Cell-Type Specific Determinants of NRAMP1 Expression in Professional Phagocytes

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. The Marine Origins of Nramp1

3. SLC11A1 Genetic Factor of Resistance to Tuberculosis
3.1. Host-Pathogen Co-Adaptation
3.2. SLC11A1 Candidate Functional Polymorphisms
3.3. Up-Regulation of SLC11A1 Expression by Hypoxia-Induced Factor 1α
4. SLC11A1 Proximal Promoter Controls Myelo-Monocytic Expression
4.1. Expression in Mature Mononuclear and Polynuclear Phagocytes
4.2. Vitamin D and Host Defense against Tuberculosis
4.3. Regulation of SLC11A1 Proximal Promoter by Vitamin D in HL-60 Cells
4.4. Transcriptional and Post-Transcriptional Determinants of SLC11A1 Expression in Macrophage-Like HL-60 Cells
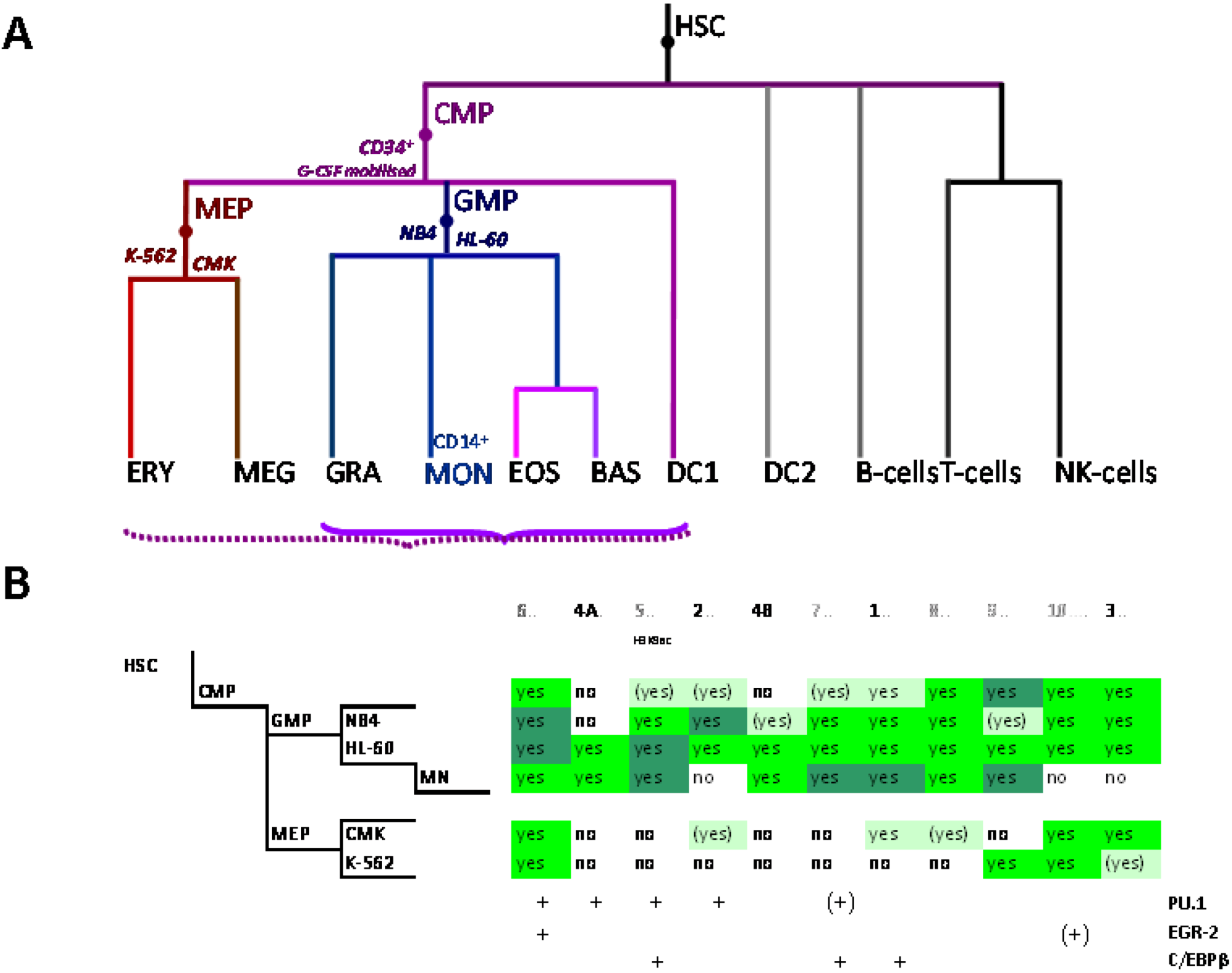
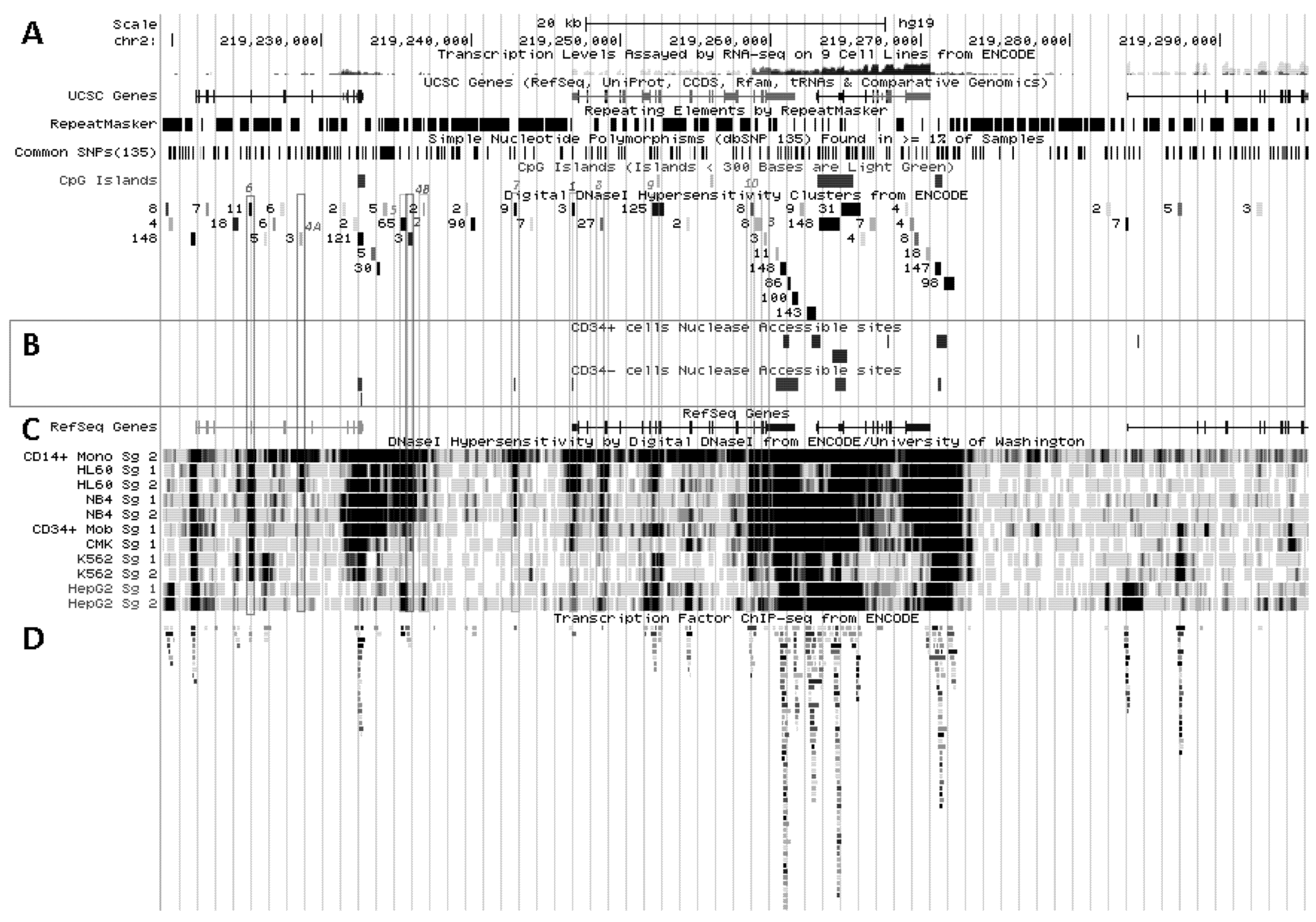
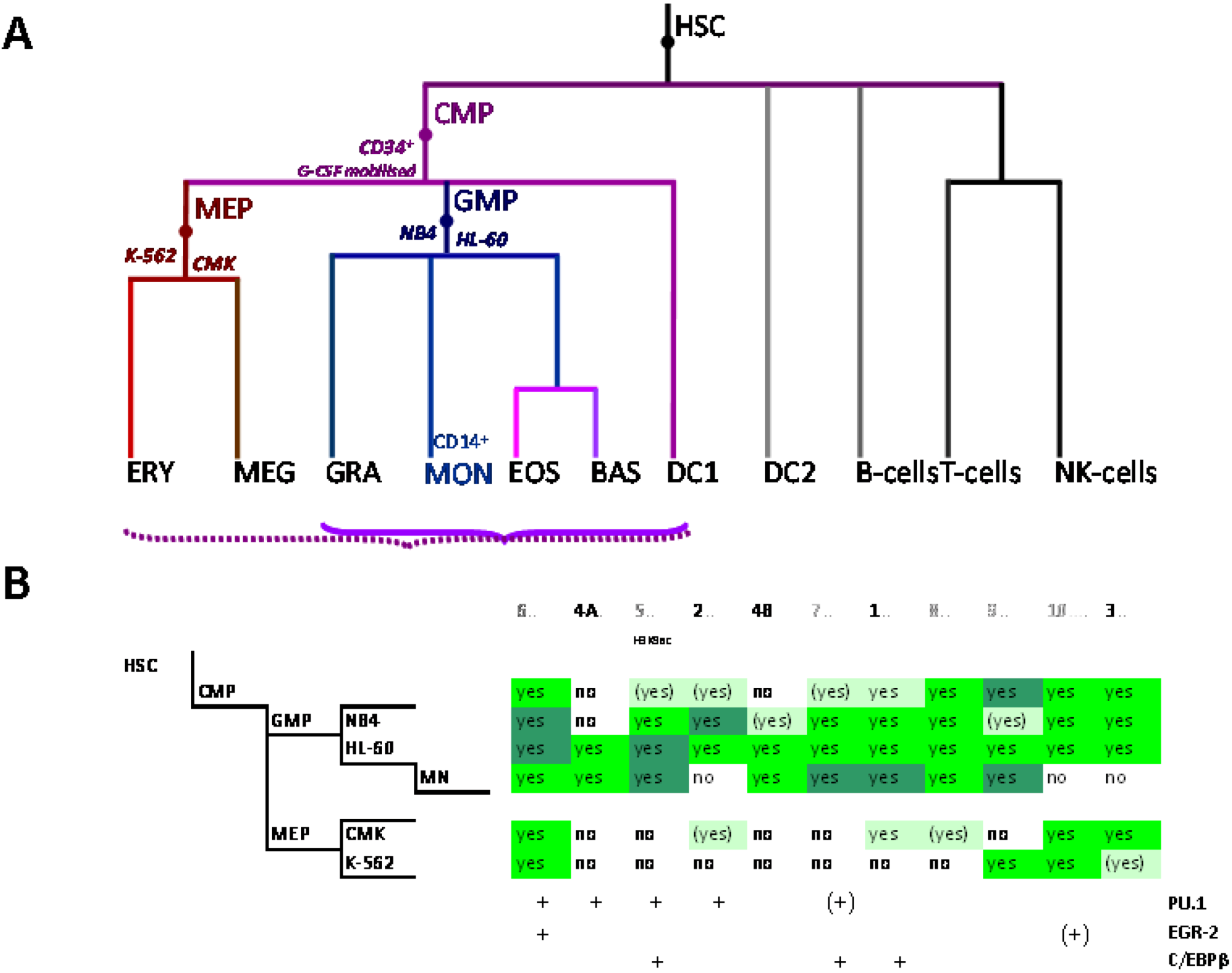
5. Delineation of SLC11A1 Distal Elements Mobilized During Myelo-Monocytic Development
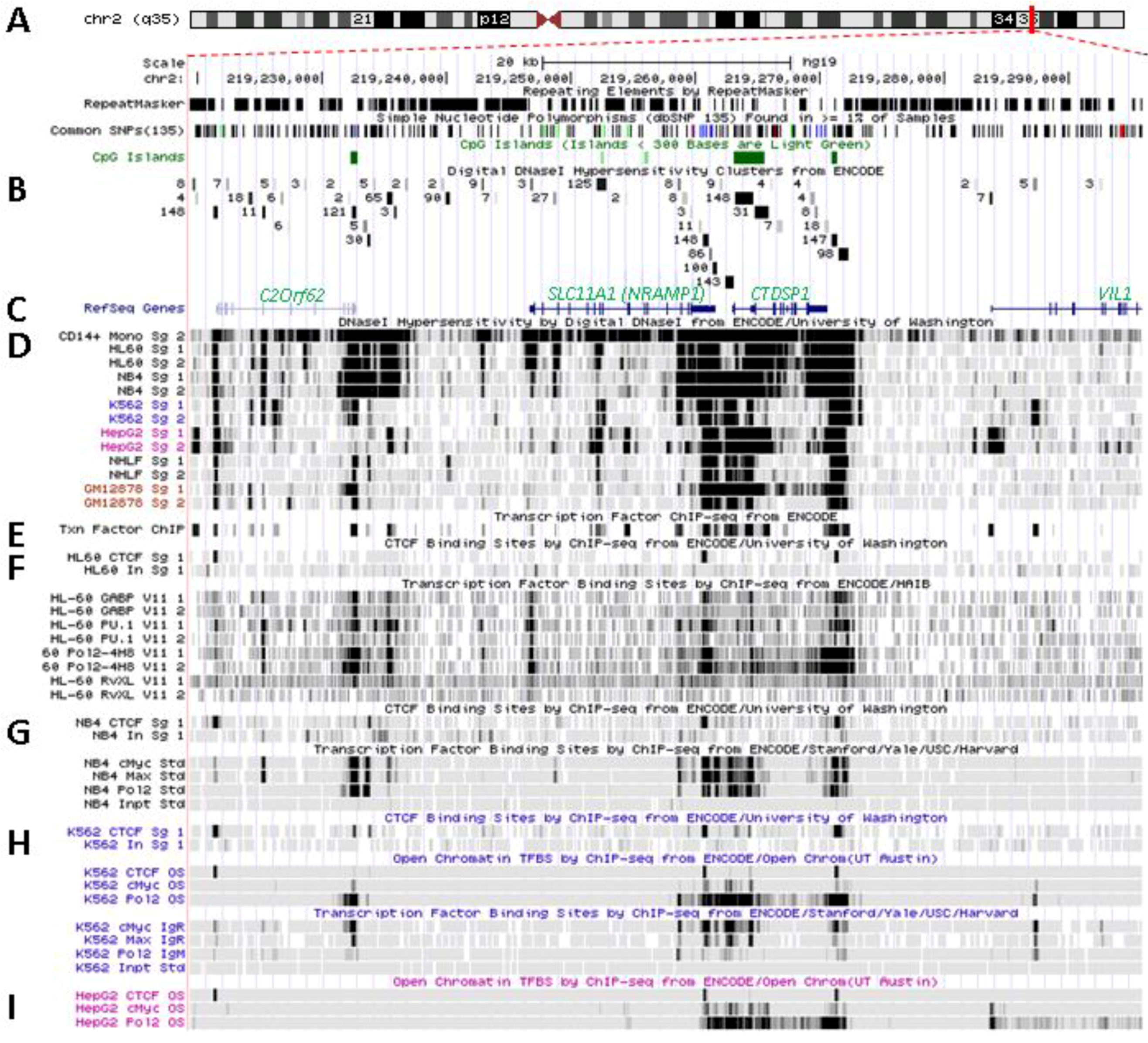
5.1. Physical Organization of SLC11A1 Locus
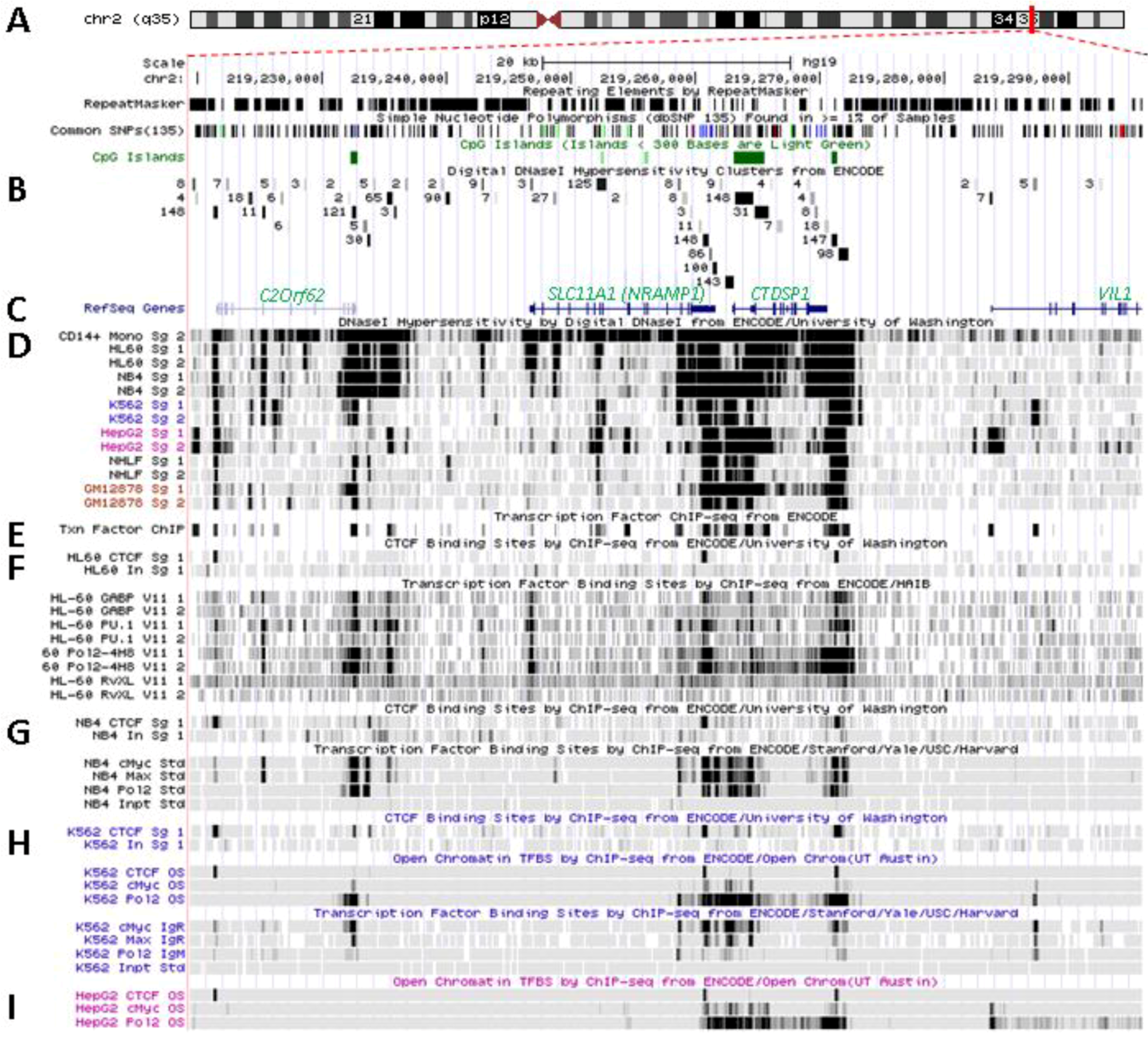
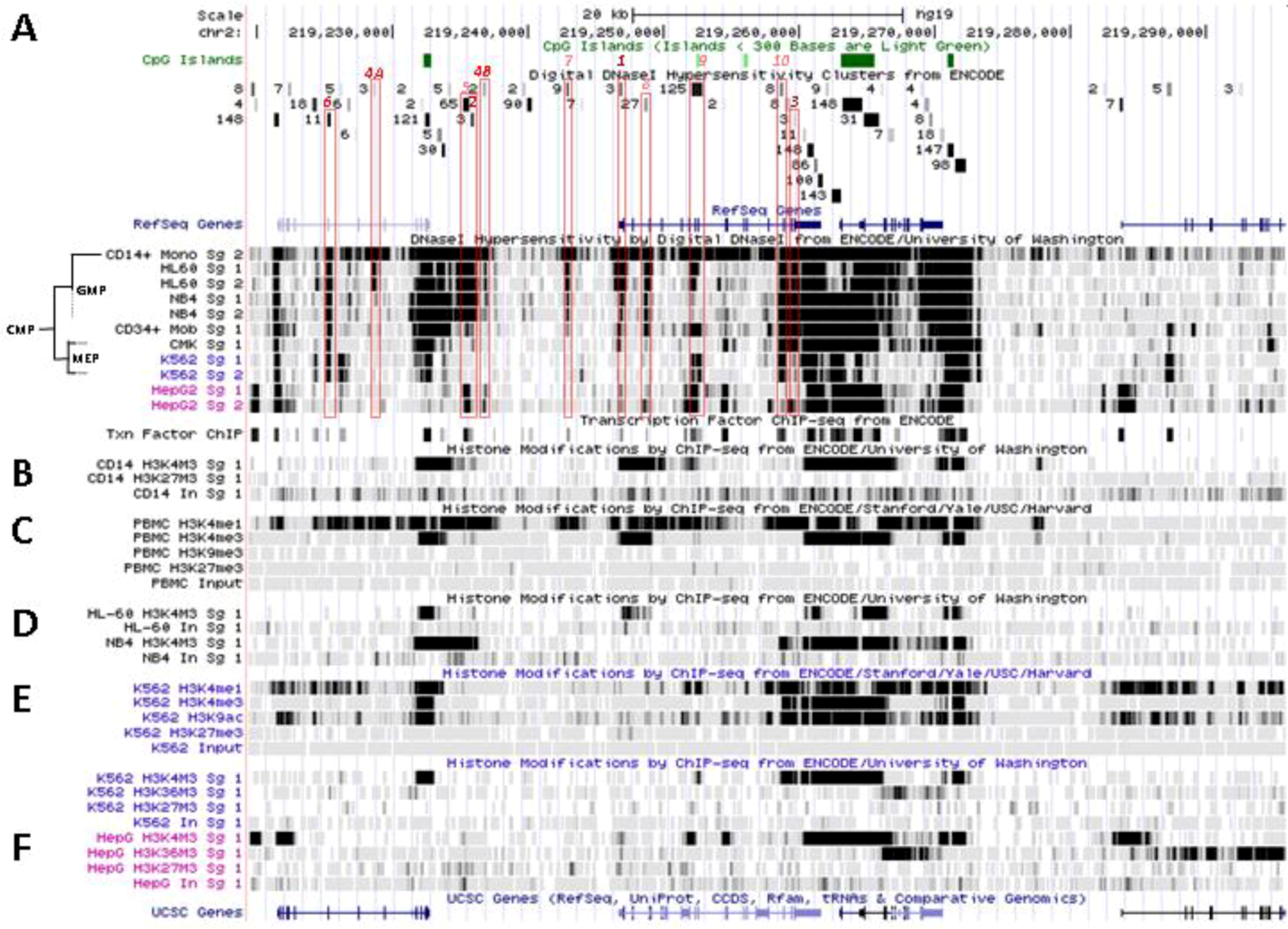
5.2. In Situ DNAse I Footprinting and Transcription Factor-Specific ChIP-Seq Studies
5.2.1. Myelo-Monocytic-Specific Signals
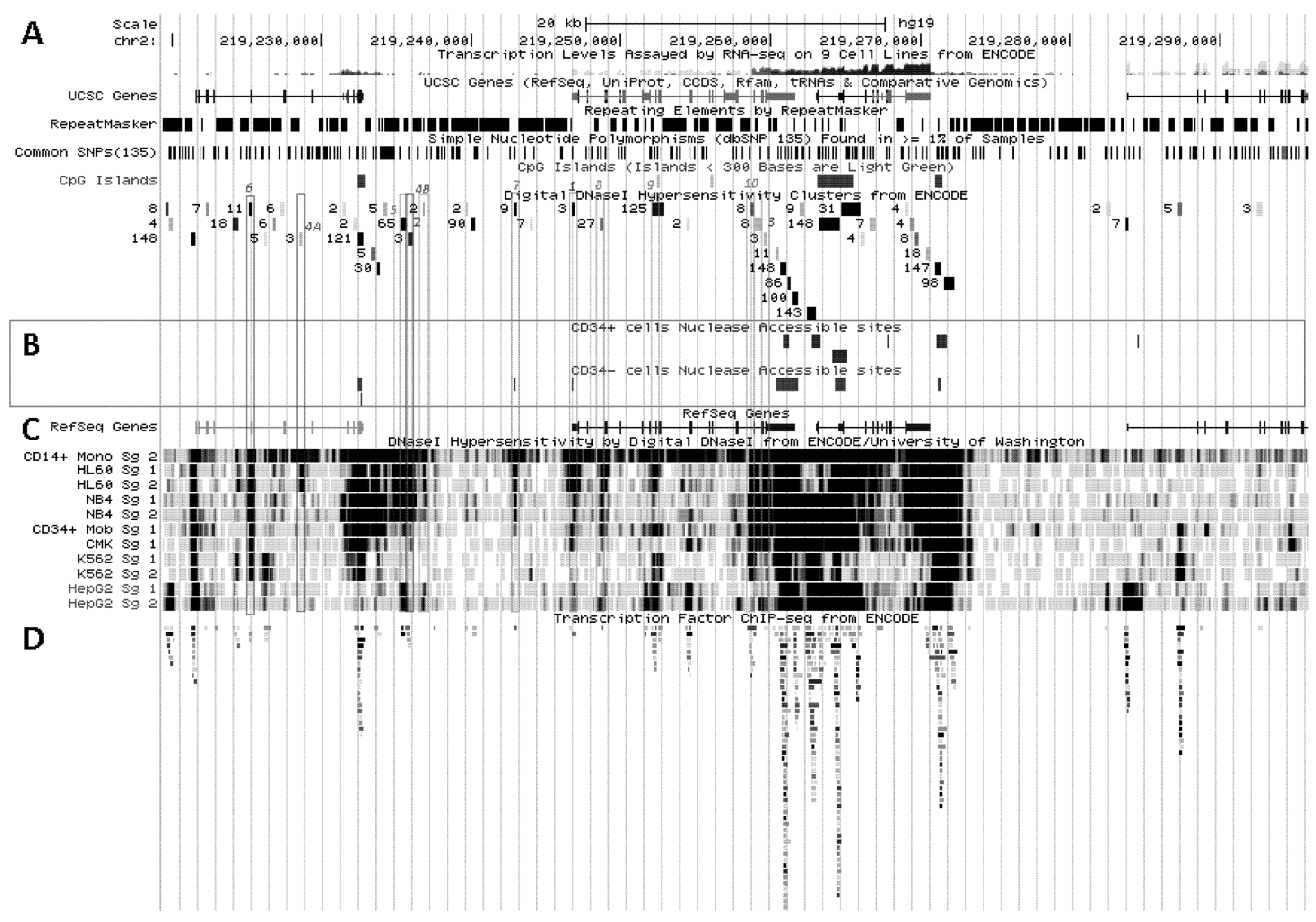

5.2.2. Signals Enriched in Myelo-Monocytic Cells
5.2.3. Other Strong Signals in Myelo-Monocytic Cells
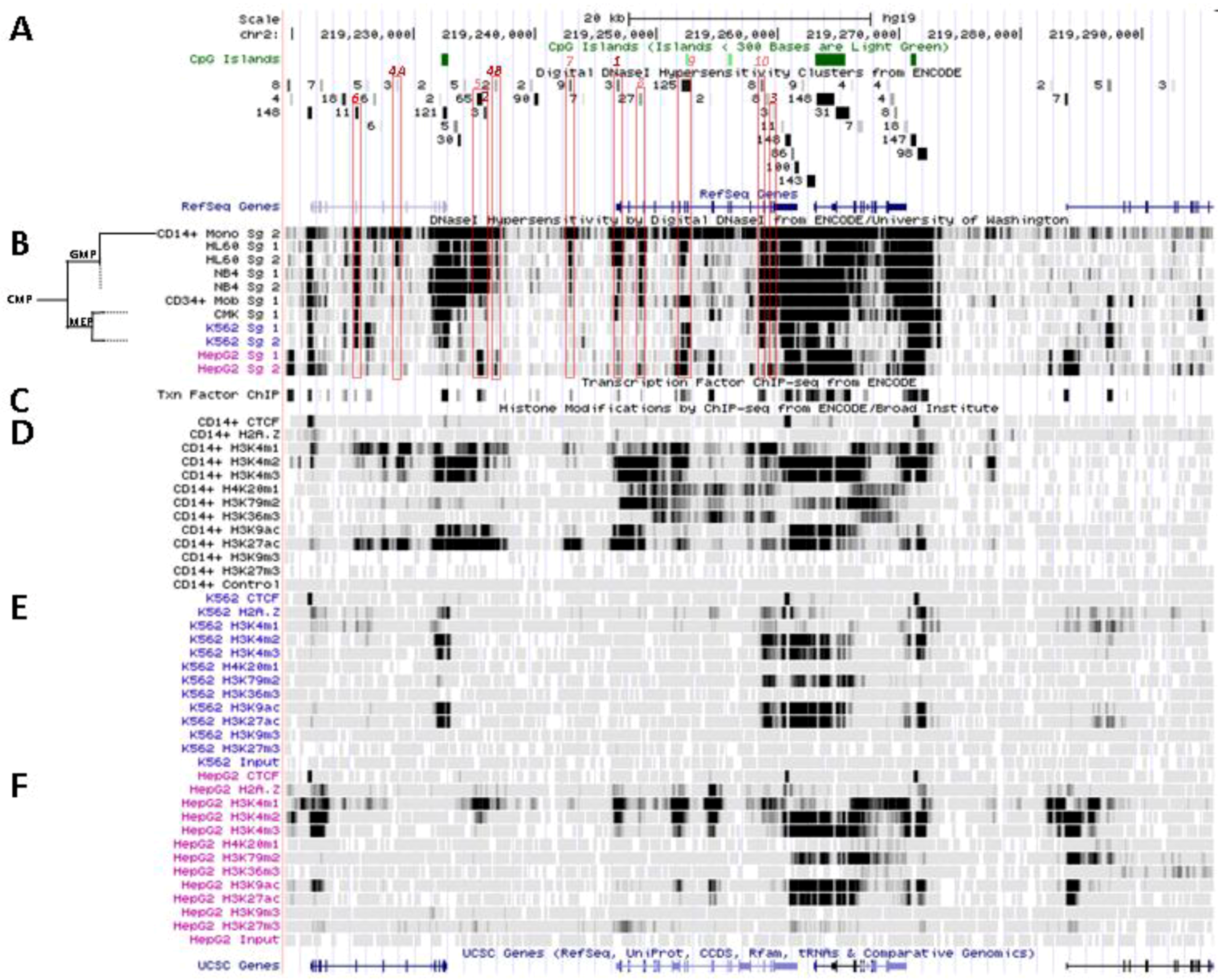
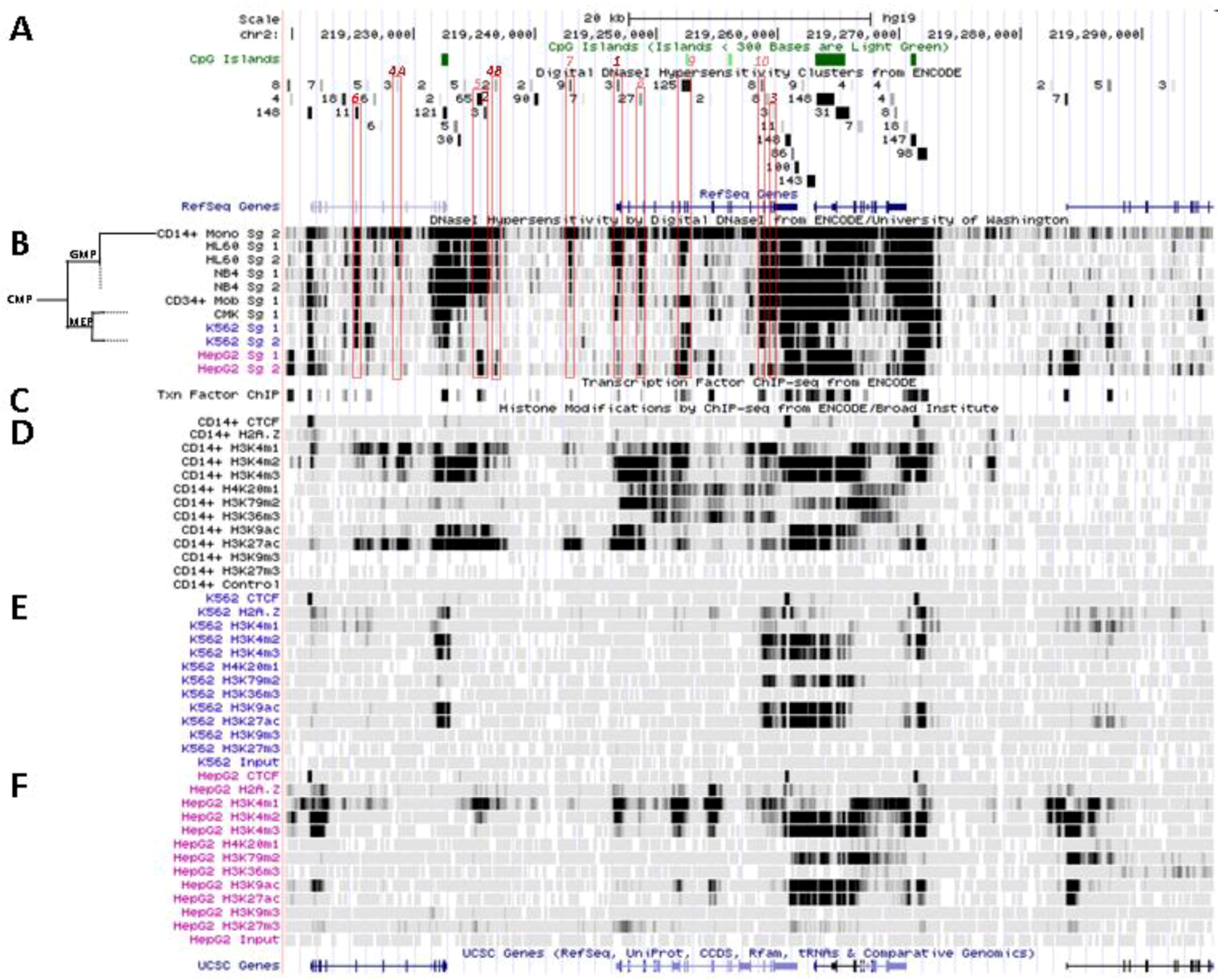
6. Patterns of Histone Marks and Transcription Factor Binding at SLC11A1 Locus
6.1. Summary of Chromatin State Segmentation
6.2. Histone Modifications at SLC11A1 Locus in CD14+ MNs
6.3. SLC11A1 Locus Predicted cis-Acting Determinants
6.3.1. 3' Distal Regulatory Element
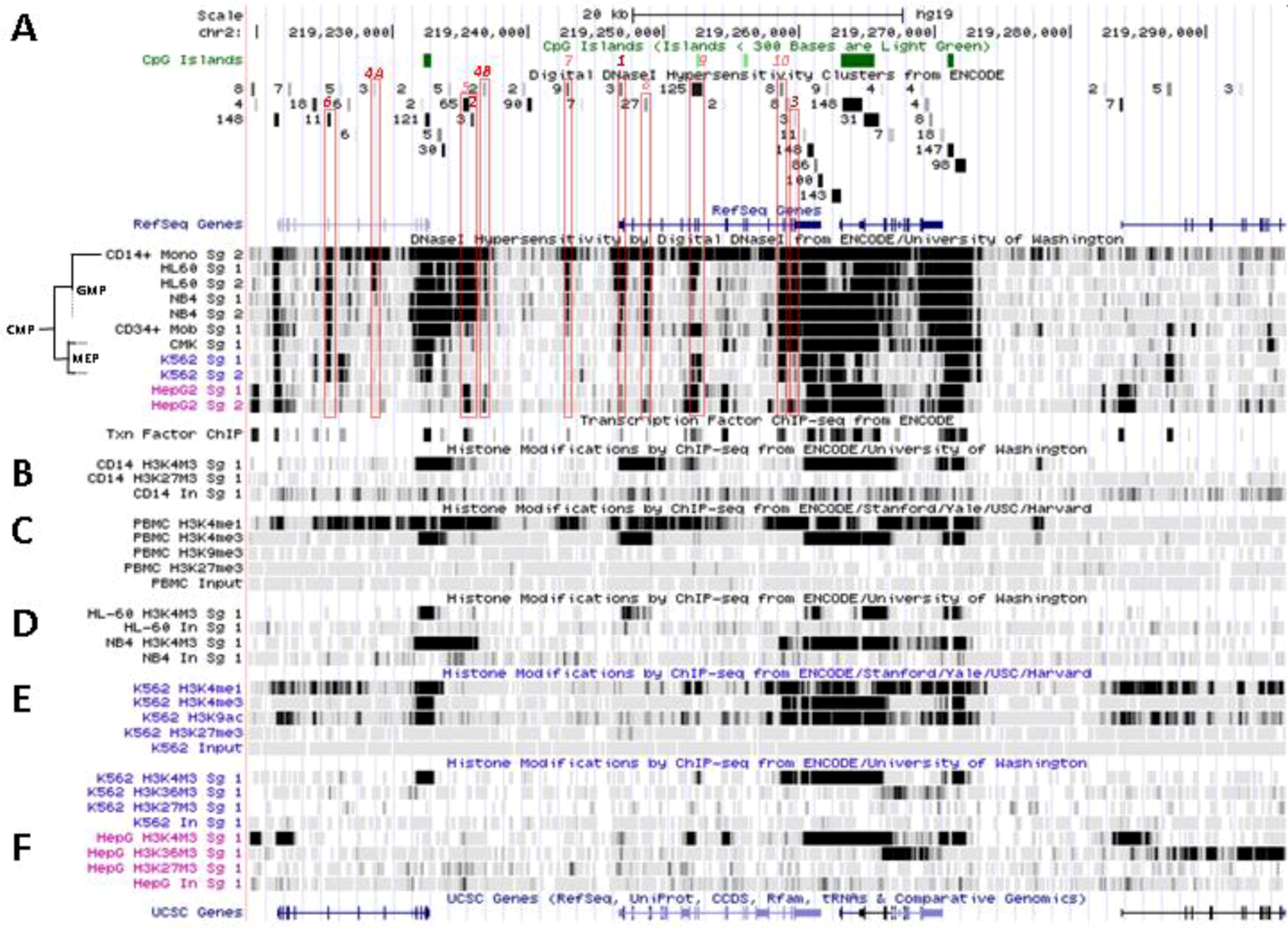
6.3.2. 5' Distal Regulatory Element
6.3.3. Transcription Start Site and Gene Body
6.3.4. Distal Promoter
7. Hypothesis on the Developmental Control of NRAMP1 Expression in Myeloid Cells
8. Conclusion
Acknowledgments
References and Notes
- Vidal, S.M.; Malo, D.; Vogan, K.; Skamene, E.; Gros, P. Natural resistance to infection with intracellular parasites: Isolation of a candidate for Bcg. Cell 1993, 73, 469–485. [Google Scholar] [CrossRef]
- Schlessinger, A.; Matsson, P.; Shima, J.E.; Pieper, U.; Yee, S.W.; Kelly, L.; Apeltsin, L.; Stroud, R.M.; Ferrin, T.E.; Giacomini, K.M.; et al. Comparison of human solute carriers. Protein Sci. 2010, 19, 412–428. [Google Scholar]
- Cellier, M.; Prive, G.; Belouchi, A.; Kwan, T.; Rodrigues, V.; Chia, W.; Gros, P. Nramp defines a family of membrane proteins. Proc. Natl. Acad. Sci. USA 1995, 92, 10089–10093. [Google Scholar]
- Courville, P.; Urbankova, E.; Rensing, C.; Chaloupka, R.; Quick, M.; Cellier, M.F. Solute carrier 11 cations symport requires distinct residues in transmembrane helices 1 and 6. J. Biol. Chem. 2008, 283, 9651–9658. [Google Scholar]
- Cellier, M.F. Nramp: From sequence to structure and mechanism of divalent metal import. Curr. Top. Membr. 2012, 69, 249–293. [Google Scholar] [CrossRef]
- Richer, E.; Courville, P.; Bergevin, I.; Cellier, M.F. Horizontal gene transfer of "prototype" Nramp in bacteria. J. Mol. Evol. 2003, 57, 363–376. [Google Scholar] [CrossRef]
- Cellier, M.F.; Courville, P.; Campion, C. Nramp1 phagocyte intracellular metal withdrawal defense. Microbes Infect. 2007, 9, 1662–1670. [Google Scholar] [CrossRef]
- Peracino, B.; Buracco, S.; Bozzaro, S. The Nramp (Slc11) proteins regulate development, resistance to pathogenic bacteria and iron homeostasis in Dictyostelium discoideum. J. Cell Sci. 2012, 19, 10–1242. [Google Scholar]
- Peracino, B.; Wagner, C.; Balest, A.; Balbo, A.; Pergolizzi, B.; Noegel, A.A.; Steinert, M.; Bozzaro, S. Function and mechanism of action of Dictyostelium Nramp1 (Slc11a1) in bacterial infection. Traffic 2006, 7, 22–38. [Google Scholar] [CrossRef]
- Forbes, J.R.; Gros, P. Divalent-metal transport by NRAMP proteins at the interface of host-pathogen interactions. Trends Microbiol. 2001, 9, 397–403. [Google Scholar] [CrossRef]
- Gruenheid, S.; Pinner, E.; Desjardins, M.; Gros, P. Natural resistance to infection with intracellular pathogens: The Nramp1 protein is recruited to the membrane of the phagosome. J. Exp. Med. 1997, 185, 717–730. [Google Scholar] [CrossRef]
- Gunshin, H.; Fujiwara, Y.; Custodio, A.O.; Direnzo, C.; Robine, S.; Andrews, N.C. Slc11a2 is required for intestinal iron absorption and erythropoiesis but dispensable in placenta and liver. J. Clin. Invest. 2005, 115, 1258–1266. [Google Scholar]
- Gunshin, H.; Mackenzie, B.; Berger, U.V.; Gunshin, Y.; Romero, M.F.; Boron, W.F.; Nussberger, S.; Gollan, J.L.; Hediger, M.A. Cloning and characterization of a mammalian proton-coupled metal-ion transporter. Nature 1997, 388, 482–488. [Google Scholar] [CrossRef]
- McKie, A.T.; Barrow, D.; Latunde-Dada, G.O.; Rolfs, A.; Sager, G.; Mudaly, E.; Mudaly, M.; Richardson, C.; Barlow, D.; Bomford, A.; et al. An iron-regulated ferric reductase associated with the absorption of dietary iron. Science 2001, 291, 1755–1759. [Google Scholar]
- Bardou-Jacquet, E.; Island, M.L.; Jouanolle, A.M.; Detivaud, L.; Fatih, N.; Ropert, M.; Brissot, E.; Mosser, A.; Maisonneuve, H.; Brissot, P.; et al. A novel N491S mutation in the human SLC11A2 gene impairs protein trafficking and in association with the G212V mutation leads to microcytic anemia and liver iron overload. Blood Cells Mol. Dis. 2011, 47, 243–248. [Google Scholar] [CrossRef]
- Czachorowski, M.; Lam-Yuk-Tseung, S.; Cellier, M.; Gros, P. Transmembrane Topology of the Mammalian Slc11a2 Iron Transporter. Biochemistry 2009, 48, 8422–8434. [Google Scholar] [CrossRef]
- Lecointre, G.; Le, G. Classification Phylogenetique du Vivant; Belin: Paris, France, 2006. [Google Scholar]
