
Maternal Nutrient Excess Induces Stress Signaling and Decreases Mitochondrial Number in Term Fetal Baboon Skeletal Muscle

 ,
,  ,
, 
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
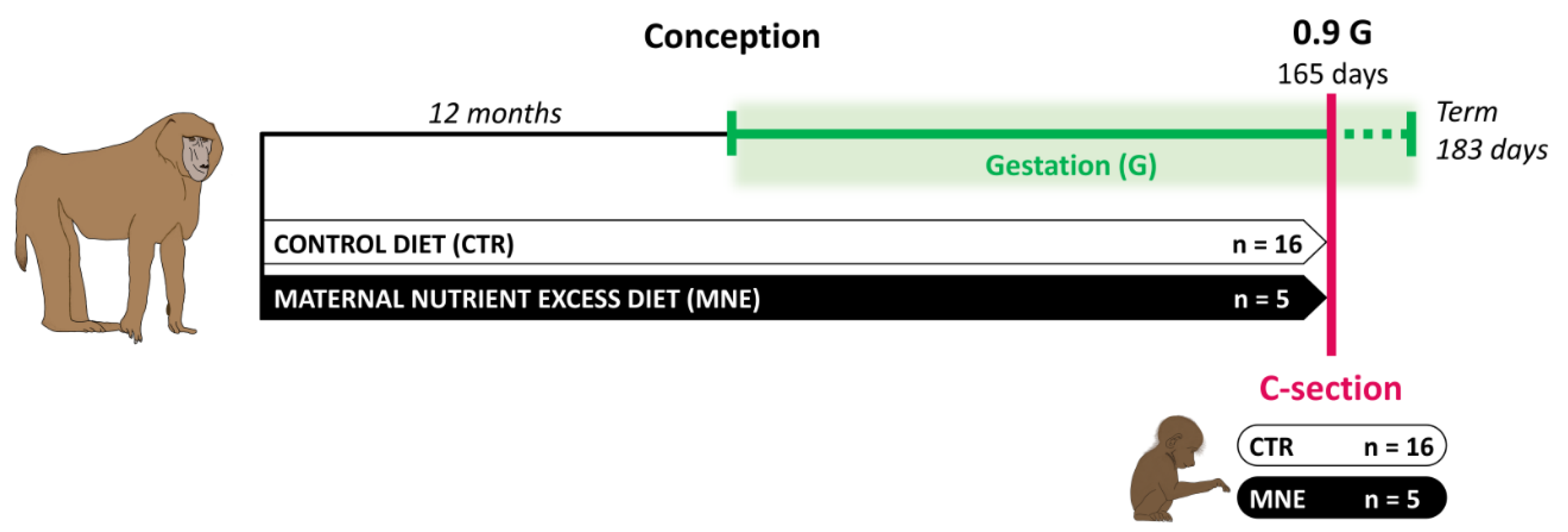
2.1. Care and Use of Animals
2.2. Cesarean Sections and Tissue Sampling
2.3. Dual-Energy X-Ray Absorptiometry (DEXA)
2.4. Immunoblotting Analysis
2.5. Analysis of mtDNA Copy Number by Quantitative Real-Time PCR
2.6. Real-Time Quantitative PCR (RT-PCR)
2.7. Measurement of Citrate Synthase and β-Hydroxyacyl-CoA Dehydrogenase Enzyme Activity
2.8. Statistical Analysis
3. Results
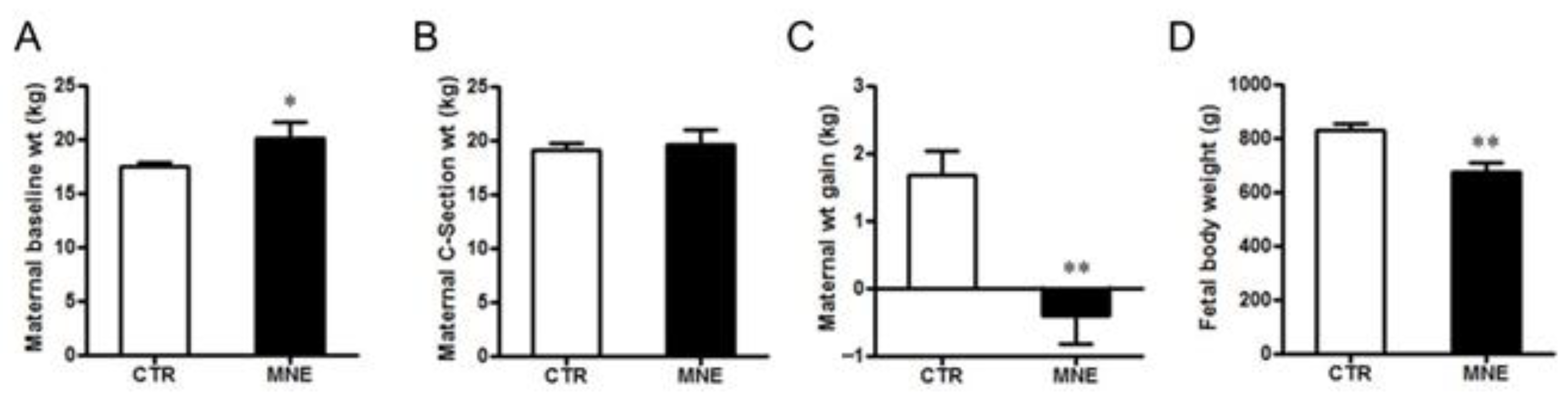
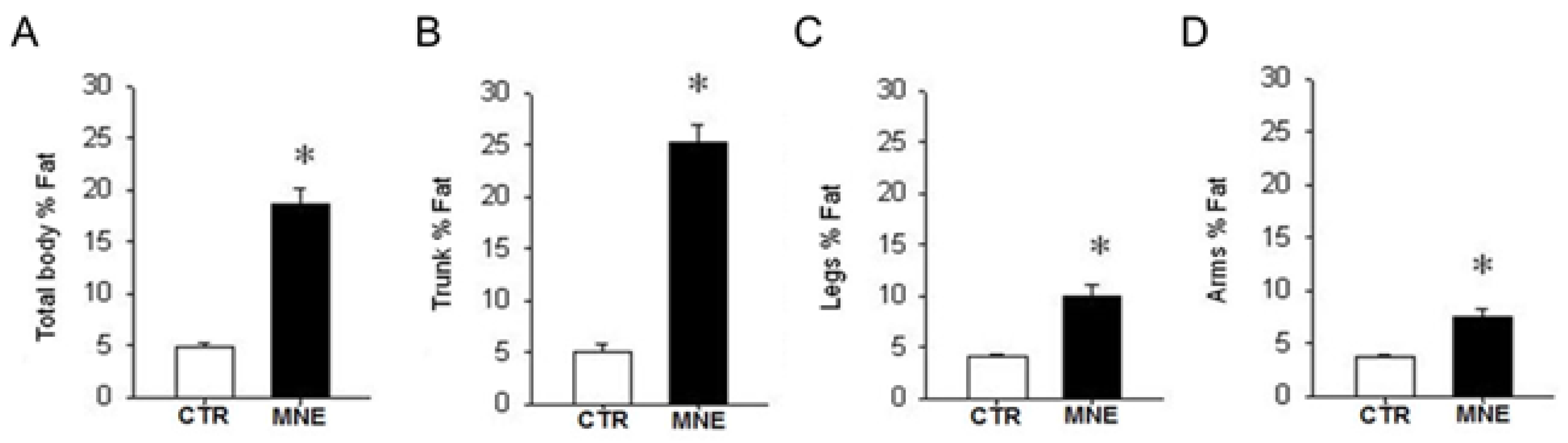
3.1. Maternal and Fetal Body Weight
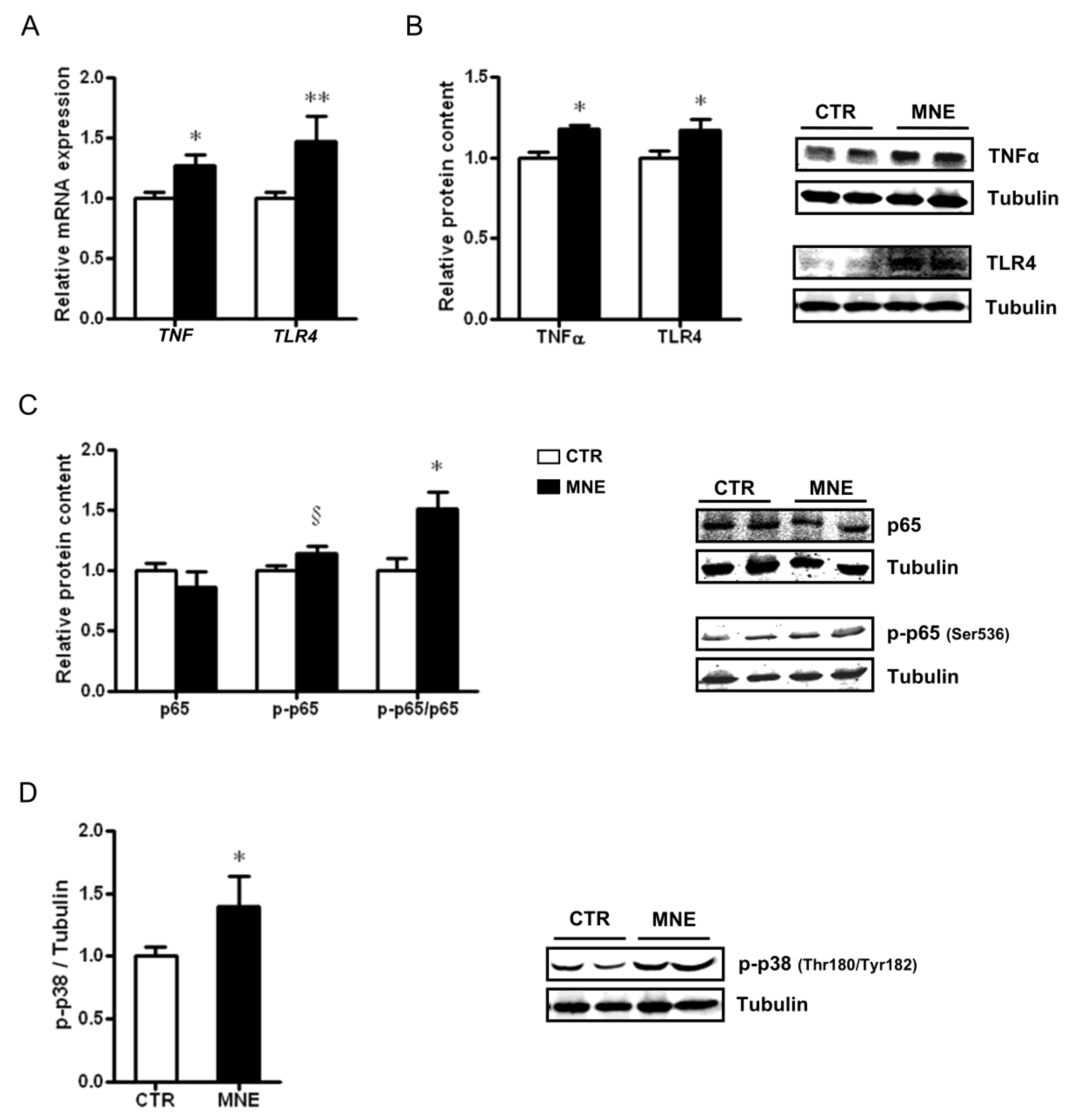
3.2. MNE Increased Stress and Pro-Inflammatory Markers in Fetal Soleus Muscle
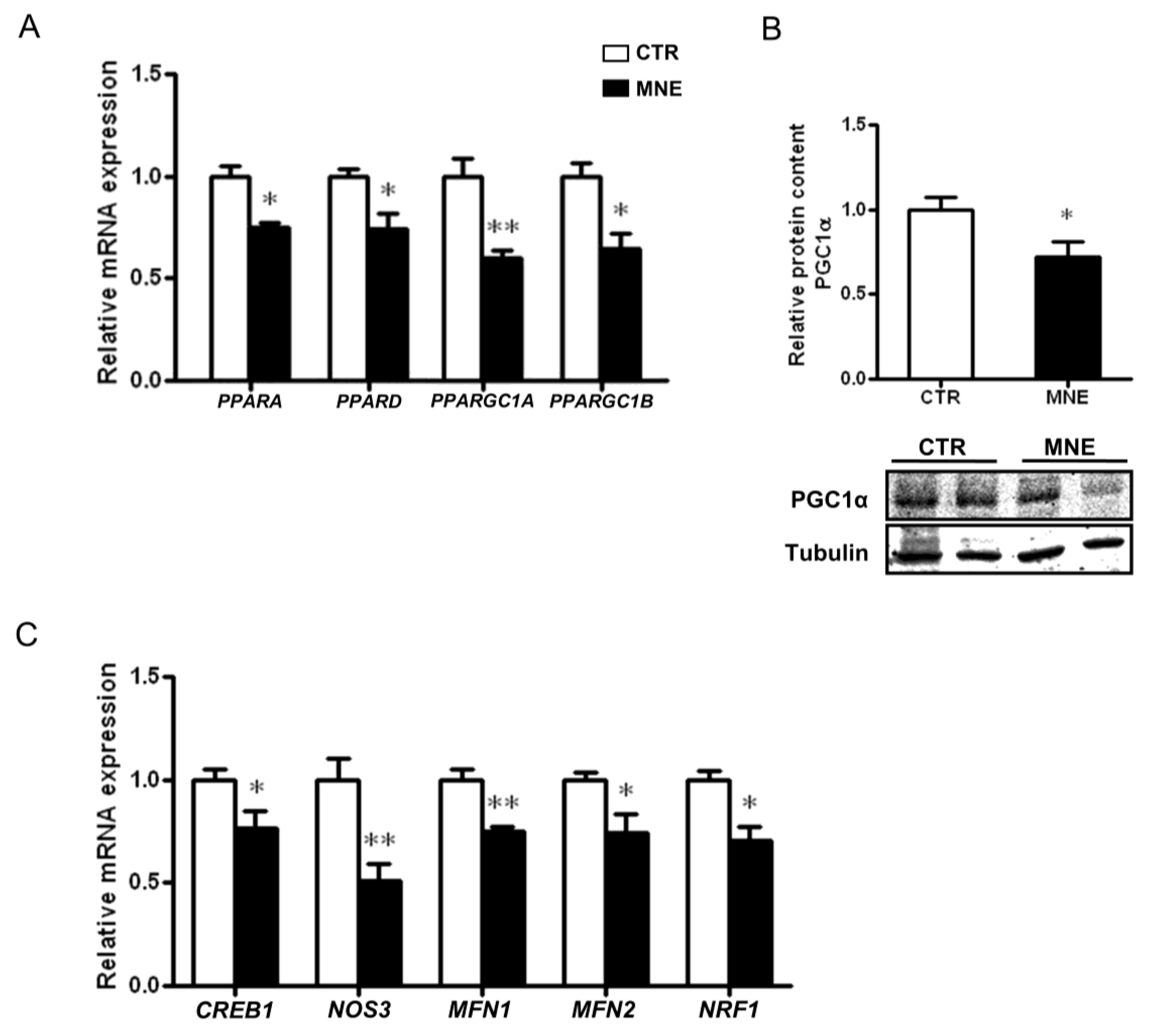
3.3. Mitochondrial Biogenesis and Mitochondrial Fusion Markers Are Decreased in the Soleus Muscle of MNE Fetuses
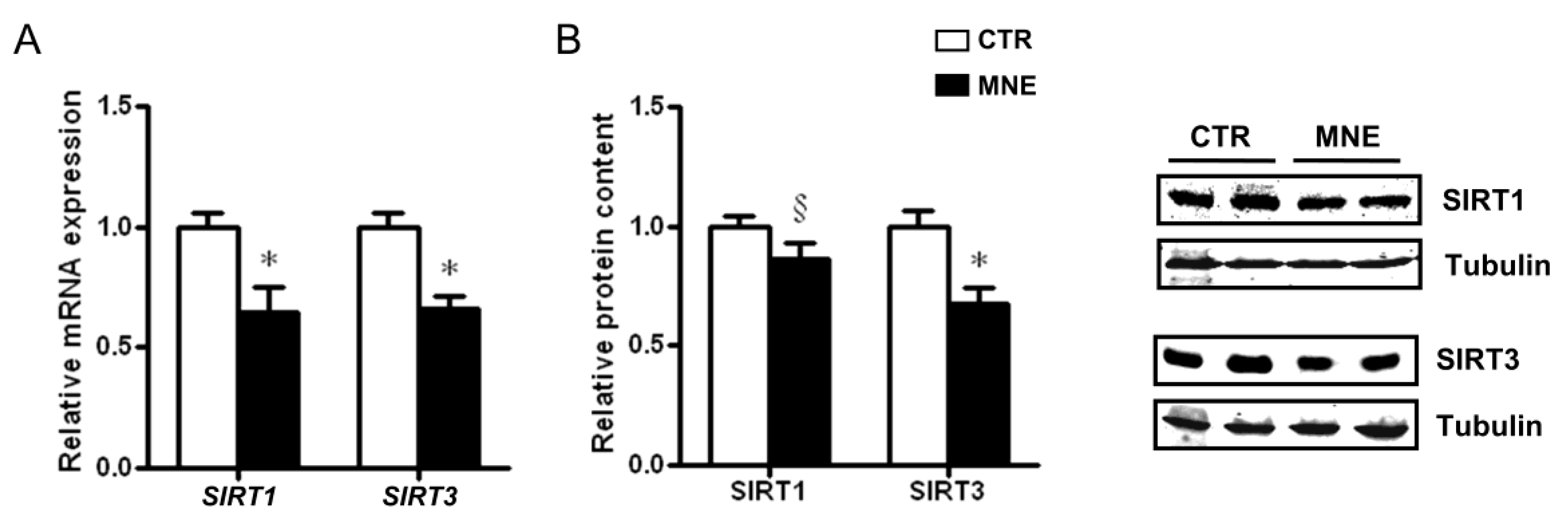
3.4. Fetal Soleus Muscle Expression of SIRT1 and SIRT3
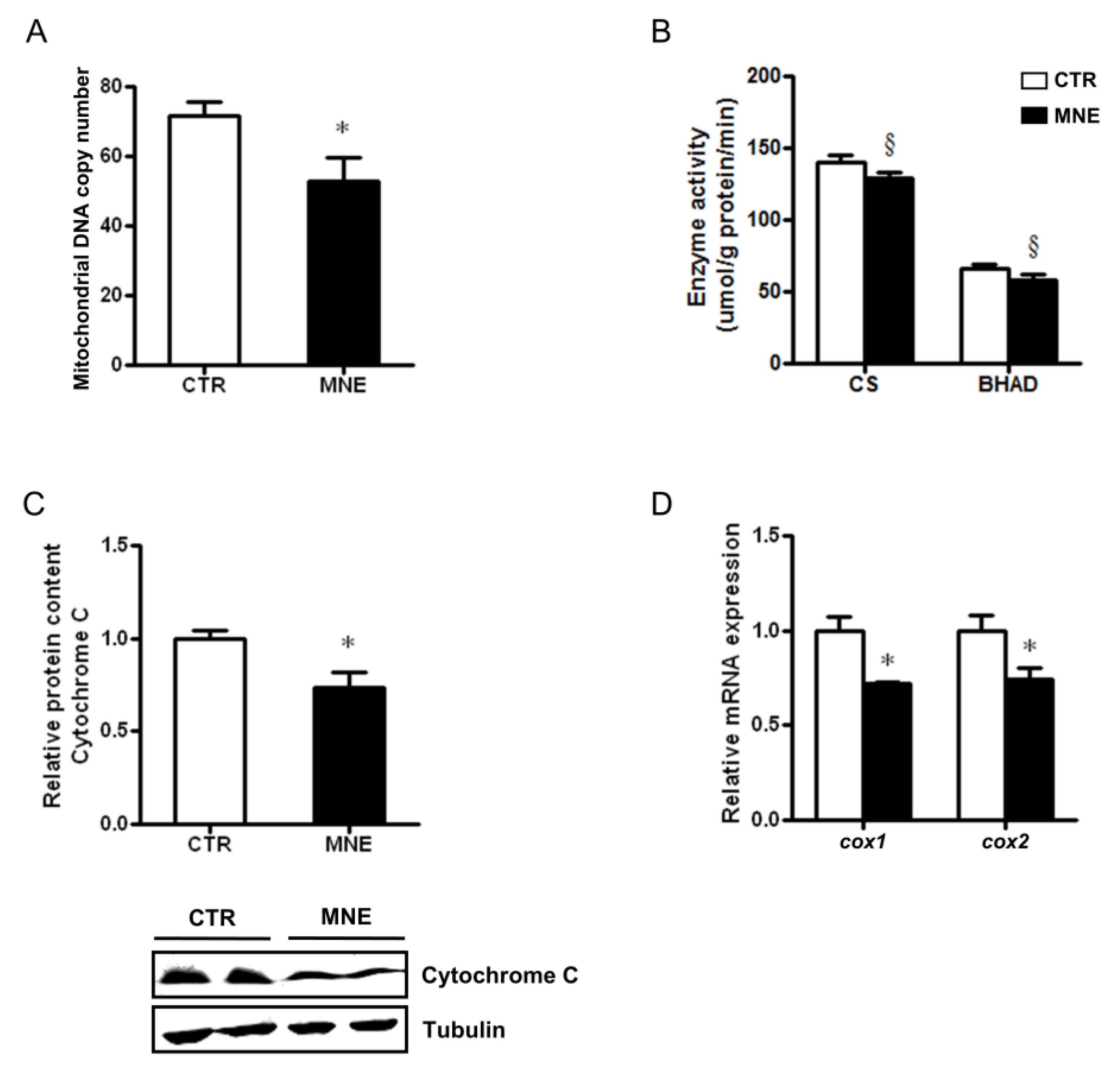
3.5. Decreased Fetal Skeletal Muscle Mitochondrial Abundance and Function
4. Discussion
5. Conclusions
6. Study Limitations
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Blüher, M. Obesity: Global Epidemiology and Pathogenesis. Nat. Rev. Endocrinol. 2019, 15, 288–298. [Google Scholar] [CrossRef] [PubMed]
- Godfrey, K.M.; Reynolds, R.M.; Prescott, S.L.; Nyirenda, M.; Jaddoe, V.W.V.; Eriksson, J.G.; Broekman, B.F.P. Influence of Maternal Obesity on the Long-Term Health of Offspring. Lancet Diabetes Endocrinol. 2017, 5, 53–64. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. World Health Statistics 2024: Monitoring Health for the SDGs, Sustainable Development Goals; World Health Organization: Geneva, Switzerland, 2024. [Google Scholar]
