. Concentrations of glycolytic enzymes and other cytosolic proteins in the diffusible fraction of a vertebrate muscle proteome. Mol. Cell. Proteomics 2005, 4, 1541–1549. [Google Scholar] [CrossRef] [PubMed]
- Toigo, M.; Donohoe, S.; Sperrazzo, G.; Jarrold, B.; Wang, F.; Hinkle, R.; Dolan, E.; Isfort, R.J.; Aebersold, R. ICAT-MS-MS time course analysis of atrophying mouse skeletal muscle cytosolic subproteome. Mol. Biosyst. 2005, 1, 229–241. [Google Scholar] [CrossRef] [PubMed]
- Lewis, C.; Ohlendieck, K. Mass spectrometric identification of dystrophin isoform Dp427 by on-membrane digestion of sarcolemma from skeletal muscle. Anal. Biochem. 2010, 404, 197–203. [Google Scholar] [CrossRef] [PubMed]
- Staunton, L.; Ohlendieck, K. Mass spectrometric characterization of the sarcoplasmic reticulum from rabbit skeletal muscle by on-membrane digestion. Protein Pept. Lett. 2012, 19, 252–263. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Du, X.; Yin, C.; Chang, Z. Shotgun proteomic analysis of sarcoplasmic reticulum preparations from rabbit skeletal muscle. Proteomics 2013, 13, 2335–2338. [Google Scholar] [CrossRef] [PubMed]
- Lefort, N.; Yi, Z.; Bowen, B.; Glancy, B.; de Filippis, E.A.; Mapes, R.; Hwang, H.; Flynn, C.R.; Willis, W.T.; Civitarese, A.; et al. Proteome profile of functional mitochondria from human skeletal muscle using one-dimensional gel electrophoresis and HPLC-ESI-MS/MS. J. Proteomics 2009, 72, 1046–1060. [Google Scholar] [CrossRef] [PubMed]
- O’Connell, K.; Ohlendieck, K. Proteomic DIGE analysis of the mitochondria-enriched fraction from aged rat skeletal muscle. Proteomics 2009, 9, 5509–5524. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, R.; Vitorino, R.; Alves, R.M.; Appell, H.J.; Powers, S.K.; Duarte, J.A.; Amado, F. Subsarcolemmal and intermyofibrillar mitochondria proteome differences disclose functional specializations in skeletal muscle. Proteomics 2010, 10, 3142–3154. [Google Scholar] [CrossRef] [PubMed]
- Carberry, S.; Zweyer, M.; Swandulla, D.; Ohlendieck, K. Comparative proteomic analysis of the contractile protein-depleted fraction from normal versus dystrophic skeletal muscle. Anal. Biochem. 2014, 446, 108–115. [Google Scholar] [CrossRef] [PubMed]
- Muntoni, F.; Torelli, S.; Ferlini, A. Dystrophin and mutations: One gene, several proteins, multiple phenotypes. Lancet Neurol. 2003, 2, 731–740. [Google Scholar] [CrossRef]
- Nakamura, A.; Takeda, S. Mammalian models of duchenne muscular dystrophy: Pathological characteristics and therapeutic applications. J. Biomed. Biotechnol. 2011. [Google Scholar] [CrossRef] [PubMed]
- Ng, R.; Banks, G.B.; Hall, J.K.; Muir, L.A.; Ramos, J.N.; Wicki, J.; Odom, G.L.; Konieczny, P.; Seto, J.; Chamberlain, J.R. Animal models of muscular dystrophy. Prog. Mol. Biol. Transl. Sci. 2012, 105, 83–111. [Google Scholar] [PubMed]
- Partridge, T.A. The mdx mouse model as a surrogate for Duchenne muscular dystrophy. FEBS J. 2013, 280, 4177–4186. [Google Scholar] [CrossRef] [PubMed]