- Shan, Y.; Gras, R. 43 genes support the lungfish-coelacanth grouping related to the closest living relative of tetrapods with the Bayesian method under the coalescence model. BMC Res. Notes 2011, 4, 49. [Google Scholar] [CrossRef]
- Yoshida, T.; Kumashiro, Y.; Iwata, T.; Ishihara, J.; Umemoto, T.; Shiratsuchi, Y.; Kawashima, N.; Sugiyama, T.; Yamato, M.; Okano, T. Requirement of Integrin beta3 for Iron Transportation during Enamel Formation. J. Dent. Res. 2012, 91, 1154–1159. [Google Scholar] [CrossRef]
- Yanagawa, T.; Itoh, K.; Uwayama, J.; Shibata, Y.; Yamaguchi, A.; Sano, T.; Ishii, T.; Yoshida, H.; Yamamoto, M. Nrf2 deficiency causes tooth decolourization due to iron transport disorder in enamel organ. Genes Cells 2004, 9, 641–651. [Google Scholar] [CrossRef]
- Harada, N.; Kanayama, M.; Maruyama, A.; Yoshida, A.; Tazumi, K.; Hosoya, T.; Mimura, J.; Toki, T.; Maher, J. M.; Yamamoto, M.; Itoh, K. Nrf2 regulates ferroportin 1-mediated iron efflux and counteracts lipopolysaccharide-induced ferroportin 1 mRNA suppression in macrophages. Arch. Biochem. Biophys. 2011, 508, 101–109. [Google Scholar] [CrossRef]
- Motohashi, H.; Kimura, M.; Fujita, R.; Inoue, A.; Pan, X.; Takayama, M.; Katsuoka, F.; Aburatani, H.; Bresnick, E.H.; Yamamoto, M. NF-E2 domination over Nrf2 promotes ROS accumulation and megakaryocytic maturation. Blood 2010, 115, 677–686. [Google Scholar] [CrossRef]
- Merchant, A.A.; Singh, A.; Matsui, W.; Biswal, S. The redox-sensitive transcription factor Nrf2 regulates murine hematopoietic stem cell survival independently of ROS levels. Blood 2011, 118, 6572–6579. [Google Scholar] [CrossRef]
- Rushworth, S.A.; Zaitseva, L.; Murray, M.Y.; Shah, N.M.; Bowles, K.M.; Macewan, D.J. The high Nrf2 expression in human acute myeloid leukemia is driven by NF-kappaB and underlies its chemo-resistance. Blood 2012, 120, 5188–5198. [Google Scholar] [CrossRef]
- Gautier, E.L.; Shay, T.; Miller, J.; Greter, M.; Jakubzick, C.; Ivanov, S.; Helft, J.; Chow, A.; Elpek, K.G.; Gordonov, S.; et al. Gene-expression profiles and transcriptional regulatory pathways that underlie the identity and diversity of mouse tissue macrophages. Nat. Immunol. 2012, 13, 1118–1128. [Google Scholar] [CrossRef]
- Suga, S.; Taki, Y.; Ogawa, M. Fluoride and iron concentrations in the enameloid of lower teleostean fish. J. Dent. Res. 1993, 72, 912–922. [Google Scholar] [CrossRef]
- Van de Peer, Y.; Maere, S.; Meyer, A. The evolutionary significance of ancient genome duplications. Nat. Rev. Genet. 2009, 10, 725–732. [Google Scholar] [CrossRef]
- Ernst, J.D. The immunological life cycle of tuberculosis. Nat. Rev. Immunol. 2012, 12, 581–591. [Google Scholar] [CrossRef]
- Ottenhoff, T.H. The knowns and unknowns of the immunopathogenesis of tuberculosis. Int. J. Tuberc. Lung Dis. 2012, 16, 1424–1432. [Google Scholar] [CrossRef]
- Ottenhoff, T.H.; Kaufmann, S.H. Vaccines against tuberculosis: Where are we and where do we need to go? PLoS Pathog. 2012, 8, e1002607. [Google Scholar] [CrossRef]
- Filipe-Santos, O.; Bustamante, J.; Chapgier, A.; Vogt, G.; de Beaucoudrey, L.; Feinberg, J.; Jouanguy, E.; Boisson-Dupuis, S.; Fieschi, C.; Picard, C.; et al. Inborn errors of IL-12/23- and IFN-gamma-mediated immunity: Molecular, cellular, and clinical features. Semin. Immunol. 2006, 18, 347–361. [Google Scholar] [CrossRef]
- Alcais, A.; Abel, L.; Casanova, J.L. Human genetics of infectious diseases: Between proof of principle and paradigm. J. Clin. Invest. 2009, 119, 2506–2514. [Google Scholar] [CrossRef]
- Flynn, J.L.; Chan, J.; Lin, P.L. Macrophages and control of granulomatous inflammation in tuberculosis. Mucosal. Immunol. 2011, 4, 271–278. [Google Scholar] [CrossRef]
- Moller, M.; Hoal, E.G. Current findings, challenges and novel approaches in human genetic susceptibility to tuberculosis. Tuberculosis (Edinb.) 2010, 90, 71–83. [Google Scholar] [CrossRef]
- Brites, D.; Gagneux, S. Old and new selective pressures on Mycobacterium tuberculosis. Infect. Genet. Evol. 2012, 12, 678–685. [Google Scholar] [CrossRef]
- Azad, A.K.; Sadee, W.; Schlesinger, L.S. Innate immune gene polymorphisms in tuberculosis. Infect. Immun. 2012, 80, 3343–3359. [Google Scholar] [CrossRef]
- Li, X.; Yang, Y.; Zhou, F.; Zhang, Y.; Lu, H.; Jin, Q.; Gao, L. SLC11A1 (NRAMP1) polymorphisms and tuberculosis susceptibility: Updated systematic review and meta-analysis. PLoS One 2011, 6, e15831. [Google Scholar]
- Stein, C.M.; Baker, A.R. Tuberculosis as a complex trait: Impact of genetic epidemiological study design. Mamm. Genome 2011, 22, 91–99. [Google Scholar] [CrossRef]
- Malik, S.; Abel, L.; Tooker, H.; Poon, A.; Simkin, L.; Girard, M.; Adams, G.J.; Starke, J.R.; Smith, K.C.; Graviss, E.A.; et al. Alleles of the NRAMP1 gene are risk factors for pediatric tuberculosis disease. Proc. Natl. Acad. Sci. USA 2005, 102, 12183–12188. [Google Scholar]
- Meilang, Q.; Zhang, Y.; Zhang, J.; Zhao, Y.; Tian, C.; Huang, J.; Fan, H. Polymorphisms in the SLC11A1 gene and tuberculosis risk: A meta-analysis update. Int. J. Tuberc. Lung Dis. 2012, 16, 437–446. [Google Scholar] [CrossRef]
- Liu, J.; Fujiwara, T.M.; Buu, N.T.; Sanchez, F.O.; Cellier, M.; Paradis, A.J.; Frappier, D.; Skamene, E.; Gros, P.; Morgan, K. Identification of polymorphisms and sequence variants in the human homologue of the mouse natural resistance-associated macrophage protein gene. Am. J. Hum. Genet. 1995, 56, 845–853. [Google Scholar]
- Barnes, I.; Duda, A.; Pybus, O.G.; Thomas, M.G. Ancient urbanization predicts genetic resistance to tuberculosis. Evolution 2011, 65, 842–848. [Google Scholar] [CrossRef]
- Donoghue, H.D. Insights gained from palaeomicrobiology into ancient and modern tuberculosis. Clin. Microbiol. Infect. 2011, 17, 821–829. [Google Scholar] [CrossRef]
- Cuellar-Mata, P.; Jabado, N.; Liu, J.; Furuya, W.; Finlay, B.B.; Gros, P.; Grinstein, S. Nramp1 modifies the fusion of Salmonella typhimurium-containing vacuoles with cellular endomembranes in macrophages. J. Biol. Chem. 2002, 277, 2258–2265. [Google Scholar]