- Stierman, B.; Afful, J.; Carroll, M.D.; Chen, T.-C.; Davy, O.; Fink, S.; Fryar, C.D.; Gu, Q.; Hales, C.M.; Hughes, J.P.; et al. National Health and Nutrition Examination Survey 2017–March 2020 Prepandemic Data Files—Development of Files and Prevalence Estimates for Selected Health Outcomes. Natl. Health Stat. Rep. 2021, 158, 10-15620. [Google Scholar] [CrossRef]
- Driscoll, A.K.; Gregory, E.C.W. Increases in Prepregnancy Obesity: United States, 2016–2019; NCHS: Hyattsville, MD, USA, 2020. [Google Scholar]
- Bass, R.; Eneli, I. Severe Childhood Obesity: An under-Recognised and Growing Health Problem. Postgrad. Med. J. 2015, 91, 639–645. [Google Scholar] [CrossRef] [PubMed]
- Vats, H.; Saxena, R.; Sachdeva, M.P.; Walia, G.K.; Gupta, V. Impact of Maternal Pre-Pregnancy Body Mass Index on Maternal, Fetal and Neonatal Adverse Outcomes in the Worldwide Populations: A Systematic Review and Meta-Analysis. Obes. Res. Clin. Pract. 2021, 15, 536–545. [Google Scholar] [CrossRef] [PubMed]
- Voerman, E.; Santos, S.; Golab, B.P.; Amiano, P.; Ballester, F.; Barros, H.; Bergström, A.; Charles, M.A.; Chatzi, L.; Chevrier, C.; et al. Maternal Body Mass Index, Gestational Weight Gain, and the Risk of Overweight and Obesity across Childhood: An Individual Participant Data Meta-Analysis. PLoS Med. 2019, 16, e1002744. [Google Scholar] [CrossRef] [PubMed]