- Doran, P.; Wilton, S.D.; Fletcher, S.; Ohlendieck, K. Proteomic profiling of antisense-induced exon skipping reveals reversal of pathobiochemical abnormalities in dystrophic mdx diaphragm. Proteomics 2009, 9, 671–685. [Google Scholar] [CrossRef] [PubMed]
- Danko, I.; Chapman, V.; Wolff, J.A. The frequency of revertants in mdx mouse genetic models for Duchenne muscular dystrophy. Pediatr. Res. 1992, 32, 128–131. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.H.; Hakim, C.H.; Zhang, K.; Duan, D. Genotyping mdx, mdx3cv, and mdx4cv mice by primer competition polymerase chain reaction. Muscle Nerve 2011, 43, 283–286. [Google Scholar] [CrossRef] [PubMed]
- Doran, P.; Martin, G.; Dowling, P.; Jockusch, H.; Ohlendieck, K. Proteome analysis of the dystrophin-deficient MDX diaphragm reveals a drastic increase in the heat shock protein cvHSP. Proteomics 2006, 6, 4610–4621. [Google Scholar] [CrossRef] [PubMed]
- Guevel, L.; Lavoie, J.R.; Perez-Iratxeta, C.; Rouger, K.; Dubreil, L.; Feron, M.; Talon, S.; Brand, M.; Megeney, L.A. Quantitative proteomic analysis of dystrophic dog muscle. J. Proteome Res. 2011, 10, 2465–2478. [Google Scholar] [CrossRef] [PubMed]
- Carberry, S.; Zweyer, M.; Swandulla, D.; Ohlendieck, K. Proteomics reveals drastic increase of extracellular matrix proteins collagen and dermatopontin in aged mdx diaphragm muscle. Int. J. Mol. Med. 2012, 30, 229–234. [Google Scholar] [PubMed]
- Rayavarapu, S.; Coley, W.; Cakir, E.; Jahnke, V.; Takeda, S.; Aoki, Y.; Grodish-Dressman, H.; Jaiswal, J.K.; Hoffman, E.P.; Brown, K.J.; et al. Identification of disease specific pathways using in vivo SILAC proteomics in dystrophin deficient mdx mouse. Mol. Cell. Proteomics 2013, 12, 1061–1073. [Google Scholar] [CrossRef] [PubMed]
- Duguez, S.; Duddy, W.; Johnston, H.; Lainé, J.; le Bihan, M.C.; Brown, K.J.; Bigot, A.; Hathout, Y.; Butler-Browne, G.; Partridge, T. Dystrophin deficiency leads to disturbance of LAMP1-vesicle-associated protein secretion. Cell. Mol. Life Sci. 2013, 70, 2159–2174. [Google Scholar] [CrossRef] [PubMed]
- Holland, A.; Carberry, S.; Ohlendieck, K. Proteomics of the dystrophin-glycoprotein complex and dystrophinopathy. Curr. Protein Pept. Sci. 2013, 14, 680–697. [Google Scholar] [CrossRef] [PubMed]
- Chapman, V.M.; Miller, D.R.; Armstrong, D.; Caskey, C.T. Recovery of induced mutations for X chromosome-linked muscular dystrophy in mice. Proc. Natl. Acad. Sci. USA 1989, 86, 1292–1296. [Google Scholar] [CrossRef] [PubMed]
- Im, W.B.; Phelps, S.F.; Copen, E.H.; Adams, E.G.; Slightom, J.L.; Chamberlain, J.S. Differential expression of dystrophin isoforms in strains of mdx mice with different mutations. Hum. Mol. Genet. 1996, 5, 1149–1153. [Google Scholar] [CrossRef] [PubMed]
- Judge, L.M.; Haraguchiln, M.; Chamberlain, J.S. Dissecting the signaling and mechanical functions of the dystrophin-glycoprotein complex. J. Cell Sci. 2006, 119, 1537–1546. [Google Scholar] [CrossRef] [PubMed]
- Mitrpant, C.; Fletcher, S.; Iversen, P.L.; Wilton, S.D. By-passing the nonsense mutation in the 4 CV mouse model of muscular dystrophy by induced exon skipping. J. Gene Med. 2009, 11, 46–56. [Google Scholar] [CrossRef] [PubMed]