- Gallant, C.J.; Malik, S.; Jabado, N.; Cellier, M.; Simkin, L.; Finlay, B.B.; Graviss, E.A.; Gros, P.; Musser, J.M.; Schurr, E. Reduced in vitro functional activity of human NRAMP1 (SLC11A1) allele that predisposes to increased risk of pediatric tuberculosis disease. Genes Immun. 2007, 8, 691–698. [Google Scholar] [CrossRef]
- Waldman, Y.Y.; Tuller, T.; Keinan, A.; Ruppin, E. Selection for translation efficiency on synonymous polymorphisms in recent human evolution. Genome Biol. Evol. 2011, 3, 749–761. [Google Scholar] [CrossRef]
- Searle, S.; Blackwell, J.M. Evidence for a functional repeat polymorphism in the promoter of the human NRAMP1 gene that correlates with autoimmune versus infectious disease susceptibility. J. Med. Genet. 1999, 36, 295–299. [Google Scholar]
- Zaahl, M.G.; Robson, K.J.; Warnich, L.; Kotze, M.J. Expression of the SLC11A1 (NRAMP1) 5'-(GT)n repeat: Opposite effect in the presence of -237C-->T. Blood Cells Mol. Dis. 2004, 33, 45–50. [Google Scholar] [CrossRef]
- Bayele, H.K.; Peyssonnaux, C.; Giatromanolaki, A.; Arrais-Silva, W.W.; Mohamed, H.S.; Collins, H.; Giorgio, S.; Koukourakis, M.; Johnson, R.S.; Blackwell, J.M.; et al. HIF-1 regulates heritable variation and allele expression phenotypes of the macrophage immune response gene SLC11A1 from a Z-DNA forming microsatellite. Blood 2007, 110, 3039–3048. [Google Scholar]
- Taka, S.; Gazouli, M.; Politis, P.K.; Pappa, K.I.; Anagnou, N.P. Transcription factor ATF-3 regulates allele variation phenotypes of the human SLC11A1 gene. Mol. Biol. Rep. 2012, 27. [Google Scholar] [CrossRef]
- Mulholland, N.; Xu, Y.; Sugiyama, H.; Zhao, K. SWI/SNF-mediated chromatin remodeling induces Z-DNA formation on a nucleosome. Cell Biosci. 2012, 2, 3. [Google Scholar] [CrossRef]
- Nairz, M.; Fritsche, G.; Crouch, M.L.; Barton, H.C.; Fang, F.C.; Weiss, G. Slc11a1 limits intracellular growth of Salmonella enterica sv. Typhimurium by promoting macrophage immune effector functions and impairing bacterial iron acquisition. Cell Microbiol. 2009, 11, 1365–1381. [Google Scholar] [CrossRef]
- Rodrigues, P.N.; Gomes, S.S.; Neves, J.V.; Gomes-Pereira, S.; Correia-Neves, M.; Nunes-Alves, C.; Stolte, J.; Sanchez, M.; Appelberg, R.; Muckenthaler, M.U.; et al. Mycobacteria-induced anaemia revisited: A molecular approach reveals the involvement of NRAMP1 and lipocalin-2, but not of hepcidin. Immunobiology 2011, 216, 1127–1134. [Google Scholar] [CrossRef]
- Formica, S.; Roach, T.I.; Blackwell, J.M. Interaction with extracellular matrix proteins influences Lsh/Ity/Bcg (candidate Nramp) gene regulation of macrophage priming/activation for tumour necrosis factor-alpha and nitrite release. Immunology 1994, 82, 42–50. [Google Scholar]
- Fritsche, G.; Dlaska, M.; Barton, H.; Theurl, I.; Garimorth, K.; Weiss, G. Nramp1 functionality increases inducible nitric oxide synthase transcription via stimulation of IFN regulatory factor 1 expression. J. Immunol. 2003, 171, 1994–1998. [Google Scholar]
- Roach, T.I.; Chatterjee, D.; Blackwell, J.M. Induction of early-response genes KC and JE by mycobacterial lipoarabinomannans: Regulation of KC expression in murine macrophages by Lsh/Ity/Bcg (candidate Nramp). Infect. Immun. 1994, 62, 1176–1184. [Google Scholar]
- Gomez, M.A.; Li, S.; Tremblay, M.L.; Olivier, M. NRAMP-1 expression modulates protein-tyrosine phosphatase activity in macrophages: Impact on host cell signaling and functions. J. Biol. Chem. 2007, 282, 36190–36198. [Google Scholar]
- Fritsche, G.; Nairz, M.; Libby, S.J.; Fang, F.C.; Weiss, G. Slc11a1 (Nramp1) impairs growth of Salmonella enterica serovar typhimurium in macrophages via stimulation of lipocalin-2 expression. J. Leukoc. Biol. 2012, 92, 353–359. [Google Scholar] [CrossRef]
- Fritsche, G.; Nairz, M.; Werner, E.R.; Barton, H.C.; Weiss, G. Nramp1-functionality increases iNOS expression via repression of IL-10 formation. Eur. J. Immunol. 2008, 38, 3060–3067. [Google Scholar] [CrossRef]
- Awomoyi, A.A.; Marchant, A.; Howson, J.M.; McAdam, K.P.; Blackwell, J.M.; Newport, M.J. Interleukin-10, polymorphism in SLC11A1 (formerly NRAMP1), and susceptibility to tuberculosis. J. Infect. Dis. 2002, 186, 1808–1814. [Google Scholar] [CrossRef]
- Weinberg, E.D. Iron availability and infection. Biochim. Biophys. Acta 2009, 1790, 600–605. [Google Scholar] [CrossRef]
- Oexle, H.; Kaser, A.; Most, J.; Bellmann-Weiler, R.; Werner, E.R.; Werner-Felmayer, G.; Weiss, G. Pathways for the regulation of interferon-gamma-inducible genes by iron in human monocytic cells. J. Leukoc. Biol. 2003, 74, 287–294. [Google Scholar] [CrossRef]
- Schaible, U.E.; Kaufmann, S.H. Iron and microbial infection. Nat. Rev. Microbiol. 2004, 2, 946–953. [Google Scholar] [CrossRef]
- Collins, H.L. Withholding iron as a cellular defence mechanism—Friend or foe? Eur. J. Immunol. 2008, 38, 1803–1806. [Google Scholar] [CrossRef]
- Nairz, M.; Schroll, A.; Sonnweber, T.; Weiss, G. The struggle for iron—A metal at the host-pathogen interface. Cell Microbiol. 2010, 12, 1691–1702. [Google Scholar] [CrossRef]
- O’Brien, B.A.; Archer, N.S.; Simpson, A.M.; Torpy, F.R.; Nassif, N.T. Association of SLC11A1 promoter polymorphisms with the incidence of autoimmune and inflammatory diseases: A meta-analysis. J. Autoimmun. 2008, 31, 42–51. [Google Scholar] [CrossRef]
- Shay, J.E.; Celeste, S.M. Hypoxia-inducible factors: Crosstalk between inflammation and metabolism. Semin. Cell Dev. Biol. 2012, 23, 389–394. [Google Scholar] [CrossRef]
- Nizet, V.; Johnson, R.S. Interdependence of hypoxic and innate immune responses. Nat. Rev. Immunol. 2009, 9, 609–617. [Google Scholar] [CrossRef]
- Aragones, J.; Elorza, A.; Acosta-Iborra, B.; Landazuri, M.O. Myeloid hypoxia-inducible factors in inflammatory diseases. Crit. Rev. Immunol. 2011, 31, 1–13. [Google Scholar] [CrossRef]