- Ijäs, H.; Koivunen, S.; Raudaskoski, T.; Kajantie, E.; Gissler, M.; Vääräsmäki, M. Independent and Concomitant Associations of Gestational Diabetes and Maternal Obesity to Perinatal Outcome: A Register-Based Study. PLoS ONE 2019, 14, e0221549. [Google Scholar] [CrossRef] [PubMed]
- Eriksson, J.G.; Sandboge, S.; Salonen, M.K.; Kajantie, E.; Osmond, C. Long-Term Consequences of Maternal Overweight in Pregnancy on Offspring Later Health: Findings from the Helsinki Birth Cohort Study. Ann. Med. 2014, 46, 434–438. [Google Scholar] [CrossRef] [PubMed]
- Grilo, L.F.; Diniz, M.S.; Tocantins, C.; Areia, A.L.; Pereira, S.P. The Endocrine–Metabolic Axis Regulation in Offspring Exposed to Maternal Obesity—Cause or Consequence in Metabolic Disease Programming? Obesities 2022, 2, 236–255. [Google Scholar] [CrossRef]
- Parrettini, S.; Caroli, A.; Torlone, E. Nutrition and Metabolic Adaptations in Physiological and Complicated Pregnancy: Focus on Obesity and Gestational Diabetes. Front. Endocrinol. 2020, 11, 611929. [Google Scholar] [CrossRef] [PubMed]
- Du, M.; Yan, X.; Tong, J.F.; Zhao, J.; Zhu, M.J. Maternal Obesity, Inflammation, and Fetal Skeletal Muscle Development. Biol. Reprod. 2010, 82, 4–12. [Google Scholar] [CrossRef] [PubMed]
- Haines, M.S.; Leong, A.; Porneala, B.C.; Meigs, J.B.; Miller, K.K. Association between Muscle Mass and Diabetes Prevalence Independent of Body Fat Distribution in Adults under 50 Years Old. Nutr. Diabetes 2022, 12, 29. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Hu, T.; Shen, Y.; Wang, Y.; Bao, Y.; Ma, X. Association of Skeletal Muscle Mass and Its Change with Diabetes Occurrence: A Population-Based Cohort Study. Diabetol. Metab. Syndr. 2023, 15, 53. [Google Scholar] [CrossRef] [PubMed]