- Kimura, E.; Li, S.; Gregorevic, P.; Fall, B.M.; Chamberlain, J.S. Dystrophin delivery to muscles of mdx mice using lentiviral vectors leads to myogenic progenitor targeting and stable gene expression. Mol. Ther. 2010, 18, 206–213. [Google Scholar] [CrossRef] [PubMed]
- Holland, A.; Dowling, P.; Zweyer, M.; Swandulla, D.; Henry, M.; Clynes, M.; Ohlendieck, K. Proteomic profiling of cardiomyopathic tissue from the aged mdx model of Duchenne muscular dystrophy reveals a drastic decrease in laminin, nidogen and annexin. Proteomics 2013, 13, 2312–2323. [Google Scholar] [CrossRef] [PubMed]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Meleady, P.; Gallagher, M.; Clarke, C.; Henry, M.; Sanchez, N.; Barron, N.; Clynes, M. Impact of miR-7 over-expression on the proteome of Chinese hamster ovary cells. J. Biotechnol. 2012, 160, 251–262. [Google Scholar] [CrossRef] [PubMed]
- Meleady, P.; Hoffrogge, R.; Henry, M.; Rupp, O.; Bort, J.H.; Clarke, C.; Brinkrolf, K.; Kelly, S.; Müller, B.; Doolan, P.; et al. Utilization and evaluation of CHO-specific sequence databases for mass spectrometry based proteomics. Biotechnol. Bioeng. 2012, 109, 1386–1394. [Google Scholar] [CrossRef] [PubMed]
- Holland, A.; Schmitt-John, T.; Dowling, P.; Meleady, P.; Henry, M.; Clynes, M.; Ohlendieck, K. Intricate effects of primary motor neuronopathy on contractile proteins and metabolic muscle enzymes as revealed by label-free mass spectrometry. Biosci. Rep. 2014, 34, e00119. [Google Scholar] [CrossRef] [PubMed]
- PANTHER Gene List Analysis. Available online: http://pantherdb.org/ (accessed on 4 May 2015).
- Mi, H.; Muruganujan, A.; Thomas, P.D. PANTHER in 2013: Modeling the evolution of gene function, and other gene attributes, in the context of phylogenetic trees. Nucleic Acids Res. 2013, 41, D377–D386. [Google Scholar] [CrossRef] [PubMed]
- STRING Functional Protein Association Network. Available online: http://string-db.org/ (accessed on 3 February 2015).
- Franceschini, A.; Szklarczyk, D.; Frankild, S.; Kuhn, M.; Simonovic, M.; Roth, A.; Lin, J.; Minguez, P.; Bork, P.; von Mering, C.; et al. STRING v9.1: Protein-protein interaction networks, with increased coverage and integration. Nucleic Acids Res. 2013, 41, D808–D815. [Google Scholar] [CrossRef] [PubMed]
- Mundegar, R.R.; Franke, E.; Schäfer, R.; Zweyer, M.; Wernig, A. Reduction of high background staining by heating unfixed mouse skeletal muscle tissue sections allows for detection of thermostable antigens with murine monoclonal antibodies. J. Histochem. Cytochem. 2008, 56, 969–975. [Google Scholar] [CrossRef] [PubMed]
- Gardan-Salmon, D.; Dixon, J.M.; Lonergan, S.M.; Selsby, J.T. Proteomic assessment of the acute phase of dystrophin deficiency in mdx mice. Eur. J. Appl. Physiol. 2011, 111, 2763–2773. [Google Scholar] [CrossRef] [PubMed]
- Carberry, S.; Brinkmeier, H.; Zhang, Y.; Winkler, C.K.; Ohlendieck, K. Comparative proteomic profiling of soleus, extensor digitorum longus, flexor digitorum brevis and interosseus muscle from the mdx mouse model of Duchenne muscular dystrophy. Int. J. Mol. Med. 2013, 32, 544–556. [Google Scholar] [PubMed]