- Cellier, M.; Shustik, C.; Dalton, W.; Rich, E.; Hu, J.; Malo, D.; Schurr, E.; Gros, P. Expression of the human NRAMP1 gene in professional primary phagocytes: Studies in blood cells and in HL-60 promyelocytic leukemia. J. Leukoc. Biol. 1997, 61, 96–105. [Google Scholar]
- Roig, E.A.; Richer, E.; Canonne-Hergaux, F.; Gros, P.; Cellier, M.F. Regulation of NRAMP1 gene expression by 1alpha,25-dihydroxy-vitamin D(3) in HL-60 phagocytes. J. Leukoc. Biol. 2002, 71, 890–904. [Google Scholar]
- Canonne-Hergaux, F.; Calafat, J.; Richer, E.; Cellier, M.; Grinstein, S.; Borregaard, N.; Gros, P. Expression and subcellular localization of NRAMP1 in human neutrophil granules. Blood 2002, 100, 268–275. [Google Scholar] [CrossRef]
- Soe-Lin, S.; Apte, S.S.; Andriopoulos, B., Jr.; Andrews, M.C.; Schranzhofer, M.; Kahawita, T.; Garcia-Santos, D.; Ponka, P. Nramp1 promotes efficient macrophage recycling of iron following erythrophagocytosis in vivo. Proc. Natl. Acad. Sci. USA 2009, 106, 5960–5965. [Google Scholar]
- Delaby, C.; Rondeau, C.; Pouzet, C.; Willemetz, A.; Pilard, N.; Desjardins, M.; Canonne-Hergaux, F. Subcellular localization of iron and heme metabolism related proteins at early stages of erythrophagocytosis. PLoS One 2012, 7, e42199. [Google Scholar]
- Cellier, M.F. Nutritional immunity: Homology modeling of Nramp metal import. Adv. Exp. Med. Biol. 2012, 946, 335–351. [Google Scholar] [CrossRef]
- Novershtern, N.; Subramanian, A.; Lawton, L.N.; Mak, R.H.; Haining, W.N.; McConkey, M.E.; Habib, N.; Yosef, N.; Chang, C.Y.; Shay, T.; et al. Densely interconnected transcriptional circuits control cell states in human hematopoiesis. Cell 2011, 144, 296–309. [Google Scholar] [CrossRef]
- Cedar, H.; Bergman, Y. Epigenetics of haematopoietic cell development. Nat. Rev. Immunol. 2011, 11, 478–488. [Google Scholar] [CrossRef]
- Fiedler, K.; Brunner, C. The role of transcription factors in the guidance of granulopoiesis. Am. J. Blood Res. 2012, 2, 57–65. [Google Scholar]
- Dorshkind, K. Not a split decision for human hematopoiesis. Nat. Immunol. 2010, 11, 569–570. [Google Scholar] [CrossRef]
- Pham, T.H.; Benner, C.; Lichtinger, M.; Schwarzfischer, L.; Hu, Y.; Andreesen, R.; Chen, W.; Rehli, M. Dynamic epigenetic enhancer signatures reveal key transcription factors associated with monocytic differentiation states. Blood 2012, 119, e161–e171. [Google Scholar] [CrossRef]
- Nowak, D.; Stewart, D.; Koeffler, H.P. Differentiation therapy of leukemia: 3 Decades of development. Blood 2009, 113, 3655–3665. [Google Scholar] [CrossRef]
- Schrumpf, J.A.; van Sterkenburg, M.A.; Verhoosel, R.M.; Zuyderduyn, S.; Hiemstra, P.S. Interleukin 13 Exposure Enhances Vitamin D-Mediated Expression of the Human Cathelicidin Antimicrobial Peptide 18/LL-37 in Bronchial Epithelial Cells. Infect. Immun. 2012, 80, 4485–4494. [Google Scholar] [CrossRef]
- Kim, R.H.; Li, J.J.; Ogata, Y.; Yamauchi, M.; Freedman, L.P.; Sodek, J. Identification of a vitamin D3-response element that overlaps a unique inverted TATA box in the rat bone sialoprotein gene. Biochem. J. 1996, 318, 219–226. [Google Scholar]
- Shaffer, P.L.; Gewirth, D.T. Structural basis of VDR-DNA interactions on direct repeat response elements. EMBO J. 2002, 21, 2242–2252. [Google Scholar] [CrossRef]
- Rosen, C.J.; Adams, J.S.; Bikle, D.D.; Black, D.M.; Demay, M.B.; Manson, J.E.; Murad, M.H.; Kovacs, C.S. The nonskeletal effects of vitamin D: An Endocrine Society scientific statement. Endocr. Rev. 2012, 33, 456–492. [Google Scholar] [CrossRef]
- Gombart, A.F.; O'Kelly, J.; Saito, T.; Koeffler, H.P. Regulation of the CAMP gene by 1,25(OH)2D3 in various tissues. J. Steroid Biochem. Mol. Biol. 2007, 103, 552–557. [Google Scholar] [CrossRef]
- Carlberg, C.; Dunlop, T.W. The impact of chromatin organization of vitamin D target genes. Anticancer Res. 2006, 26, 2637–2645. [Google Scholar]
- Lips, P. Vitamin D physiology. Prog. Biophys. Mol. Biol. 2006, 92, 4–8. [Google Scholar] [CrossRef]
- Mantovani, A.; Sica, A.; Locati, M. New vistas on macrophage differentiation and activation. Eur. J. Immunol. 2007, 37, 14–16. [Google Scholar] [CrossRef]
- Mantovani, A.; Sica, A.; Locati, M. Macrophage polarization comes of age. Immunity 2005, 23, 344–346. [Google Scholar] [CrossRef]
- Martineau, A.R.; Wilkinson, K.A.; Newton, S.M.; Floto, R.A.; Norman, A.W.; Skolimowska, K.; Davidson, R.N.; Sorensen, O.E.; Kampmann, B.; Griffiths, C.J.; et al. IFN-gamma- and TNF-independent vitamin D-inducible human suppression of mycobacteria: The role of cathelicidin LL-37. J. Immunol. 2007, 178, 7190–7198. [Google Scholar]
- Imazeki, I.; Matsuzaki, J.; Tsuji, K.; Nishimura, T. Immunomodulating effect of vitamin D3 derivatives on type-1 cellular immunity. Biomed. Res. 2006, 27, 1–9. [Google Scholar] [CrossRef]
- White, J.H. Vitamin D signaling, infectious diseases, and regulation of innate immunity. Infect. Immun. 2008, 76, 3837–3843. [Google Scholar] [CrossRef]
- Ralph, A.P.; Kelly, P.M.; Anstey, N.M. L-arginine and vitamin D: Novel adjunctive immunotherapies in tuberculosis. Trends Microbiol. 2008, 16, 336–344. [Google Scholar] [CrossRef]
- Hughes, P.J.; Steinmeyer, A.; Chandraratna, R.A.; Brown, G. 1alpha,25-dihydroxyvitamin D3 stimulates steroid sulphatase activity in HL60 and NB4 acute myeloid leukaemia cell lines by different receptor-mediated mechanisms. J. Cell Biochem. 2005, 94, 1175–1189. [Google Scholar] [CrossRef]
- Guillot, X.; Semerano, L.; Saidenberg-Kermanac'h, N.; Falgarone, G.; Boissier, M.C. Vitamin D and inflammation. Joint Bone Spine 2010, 77, 552–557. [Google Scholar] [CrossRef]
- Schauber, J.; Oda, Y.; Buchau, A.S.; Yun, Q.C.; Steinmeyer, A.; Zugel, U.; Bikle, D.D.; Gallo, R.L. Histone acetylation in keratinocytes enables control of the expression of cathelicidin and CD14 by 1,25-dihydroxyvitamin D3. J. Invest. Dermatol. 2008, 128, 816–824. [Google Scholar] [CrossRef]