- Swanson, A.M.; David, A.L. Animal Models of Fetal Growth Restriction: Considerations for Translational Medicine. Placenta 2015, 36, 623–630. [Google Scholar] [CrossRef] [PubMed]
- Tarantal, A.F.; Noctor, S.C.; Hartigan-O’connor, D.J. Nonhuman Primates in Translational Research. Annu. Rev. Anim. Biosci. 2022, 10, 441–468. [Google Scholar] [CrossRef] [PubMed]
- Campodonico-Burnett, W.; Hetrick, B.; Wesolowski, S.R.; Schenk, S.; Takahashi, D.L.; Dean, T.A.; Sullivan, E.L.; Kievit, P.; Gannon, M.; Aagaard, K.; et al. Maternal Obesity and Western-Style Diet Impair Fetal and Juvenile Offspring Skeletal Muscle Insulin-Stimulated Glucose Transport in Nonhuman Primates. Diabetes 2020, 69, 1389–1400. [Google Scholar] [CrossRef] [PubMed]
- Yan, X.; Zhu, M.J.; Xu, W.; Tong, J.F.; Ford, S.P.; Nathanielsz, P.W.; Du, M. Up-Regulation of Toll-like Receptor 4/Nuclear Factor-ΚB Signaling Is Associated with Enhanced Adipogenesis and Insulin Resistance in Fetal Skeletal Muscle of Obese Sheep at Late Gestation. Endocrinology 2010, 151, 380–387. [Google Scholar] [CrossRef] [PubMed]
- Omar, A.K.; Li Puma, L.C.; Whitcomb, L.A.; Risk, B.D.; Witt, A.C.; Bruemmer, J.E.; Winger, Q.A.; Bouma, G.J.; Chicco, A.J. High-Fat Diet during Pregnancy Promotes Fetal Skeletal Muscle Fatty Acid Oxidation and Insulin Resistance in an Ovine Model. Am. J. Physiol. Integr. Comp. Physiol. 2023, 325, R523–R533. [Google Scholar] [CrossRef] [PubMed]
- Rogero, M.M.; Calder, P.C. Obesity, Inflammation, Toll-like Receptor 4 and Fatty Acids. Nutrients 2018, 10, 432. [Google Scholar] [CrossRef] [PubMed]
- Parisi, F.; Milazzo, R.; Savasi, V.M.; Cetin, I. Maternal Low-Grade Chronic Inflammation and Intrauterine Programming of Health and Disease. Int. J. Mol. Sci. 2021, 22, 1732. [Google Scholar] [CrossRef] [PubMed]