- Yoon, J.H.; Johnson, E.; Xu, R.; Martin, L.T.; Martin, P.T.; Montanaro, F. Comparative proteomic profiling of dystroglycan-associated proteins in wild type, mdx, and Galgt2 transgenic mouse skeletal muscle. J. Proteome Res. 2012, 11, 4413–4424. [Google Scholar] [CrossRef] [PubMed]
- Johnson, E.K.; Zhang, L.; Adams, M.E.; Phillips, A.; Freitas, M.A.; Froehner, S.C.; Green-Church, K.B.; Montanaro, F. Proteomic analysis reveals new cardiac-specific dystrophin-associated proteins. PLoS ONE 2012, 7, e43515. [Google Scholar] [CrossRef] [PubMed]
- Swiderski, K.; Shaffer, S.A.; Gallis, B.; Odom, G.L.; Arnett, A.L.; Edgar, S.J.; Baum, D.M.; Chee, A.; Naim, T.; Gregorevic, P.; et al. Phosphorylation within the cysteine-rich region of dystrophin enhances its association with β-dystroglycan and identifies a potential novel therapeutic target for skeletal muscle wasting. Hum. Mol. Genet. 2014, 23, 6697–6711. [Google Scholar] [CrossRef] [PubMed]
- Brown, K.J.; Marathi, R.; Fiorillo, A.A.; Ciccimaro, E.F.; Sharma, S.; Rowlands, D.S.; Rayavarapu, S.; Nagaraju, K.; Hoffman, E.P.; Hathout, Y. Accurate quantitation of dystrophin protein in human skeletal muscle using mass spectrometry. J. Bioanal. Biomed. 2012. [Google Scholar] [CrossRef] [PubMed]
- Ohlendieck, K. Proteomic identification of biomarkers of skeletal muscle disorders. Biomark. Med. 2013, 7, 169–186. [Google Scholar] [CrossRef] [PubMed]
- Dowling, P.; Holland, A.; Ohlendieck, K. Mass spectrometry-based identification of muscle-associated and muscle-derived proteomic biomarkers of dystrophinopathies. J. Neuromusc. Dis. 2014, 1, 15–40. [Google Scholar]
- Gannon, J.; Doran, P.; Kirwan, A.; Ohlendieck, K. Drastic increase of myosin light chain MLC-2 in senescent skeletal muscle indicates fast-to-slow fibre transition in sarcopenia of old age. Eur. J. Cell Biol. 2009, 88, 685–700. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, D.; Ohlendieck, K. Oligomerisation of sarcoplasmic reticulum Ca2+-ATPase monomers from skeletal muscle. Protein Pept. Lett. 2007, 14, 219–226. [Google Scholar] [CrossRef] [PubMed]
- Ohlendieck, K.; Campbell, K.P. Dystrophin-associated proteins are greatly reduced in skeletal muscle from mdx mice. J. Cell Biol. 1991, 115, 1685–1694. [Google Scholar] [CrossRef] [PubMed]
- Ohlendieck, K.; Matsumura, K.; Ionasescu, V.V.; Towbin, J.A.; Bosch, E.P.; Weinstein, S.L.; Sernett, S.W.; Campbell, K.P. Duchenne muscular dystrophy: Deficiency of dystrophin-associated proteins in the sarcolemma. Neurology 1993, 43, 795–800. [Google Scholar] [CrossRef] [PubMed]
- Fumagalli, L.; Businaro, R.; Nori, S.L.; Toesca, A.; Pompili, E.; Evangelisti, E.; Giannetti, S.; Ippoliti, F.; de Renzis, G. Protease inhibitors in mouse skeletal muscle: Tissue-associated components of serum inhibitors and calpastatin. Cell. Mol. Biol. 1996, 42, 535–546. [Google Scholar] [PubMed]