- Liu, P.T.; Stenger, S.; Tang, D.H.; Modlin, R.L. Cutting edge: Vitamin D-mediated human antimicrobial activity against Mycobacterium tuberculosis is dependent on the induction of cathelicidin. J. Immunol. 2007, 179, 2060–2063. [Google Scholar]
- Ding, C.; Gao, D.; Wilding, J.; Trayhurn, P.; Bing, C. Vitamin D signalling in adipose tissue. Br. J. Nutr. 2012, 108, 1915–1923. [Google Scholar] [CrossRef]
- Nestle, F.O.; di, M.P.; Qin, J.Z.; Nickoloff, B.J. Skin immune sentinels in health and disease. Nat. Rev. Immunol. 2009, 9, 679–691. [Google Scholar]
- Miller, J.; Gallo, R.L. Vitamin D and innate immunity. Dermatol. Ther. 2010, 23, 13–22. [Google Scholar] [CrossRef]
- Hoyer-Hansen, M.; Nordbrandt, S.P.; Jaattela, M. Autophagy as a basis for the health-promoting effects of vitamin D. Trends Mol. Med. 2010, 16, 295–302. [Google Scholar] [CrossRef]
- Hewison, M. Antibacterial effects of vitamin D. Nat. Rev. Endocrinol. 2011, 7, 337–345. [Google Scholar] [CrossRef]
- Campbell, G.R.; Spector, S.A. Vitamin D inhibits human immunodeficiency virus type 1 and Mycobacterium tuberculosis infection in macrophages through the induction of autophagy. PLoS Pathog. 2012, 8, e1002689. [Google Scholar] [CrossRef]
- Fabri, M.; Stenger, S.; Shin, D.M.; Yuk, J.M.; Liu, P.T.; Realegeno, S.; Lee, H.M.; Krutzik, S.R.; Schenk, M.; Sieling, P.A.; et al. Vitamin D is required for IFN-gamma-mediated antimicrobial activity of human macrophages. Sci. Transl. Med. 2011, 3, 104–104ra102. [Google Scholar] [CrossRef]
- Greer, E.L.; Shi, Y. Histone methylation: a dynamic mark in health, disease and inheritance. Nat. Rev. Genet. 2012, 13, 343–357. [Google Scholar] [CrossRef]
- Valen, E.; Sandelin, A. Genomic and chromatin signals underlying transcription start-site selection. Trends Genet. 2011, 27, 475–485. [Google Scholar] [CrossRef]
- Blackwell, J.M.; Barton, C.H.; White, J.K.; Searle, S.; Baker, A.M.; Williams, H.; Shaw, M.A. Genomic organization and sequence of the human NRAMP gene: Identification and mapping of a promoter region polymorphism. Mol. Med. 1995, 1, 194–205. [Google Scholar]
- Richer, E.; Campion, C.G.; Dabbas, B.; White, J.H.; Cellier, M.F. Transcription factors Sp1 and C/EBP regulate NRAMP1 gene expression. FEBS J. 2008, 275, 5074–5089. [Google Scholar] [CrossRef]
- Lawrence, T.; Natoli, G. Transcriptional regulation of macrophage polarization: Enabling diversity with identity. Nat. Rev. Immunol. 2011, 11, 750–761. [Google Scholar] [CrossRef]
- Huber, R.; Pietsch, D.; Panterodt, T.; Brand, K. Regulation of C/EBPbeta and resulting functions in cells of the monocytic lineage. Cell Signal. 2012, 24, 1287–1296. [Google Scholar] [CrossRef]
- Spitz, F.; Furlong, E.E. Transcription factors: From enhancer binding to developmental control. Nat. Rev. Genet. 2012, 13, 613–626. [Google Scholar] [CrossRef]
- Feng, R.; Desbordes, S.C.; Xie, H.; Tillo, E.S.; Pixley, F.; Stanley, E.R.; Graf, T. PU.1 and C/EBPalpha/beta convert fibroblasts into macrophage-like cells. Proc. Natl. Acad. Sci. USA 2008, 105, 6057–6062. [Google Scholar]
- Corna, G.; Campana, L.; Pignatti, E.; Castiglioni, A.; Tagliafico, E.; Bosurgi, L.; Campanella, A.; Brunelli, S.; Manfredi, A.A.; Apostoli, P.; et al. Polarization dictates iron handling by inflammatory and alternatively activated macrophages. Haematologica 2010, 95, 1814–1822. [Google Scholar] [CrossRef]
- Recalcati, S.; Locati, M.; Marini, A.; Santambrogio, P.; Zaninotto, F.; de, P.M.; Zammataro, L.; Girelli, D.; Cairo, G. Differential regulation of iron homeostasis during human macrophage polarized activation. Eur. J. Immunol. 2010, 40, 824–835. [Google Scholar] [CrossRef]
- Fernandes de Abreu, D.A.; Eyles, D.; Feron, F. Vitamin D, a neuro-immunomodulator: implications for neurodegenerative and autoimmune diseases. Psychoneuroendocrinology 2009, 34, S265–S277. [Google Scholar] [CrossRef]
- Gonzalez, N.; Castrillo, A. Liver X receptors as regulators of macrophage inflammatory and metabolic pathways. Biochim. Biophys. Acta 2011, 1812, 982–994. [Google Scholar] [CrossRef]
- Wahli, W.; Michalik, L. PPARs at the crossroads of lipid signaling and inflammation. Trends Endocrinol. Metab. 2012, 23, 351–363. [Google Scholar] [CrossRef]
- Nagy, L.; Szanto, A.; Szatmari, I.; Szeles, L. Nuclear hormone receptors enable macrophages and dendritic cells to sense their lipid environment and shape their immune response. Physiol. Rev. 2012, 92, 739–789. [Google Scholar] [CrossRef]
- Resendes, K.K.; Rosmarin, A.G. Sp1 control of gene expression in myeloid cells. Crit. Rev. Eukaryot. Gene Exp. 2004, 14, 171–181. [Google Scholar] [CrossRef]
- Chen, H.; Pahl, H.L.; Scheibe, R.J.; Zhang, D.; Tenen, D.G. The Sp1 transcription factor binds the CD11b promoter specifically in myeloid cells in vivo and is essential for myeloid-specific promoter activity. J. Biol. Chem. 1993, 268, 8230–8239. [Google Scholar]
- Zhang, D.E.; Hetherington, C.J.; Gonzalez, D.A.; Chen, H.M.; Tenen, D.G. Regulation of CD14 expression during monocytic differentiation induced with 1 alpha,25-dihydroxyvitamin D3. J. Immunol. 1994, 153, 3276–3284. [Google Scholar]
- Khanna-Gupta, A.; Zibello, T.; Simkevich, C.; Rosmarin, A.G.; Berliner, N. Sp1 and C/EBP are necessary to activate the lactoferrin gene promoter during myeloid differentiation. Blood 2000, 95, 3734–3741. [Google Scholar]
- Koga, T.; Suico, M.A.; Nakamura, H.; Taura, M.; Lu, Z.; Shuto, T.; Okiyoneda, T.; Kai, H. Sp1-dependent regulation of Myeloid Elf-1 like factor in human epithelial cells. FEBS Lett. 2005, 579, 2811–2816. [Google Scholar] [CrossRef]