- Prasun, P. Mitochondrial Dysfunction in Metabolic Syndrome. Biochim. Biophys. Acta-Mol. Basis Dis. 2020, 1866, 165838. [Google Scholar] [CrossRef] [PubMed]
- de Mello, A.H.; Costa, A.B.; Engel, J.D.G.; Rezin, G.T. Mitochondrial Dysfunction in Obesity. Life Sci. 2018, 192, 26–32. [Google Scholar] [CrossRef] [PubMed]
- Blake, R.; Trounce, I.A. Mitochondrial Dysfunction and Complications Associated with Diabetes. Biochim. Biophys. Acta-Gen. Subj. 2014, 1840, 1404–1412. [Google Scholar] [CrossRef] [PubMed]
- Pazderska, A.; Wanic, K.; Nolan, J.J. Skeletal Muscle Mitochondrial Dysfunction in Type2 Diabetes. Expert Rev. Endocrinol. Metab. 2010, 5, 475–477. [Google Scholar] [CrossRef] [PubMed]
- du Sert, N.; Hurst, V.; Ahluwalia, A.; Alam, S.; Avey, M.T.; Baker, M.; Browne, W.J.; Clark, A.; Cuthill, I.C.; Dirnagl, U.; et al. The ARRIVE Guidelines 2.0: Updated Guidelines for Reporting Animal Research. PLoS Biol. 2020, 18, e3000410. [Google Scholar] [CrossRef]
- Schlabritz-Loutsevitch, N.E.; Howell, K.; Rice, K.; Glover, E.J.; Nevill, C.H.; Jenkins, S.L.; Cummins, L.B.; Frost, P.A.; McDonald, T.J.; Nathanielsz, P.W. Development of a System for Individual Feeding of Baboons Maintained in an Outdoor Group Social Environment. J. Med. Primatol. 2004, 33, 117–126. [Google Scholar] [CrossRef] [PubMed]
- Maloyan, A.; Muralimanoharan, S.; Huffman, S.; Cox, L.A.; Nathanielsz, P.W.; Myatt, L.; Nijland, M.J. Identification and Comparative Analyses of Myocardial MiRNAs Involved in the Fetal Response to Maternal Obesity. Physiol. Genom. 2013, 45, 889–900. [Google Scholar] [CrossRef] [PubMed]