- Miravitlles, M. Alpha-1-antitrypsin and other proteinase inhibitors. Curr. Opin. Pharmacol. 2012, 12, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Jonigk, D.; Al-Omari, M.; Maegel, L.; Müller, M.; Izykowski, N.; Hong, J.; Hong, K.; Kim, S.H.; Dorsch, M.; Mahadeva, R.; et al. Anti-inflammatory and immunomodulatory properties of α1-antitrypsin without inhibition of elastase. Proc. Natl. Acad. Sci. USA 2013, 110, 15007–15012. [Google Scholar] [CrossRef] [PubMed]
- Holland, A.; Dowling, P.; Meleady, P.; Henry, M.; Zweyer, M.; Mundegar, R.R.; Swandulla, D.; Ohlendieck, K. Label-free mass spectrometric analysis of the mdx-4cv diaphragm identifies the matricellular protein periostin as a potential factor involved in dystrophinopathy-related fibrosis. Proteomics 2015. [Google Scholar] [CrossRef]
- Vidal, B.; Serrano, A.L.; Tjwa, M.; Suelves, M.; Ardite, E.; de Mori, R.; Baeza-Raja, B.; de Lagrán, M.M.; Lafuste, P.; Ruiz-Bonilla, V.; et al. Fibrinogen drives dystrophic muscle fibrosis via a TGFbeta/alternative macrophage activation pathway. Genes Dev. 2008, 22, 1747–1752. [Google Scholar] [CrossRef] [PubMed]
- Selbert, S.; Fischer, P.; Menke, A.; Jockusch, H.; Pongratz, D.; Noegel, A.A. Annexin VII relocalization as a result of dystrophin deficiency. Exp. Cell Res. 1996, 222, 199–208. [Google Scholar] [CrossRef] [PubMed]
- Bizzarro, V.; Petrella, A.; Parente, L. Annexin A1: Novel roles in skeletal muscle biology. J. Cell. Physiol. 2012, 227, 3007–3015. [Google Scholar] [CrossRef] [PubMed]
- Dunn, J.F.; Burton, K.A.; Dauncey, M.J. Ouabain sensitive Na+/K+-ATPase content is elevated in mdx mice: Implications for the regulation of ions in dystrophic muscle. J. Neurol. Sci. 1995, 133, 11–15. [Google Scholar] [CrossRef]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Murphy, S.; Henry, M.; Meleady, P.; Zweyer, M.; Mundegar, R.R.; Swandulla, D.; Ohlendieck, K. Simultaneous Pathoproteomic Evaluation of the Dystrophin-Glycoprotein Complex and Secondary Changes in the mdx-4cv Mouse Model of Duchenne Muscular Dystrophy. Biology 2015, 4, 397-423. https://doi.org/10.3390/biology4020397
Murphy S, Henry M, Meleady P, Zweyer M, Mundegar RR, Swandulla D, Ohlendieck K. Simultaneous Pathoproteomic Evaluation of the Dystrophin-Glycoprotein Complex and Secondary Changes in the mdx-4cv Mouse Model of Duchenne Muscular Dystrophy. Biology. 2015; 4(2):397-423. https://doi.org/10.3390/biology4020397
Chicago/Turabian StyleMurphy, Sandra, Michael Henry, Paula Meleady, Margit Zweyer, Rustam R. Mundegar, Dieter Swandulla, and Kay Ohlendieck. 2015. "Simultaneous Pathoproteomic Evaluation of the Dystrophin-Glycoprotein Complex and Secondary Changes in the mdx-4cv Mouse Model of Duchenne Muscular Dystrophy" Biology 4, no. 2: 397-423. https://doi.org/10.3390/biology4020397
APA StyleMurphy, S., Henry, M., Meleady, P., Zweyer, M., Mundegar, R. R., Swandulla, D., & Ohlendieck, K. (2015). Simultaneous Pathoproteomic Evaluation of the Dystrophin-Glycoprotein Complex and Secondary Changes in the mdx-4cv Mouse Model of Duchenne Muscular Dystrophy. Biology, 4(2), 397-423. https://doi.org/10.3390/biology4020397