- Ceccarelli, V.; Racanicchi, S.; Martelli, M.P.; Nocentini, G.; Fettucciari, K.; Riccardi, C.; Marconi, P.; di, N.P.; Grignani, F.; Binaglia, L.; Vecchini, A. Eicosapentaenoic acid demethylates a single CpG that mediates expression of tumor suppressor CCAAT/enhancer-binding protein delta in U937 leukemia cells. J. Biol. Chem. 2011, 286, 27092–27102. [Google Scholar]
- Ajore, R.; Kumar, P.; Dhanda, R.S.; Gullberg, U.; Olsson, I. The leukemia associated nuclear corepressor ETO homologue genes MTG16 and MTGR1 are regulated differently in hematopoietic cells. BMC. Mol. Biol. 2012, 13, 11. [Google Scholar] [CrossRef]
- Xu, Y.Z.; di, M.S.; Gallouzi, I.; Rola-Pleszczynski, M.; Radzioch, D. RNA-binding protein HuR is required for stabilization of SLC11A1 mRNA and SLC11A1 protein expression. Mol. Cell Biol. 2005, 25, 8139–8149. [Google Scholar] [CrossRef]
- Yiakouvaki, A.; Dimitriou, M.; Karakasiliotis, I.; Eftychi, C.; Theocharis, S.; Kontoyiannis, D.L. Myeloid cell expression of the RNA-binding protein HuR protects mice from pathologic inflammation and colorectal carcinogenesis. J. Clin. Invest. 2012, 122, 48–61. [Google Scholar] [CrossRef]
- Xu, Y.Z.; Thuraisingam, T.; Marino, R.; Radzioch, D. Recruitment of SWI/SNF complex is required for transcriptional activation of the SLC11A1 gene during macrophage differentiation of HL-60 cells. J. Biol. Chem. 2011, 286, 12839–12849. [Google Scholar] [CrossRef]
- Xu, Y.Z.; Thuraisingam, T.; Morais, D.A.; Rola-Pleszczynski, M.; Radzioch, D. Nuclear translocation of beta-actin is involved in transcriptional regulation during macrophage differentiation of HL-60 cells. Mol. Biol. Cell 2010, 21, 811–820. [Google Scholar] [CrossRef]
- Gold, E.S.; Ramsey, S.A.; Sartain, M.J.; Selinummi, J.; Podolsky, I.; Rodriguez, D.J.; Moritz, R.L.; Aderem, A. ATF3 protects against atherosclerosis by suppressing 25-hydroxycholesterol-induced lipid body formation. J. Exp. Med. 2012, 209, 807–817. [Google Scholar] [CrossRef]
- Thompson, M.R.; Xu, D.; Williams, B.R. ATF3 transcription factor and its emerging roles in immunity and cancer. J. Mol. Med. (Berl.) 2009, 87, 1053–1060. [Google Scholar] [CrossRef]
- De Lanerolle, P.; Serebryannyy, L. Nuclear actin and myosins: Life without filaments. Nat. Cell Biol. 2011, 13, 1282–1288. [Google Scholar] [CrossRef]
- Vartiainen, M.K.; Huet, G.; Skarp, K.P. Nuclear actin levels as an important transcriptional switch. Transcription 2012, 3, 226–230. [Google Scholar] [CrossRef]
- Miyamoto, K.; Gurdon, J.B. Nuclear actin and transcriptional activation. Commun. Integr. Biol. 2011, 4, 582–583. [Google Scholar]
- Smale, S.T. Transcriptional regulation in the innate immune system. Curr. Opin. Immunol. 2012, 24, 51–57. [Google Scholar] [CrossRef]
- Janardhan, H.P. The HIF-1 alpha-C/EBP alpha axis. Sci. Signal. 2008, 1, jc2. [Google Scholar] [CrossRef]
- Zhao, X.Y.; Zhao, K.W.; Jiang, Y.; Zhao, M.; Chen, G.Q. Synergistic induction of galectin-1 by CCAAT/enhancer binding protein alpha and hypoxia-inducible factor 1alpha and its role in differentiation of acute myeloid leukemic cells. J. Biol. Chem. 2011, 286, 36808–36819. [Google Scholar]
- Heintzman, N.D.; Ren, B. Finding distal regulatory elements in the human genome. Curr. Opin. Genet. Dev. 2009, 19, 541–549. [Google Scholar] [CrossRef]
- Smale, S.T. Seq-ing LPS-induced enhancers. Immunity 2010, 32, 296–298. [Google Scholar] [CrossRef]
- Malik, S.; Roeder, R.G. The metazoan Mediator co-activator complex as an integrative hub for transcriptional regulation. Nat. Rev. Genet. 2010, 11, 761–772. [Google Scholar] [CrossRef]
- Weake, V.M.; Workman, J.L. Inducible gene expression: Diverse regulatory mechanisms. Nat. Rev. Genet. 2010, 11, 426–437. [Google Scholar] [CrossRef]
- Krysinska, H.; Hoogenkamp, M.; Ingram, R.; Wilson, N.; Tagoh, H.; Laslo, P.; Singh, H.; Bonifer, C. A two-step, PU..1-dependent mechanism for developmentally regulated chromatin remodeling and transcription of the c-fms gene. Mol. Cell Biol. 2007, 27, 878–887. [Google Scholar] [CrossRef]
- Bonifer, C. Epigenetic plasticity of hematopoietic cells. Cell Cycle 2005, 4, 211–214. [Google Scholar]
- Gozzini, A.; Rovida, E.; Dello, S.P.; Galimberti, S.; Santini, V. Butyrates, as a single drug, induce histone acetylation and granulocytic maturation: Possible selectivity on core binding factor-acute myeloid leukemia blasts. Cancer Res. 2003, 63, 8955–8961. [Google Scholar]
- Danilenko, M.; Wang, Q.; Wang, X.; Levy, J.; Sharoni, Y.; Studzinski, G.P. Carnosic acid potentiates the antioxidant and prodifferentiation effects of 1alpha,25-dihydroxyvitamin D3 in leukemia cells but does not promote elevation of basal levels of intracellular calcium. Cancer Res. 2003, 63, 1325–1332. [Google Scholar]
- Mai, A.; Massa, S.; Rotili, D.; Cerbara, I.; Valente, S.; Pezzi, R.; Simeoni, S.; Ragno, R. Histone deacetylation in epigenetics: An attractive target for anticancer therapy. Med. Res. Rev. 2005, 25, 261–309. [Google Scholar] [CrossRef]
- Savickiene, J.; Treigyte, G.; Borutinskaite, V.; Navakauskiene, R.; Magnusson, K.E. The histone deacetylase inhibitor FK228 distinctly sensitizes the human leukemia cells to retinoic acid-induced differentiation. Ann. N. Y. Acad. Sci. 2006, 1091, 368–384. [Google Scholar] [CrossRef]
- Kuhn, R.M.; Haussler, D.; Kent, W.J. The UCSC genome browser and associated tools. Brief. Bioinform. 2012, 20. [Google Scholar] [CrossRef]
- Dunham, I.; Kundaje, A.; Aldred, S.F.; Collins, P.J.; Davis, C.A.; Doyle, F.; Epstein, C.B.; Frietze, S.; Harrow, J.; Kaul, R.; et al. An integrated encyclopedia of DNA elements in the human genome. Nature 2012, 489, 57–74. [Google Scholar]
- Ecker, J.R.; Bickmore, W.A.; Barroso, I.; Pritchard, J.K.; Gilad, Y.; Segal, E. Genomics: ENCODE explained. Nature 2012, 489, 52–55. [Google Scholar]
- Thurman, R.E.; Rynes, E.; Humbert, R.; Vierstra, J.; Maurano, M.T.; Haugen, E.; Sheffield, N.C.; Stergachis, A.B.; Wang, H.; Vernot, B.; et al. The accessible chromatin landscape of the human genome. Nature 2012, 489, 75–82. [Google Scholar]