- Ford, S.P.; Zhang, L.; Zhu, M.; Miller, M.M.; Smith, D.T.; Hess, B.W.; Moss, G.E.; Nathanielsz, P.W.; Nijland, M.J. Maternal Obesity Accelerates Fetal Pancreatic β-Cell but Not α-Cell Development in Sheep: Prenatal Consequences. Am. J. Physiol.-Regul. Integr. Comp. Physiol. 2009, 297, 835–843. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2−ΔΔCT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Call, J.A.; Voelker, K.A.; Wolff, A.V.; McMillan, R.P.; Evans, N.P.; Hulver, M.W.; Talmadge, R.J.; Grange, R.W. Endurance Capacity in Maturing Mdx Mice Is Markedly Enhanced by Combined Voluntary Wheel Running and Green Tea Extract. J. Appl. Physiol. 2008, 105, 923–932. [Google Scholar] [CrossRef] [PubMed]
- Baldelli, S.; Barbato, D.L.; Tatulli, G.; Aquilano, K.; Ciriolo, M.R. The Role of NNOS and PGC-1α in Skeletal Muscle Cells. J. Cell Sci. 2014, 127, 4813–4820. [Google Scholar] [CrossRef] [PubMed]
- Palacios, O.M.; Carmona, J.J.; Michan, S.; Chen, K.Y.; Manabe, Y.; Ward, J.L.; Goodyear, L.J.; Tong, Q. Diet and Exercise Signals Regulate SIRT3 and Activate AMPK and PGC-1alpha in Skeletal Muscle. Aging 2009, 1, 771–783. [Google Scholar] [CrossRef] [PubMed]
- Lantier, L.; Williams, A.S.; Williams, I.M.; Yang, K.K.; Bracy, D.P.; Goelzer, M.; James, F.D.; Gius, D.; Wasserman, D.H. SIRT3 Is Crucial for Maintaining Skeletal Muscle Insulin Action and Protects against Severe Insulin Resistance in High-Fat-Fed Mice. Diabetes 2015, 64, 3081–3092. [Google Scholar] [CrossRef] [PubMed]
- Tocantins, C.; Diniz, M.S.; Grilo, L.F.; Pereira, S.P. The Birth of Cardiac Disease: Mechanisms Linking Gestational Diabetes Mellitus and Early Onset of Cardiovascular Disease in Offspring. WIREs Mech. Dis. 2022, 14, e1555. [Google Scholar] [CrossRef] [PubMed]
- Hill, J.H.; Solt, C.; Foster, M.T. Obesity Associated Disease Risk: The Role of Inherent Differences and Location of Adipose Depots. Horm. Mol. Biol. Clin. Investig. 2018, 33, 20180012. [Google Scholar] [CrossRef] [PubMed]
- Kwon, H.; Kim, D.; Kim, J.S. Body Fat Distribution and the Risk of Incident Metabolic Syndrome: A Longitudinal Cohort Study. Sci. Rep. 2017, 7, 10955. [Google Scholar] [CrossRef] [PubMed]
- Kang, P.S.; Neeland, I.J. Body Fat Distribution, Diabetes Mellitus, and Cardiovascular Disease: An Update. Curr. Cardiol. Rep. 2023, 25, 1555–1564. [Google Scholar] [CrossRef] [PubMed]
- Gaudet, L.; Ferraro, Z.M.; Wen, S.W.; Walker, M. Maternal Obesity and Occurrence of Fetal Macrosomia: A Systematic Review and Meta-Analysis. Biomed Res. Int. 2014, 2014, 640291. [Google Scholar] [CrossRef] [PubMed]
- Santos, S.; Voerman, E.; Amiano, P.; Barros, H.; Beilin, L.J.; Bergström, A.; Charles, M.A.; Chatzi, L.; Chevrier, C.; Chrousos, G.P.; et al. Impact of Maternal Body Mass Index and Gestational Weight Gain on Pregnancy Complications: An Individual Participant Data Meta-Analysis of European, North American and Australian Cohorts. BJOG An Int. J. Obstet. Gynaecol. 2019, 126, 984–995. [Google Scholar] [CrossRef] [PubMed]
- Champion, M.L.; Harper, L.M. Gestational Weight Gain: Update on Outcomes and Interventions. Curr. Diab. Rep. 2020, 20, 11. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Wen, Z.; Zhou, Y.; Li, D.; Luo, Z. Inadequate Weight Gain in Obese Women and the Risk of Small for Gestational Age (SGA): A Systematic Review and Meta-Analysis. J. Matern. Neonatal Med. 2017, 30, 357–367. [Google Scholar] [CrossRef] [PubMed]