- Sabo, P.J.; Hawrylycz, M.; Wallace, J.C.; Humbert, R.; Yu, M.; Shafer, A.; Kawamoto, J.; Hall, R.; Mack, J.; Dorschner, M.O.; McArthur, M.; Stamatoyannopoulos, J.A. Discovery of functional noncoding elements by digital analysis of chromatin structure. Proc. Natl. Acad. Sci. USA 2004, 101, 16837–16842. [Google Scholar]
- Shu, W.; Chen, H.; Bo, X.; Wang, S. Genome-wide analysis of the relationships between DNaseI HS, histone modifications and gene expression reveals distinct modes of chromatin domains. Nucleic Acids Res. 2011, 39, 7428–7443. [Google Scholar] [CrossRef]
- Xiao, S.; Xie, D.; Cao, X.; Yu, P.; Xing, X.; Chen, C.C.; Musselman, M.; Xie, M.; West, F.D.; Lewin, H.A.; et al. Comparative epigenomic annotation of regulatory DNA. Cell 2012, 149, 1381–1392. [Google Scholar] [CrossRef]
- Calero-Nieto, F.J.; Wood, A.D.; Wilson, N.K.; Kinston, S.; Landry, J.R.; Gottgens, B. Transcriptional regulation of Elf-1: Locus-wide analysis reveals four distinct promoters, a tissue-specific enhancer, control by PU.1 and the importance of Elf-1 downregulation for erythroid maturation. Nucleic Acids Res. 2010, 38, 6363–6374. [Google Scholar] [CrossRef]
- Takahashi, K.; Hayashi, N.; Shimokawa, T.; Umehara, N.; Kaminogawa, S.; Ra, C. Cooperative regulation of Fc receptor gamma-chain gene expression by multiple transcription factors, including Sp1, GABP, and Elf-1. J. Biol. Chem. 2008, 283, 15134–15141. [Google Scholar] [CrossRef]
- Gargiulo, G.; Levy, S.; Bucci, G.; Romanenghi, M.; Fornasari, L.; Beeson, K.Y.; Goldberg, S.M.; Cesaroni, M.; Ballarini, M.; Santoro, F.; et al. NA-Seq: A discovery tool for the analysis of chromatin structure and dynamics during differentiation. Dev. Cell 2009, 16, 466–481. [Google Scholar] [CrossRef]
- Akira, S. IL-6-regulated transcription factors. Int. J. Biochem. Cell Biol. 1997, 29, 1401–1418. [Google Scholar] [CrossRef]
- Akira, S.; Isshiki, H.; Sugita, T.; Tanabe, O.; Kinoshita, S.; Nishio, Y.; Nakajima, T.; Hirano, T.; Kishimoto, T. A nuclear factor for IL-6 expression (NF-IL6) is a member of a C/EBP family. EMBO J. 1990, 9, 1897–1906. [Google Scholar]
- Alonzi, T.; Maritano, D.; Gorgoni, B.; Rizzuto, G.; Libert, C.; Poli, V. Essential role of STAT3 in the control of the acute-phase response as revealed by inducible gene inactivation [correction of activation] in the liver. Mol. Cell Biol. 2001, 21, 1621–1632. [Google Scholar] [CrossRef]
- Verga Falzacappa, M.V.; Vujic, S.M.; Kessler, R.; Stolte, J.; Hentze, M.W.; Muckenthaler, M.U. STAT3 mediates hepatic hepcidin expression and its inflammatory stimulation. Blood 2007, 109, 353–358. [Google Scholar] [CrossRef]
- Eychene, A.; Rocques, N.; Pouponnot, C. A new MAFia in cancer. Nat. Rev. Cancer 2008, 8, 683–693. [Google Scholar] [CrossRef]
- Kelly, L.M.; Englmeier, U.; Lafon, I.; Sieweke, M.H.; Graf, T. MafB is an inducer of monocytic differentiation. EMBO J. 2000, 19, 1987–1997. [Google Scholar] [CrossRef]
- Aziz, A.; Soucie, E.; Sarrazin, S.; Sieweke, M.H. MafB/c-Maf deficiency enables self-renewal of differentiated functional macrophages. Science 2009, 326, 867–871. [Google Scholar] [CrossRef]
- Murray, P.J.; Wynn, T.A. Protective and pathogenic functions of macrophage subsets. Nat. Rev. Immunol. 2011, 11, 723–737. [Google Scholar] [CrossRef]
- Kannan, M.B.; Solovieva, V.; Blank, V. The small MAF transcription factors MAFF, MAFG and MAFK: Current knowledge and perspectives. Biochim. Biophys. Acta 2012, 1823, 1841–1846. [Google Scholar] [CrossRef]
- Dohi, Y.; Alam, J.; Yoshizumi, M.; Sun, J.; Igarashi, K. Heme oxygenase-1 gene enhancer manifests silencing activity in a chromatin environment prior to oxidative stress. Antioxid. Redox. Signal. 2006, 8, 60–67. [Google Scholar] [CrossRef]
- Ohta, K.; Ohigashi, M.; Naganawa, A.; Ikeda, H.; Sakai, M.; Nishikawa, J.; Imagawa, M.; Osada, S.; Nishihara, T. Histone acetyltransferase MOZ acts as a co-activator of Nrf2-MafK and induces tumour marker gene expression during hepatocarcinogenesis. Biochem. J. 2007, 402, 559–566. [Google Scholar] [CrossRef]
- Jyrkkanen, H.K.; Kuosmanen, S.; Heinaniemi, M.; Laitinen, H.; Kansanen, E.; Mella-Aho, E.; Leinonen, H.; Yla-Herttuala, S.; Levonen, A.L. Novel insights into the regulation of antioxidant-response-element-mediated gene expression by electrophiles: Induction of the transcriptional repressor BACH1 by Nrf2. Biochem. J. 2011, 440, 167–174. [Google Scholar] [CrossRef]
- Soares, M.P.; Marguti, I.; Cunha, A.; Larsen, R. Immunoregulatory effects of HO-1: How does it work? Curr. Opin. Pharmacol. 2009, 9, 482–489. [Google Scholar] [CrossRef]
- Egan, B.S.; Lane, K.B.; Shepherd, V.L. PU.1 and USF are required for macrophage-specific mannose receptor promoter activity. J. Biol. Chem. 1999, 274, 9098–9107. [Google Scholar] [CrossRef]
- Kamimura, M.; Viedt, C.; Dalpke, A.; Rosenfeld, M.E.; Mackman, N.; Cohen, D.M.; Blessing, E.; Preusch, M.; Weber, C.M.; Kreuzer, J.; et al. Interleukin-10 suppresses tissue factor expression in lipopolysaccharide-stimulated macrophages via inhibition of Egr-1 and a serum response element/MEK-ERK1/2 pathway. Circ. Res. 2005, 97, 305–313. [Google Scholar] [CrossRef]
- Natoli, G.; Ghisletti, S.; Barozzi, I. The genomic landscapes of inflammation. Genes Dev. 2011, 25, 101–106. [Google Scholar] [CrossRef]
- Bell, A.C.; West, A.G.; Felsenfeld, G. The protein CTCF is required for the enhancer blocking activity of vertebrate insulators. Cell 1999, 98, 387–396. [Google Scholar] [CrossRef]
- Thompson, C.B.; Wang, C.Y.; Ho, I.C.; Bohjanen, P.R.; Petryniak, B.; June, C.H.; Miesfeldt, S.; Zhang, L.; Nabel, G.J.; Karpinski, B. cis-acting sequences required for inducible interleukin-2 enhancer function bind a novel Ets-related protein, Elf-1. Mol. Cell Biol. 1992, 12, 1043–1053. [Google Scholar]
- Croker, B.A.; Mielke, L.A.; Wormald, S.; Metcalf, D.; Kiu, H.; Alexander, W.S.; Hilton, D.J.; Roberts, A.W. Socs3 maintains the specificity of biological responses to cytokine signals during granulocyte and macrophage differentiation. Exp. Hematol. 2008, 36, 786–798. [Google Scholar] [CrossRef]
- Fratkin, E.; Bercovici, S.; Stephan, D.A. The implications of ENCODE for diagnostics. Nat. Biotechnol. 2012, 30, 1064–1065. [Google Scholar] [CrossRef]
- Hubel, K.; Engert, A. Granulocyte transfusion therapy for treatment of infections after cytotoxic chemotherapy. Onkologie 2003, 26, 73–79. [Google Scholar] [CrossRef]
- Elmaagacli, A.H.; Koldehoff, M.; Zakrzewski, J.L.; Steckel, N.K.; Ottinger, H.; Beelen, D.W. Growth factor-independent 1B gene (GFI1B) is overexpressed in erythropoietic and megakaryocytic malignancies and increases their proliferation rate. Br. J. Haematol. 2007, 136, 212–219. [Google Scholar] [CrossRef]
- Wei, G.; Hu, G.; Cui, K.; Zhao, K. Genome-wide mapping of nucleosome occupancy, histone modifications, and gene expression using next-generation sequencing technolog. Methods Enzymol. 2012, 513, 297–313. [Google Scholar] [CrossRef]
- Henikoff, S.; Shilatifard, A. Histone modification: Cause or cog? Trends Genet. 2011, 27, 389–396. [Google Scholar] [CrossRef]
- Zhou, V.W.; Goren, A.; Bernstein, B.E. Charting histone modifications and the functional organization of mammalian genomes. Nat. Rev. Genet. 2011, 12, 7–18. [Google Scholar]
- Northrup, D.L.; Zhao, K. Application of ChIP-Seq and related techniques to the study of immune function. Immunity 2011, 34, 830–842. [Google Scholar] [CrossRef]
- Ning, B.; Liu, G.; Liu, Y.; Su, X.; Anderson, G.J.; Zheng, X.; Chang, Y.; Guo, M.; Liu, Y.; Zhao, Y.; et al. 5-aza-2'-deoxycytidine activates iron uptake and heme biosynthesis by increasing c-Myc nuclear localization and binding to the E-boxes of transferrin receptor 1 (TfR1) and ferrochelatase (Fech) genes. J. Biol. Chem. 2011, 286, 37196–37206. [Google Scholar]
- Kharbanda, S.; Nakamura, T.; Stone, R.; Hass, R.; Bernstein, S.; Datta, R.; Sukhatme, V.P.; Kufe, D. Expression of the early growth response 1 and 2 zinc finger genes during induction of monocytic differentiation. J. Clin. Invest. 1991, 88, 571–577. [Google Scholar] [CrossRef]
- Savickiene, J.; Treigyte, G.; Vistartaite, G.; Tunaitis, V.; Magnusson, K.E.; Navakauskiene, R. C/EBPalpha and PU.1 are involved in distinct differentiation responses of acute promyelocytic leukemia HL-60 and NB4 cells via chromatin remodeling. Differentiation 2011, 81, 57–67. [Google Scholar] [CrossRef]
- Wang, J.; Lunyak, V.V.; Jordan, I.K. Genome-wide prediction and analysis of human chromatin boundary elements. Nucleic Acids Res. 2012, 40, 511–529. [Google Scholar] [CrossRef]
- Bonn, S.; Zinzen, R.P.; Girardot, C.; Gustafson, E.H.; Perez-Gonzalez, A.; Delhomme, N.; Ghavi-Helm, Y.; Wilczynski, B.; Riddell, A.; Furlong, E.E. Tissue-specific analysis of chromatin state identifies temporal signatures of enhancer activity during embryonic development. Nat. Genet. 2012, 44, 148–156. [Google Scholar] [CrossRef]
- Zentner, G.E.; Scacheri, P.C. The chromatin fingerprint of gene enhancer elements. J. Biol. Chem. 2012, 287, 30888–30896. [Google Scholar] [CrossRef]
- Chopra, V.S.; Kong, N.; Levine, M. Transcriptional repression via antilooping in the Drosophila embryo. Proc. Natl. Acad. Sci. USA 2012, 109, 9460–9464. [Google Scholar]
- Gaston, K.; Jayaraman, P.S. Transcriptional repression in eukaryotes: Repressors and repression mechanisms. Cell Mol. Life Sci. 2003, 60, 721–741. [Google Scholar] [CrossRef]
- Wu, W.; Beilhartz, G.; Roy, Y.; Richard, C.L.; Curtin, M.; Brown, L.; Cadieux, D.; Coppolino, M.; Farach-Carson, M.C.; Nemere, I.; Meckling, K.A. Nuclear translocation of the 1,25D3-MARRS (membrane associated rapid response to steroids) receptor protein and NFkappaB in differentiating NB4 leukemia cells. Exp. Cell Res. 2010, 316, 1101–1108. [Google Scholar] [CrossRef]
- Takeuch, O.; Akira, S. Epigenetic control of macrophage polarization. Eur. J. Immunol. 2011, 41, 2490–2493. [Google Scholar] [CrossRef]
- Emre, Y.; Nubel, T. Uncoupling protein UCP2: When mitochondrial activity meets immunity. FEBS Lett. 2010, 584, 1437–1442. [Google Scholar] [CrossRef]
- Fedorenko, A.; Lishko, P.V.; Kirichok, Y. Mechanism of fatty-acid-dependent UCP1 uncoupling in brown fat mitochondria. Cell 2012, 151, 400–413. [Google Scholar] [CrossRef]
- Park, D.; Han, C.Z.; Elliott, M.R.; Kinchen, J.M.; Trampont, P.C.; Das, S.; Collins, S.; Lysiak, J.J.; Hoehn, K.L.; Ravichandran, K.S. Continued clearance of apoptotic cells critically depends on the phagocyte Ucp2 protein. Nature 2011, 477, 220–224. [Google Scholar]
- Han, C.Z.; Ravichandran, K.S. Metabolic connections during apoptotic cell engulfment. Cell 2011, 147, 1442–1445. [Google Scholar] [CrossRef]
- Maurano, M.T.; Humbert, R.; Rynes, E.; Thurman, R.E.; Haugen, E.; Wang, H.; Reynolds, A.P.; Sandstrom, R.; Qu, H.; Brody, J.; et al. Systematic localization of common disease-associated variation in regulatory DNA. Science 2012, 337, 1190–1195. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Cellier, M.F.M. Cell-Type Specific Determinants of NRAMP1 Expression in Professional Phagocytes. Biology 2013, 2, 233-283. https://doi.org/10.3390/biology2010233
Cellier MFM. Cell-Type Specific Determinants of NRAMP1 Expression in Professional Phagocytes. Biology. 2013; 2(1):233-283. https://doi.org/10.3390/biology2010233
Chicago/Turabian StyleCellier, Mathieu F. M. 2013. "Cell-Type Specific Determinants of NRAMP1 Expression in Professional Phagocytes" Biology 2, no. 1: 233-283. https://doi.org/10.3390/biology2010233
APA StyleCellier, M. F. M. (2013). Cell-Type Specific Determinants of NRAMP1 Expression in Professional Phagocytes. Biology, 2(1), 233-283. https://doi.org/10.3390/biology2010233