- Tam, C.H.T.; Ma, R.C.W.; Yuen, L.Y.; Ozaki, R.; Li, A.M.; Hou, Y.; Chan, M.H.M.; Ho, C.S.; Yang, X.; Chan, J.C.N.; et al. The Impact of Maternal Gestational Weight Gain on Cardiometabolic Risk Factors in Children. Diabetologia 2018, 61, 2539–2548. [Google Scholar] [CrossRef] [PubMed]
- Puppala, S.; Li, C.; Glenn, J.P.; Saxena, R.; Gawrieh, S.; Quinn, A.; Palarczyk, J.; Dick, E.J.; Nathanielsz, P.W.; Cox, L.A. Primate Fetal Hepatic Responses to Maternal Obesity: Epigenetic Signalling Pathways and Lipid Accumulation. J. Physiol. 2018, 596, 5823–5837. [Google Scholar] [CrossRef] [PubMed]
- Nathanielsz, P.W.; Yan, J.; Green, R.; Nijland, M.; Miller, J.W.; Wu, G.; McDonald, T.J.; Caudill, M.A. Maternal Obesity Disrupts the Methionine Cycle in Baboon Pregnancy. Physiol. Rep. 2015, 3, e12564. [Google Scholar] [CrossRef] [PubMed]
- Sanches, A.P.V.; de Oliveira, J.L.; Ferreira, M.S.; Lima, B.d.S.; Miyamoto, J.É.; Simino, L.A.d.P.; Torsoni, M.A.; Torsoni, A.S.; Milanski, M.; Ignácio-Souza, L. Obesity Phenotype Induced by High-Fat Diet Leads to Maternal-Fetal Constraint, Placental Inefficiency, and Fetal Growth Restriction in Mice. J. Nutr. Biochem. 2022, 104, 108977. [Google Scholar] [CrossRef] [PubMed]
- Mayor, R.S.; Finch, K.E.; Zehr, J.; Morselli, E.; Neinast, M.D.; Frank, A.P.; Hahner, L.D.; Wang, J.; Rakheja, D.; Palmer, B.F.; et al. Maternal High-Fat Diet Is Associated with Impaired Fetal Lung Development. Am. J. Physiol. Lung Cell. Mol. Physiol. 2015, 309, L360-8. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Jenkins, S.; Considine, M.M.; Cox, L.A.; Gerow, K.G.; Huber, H.F.; Nathanielsz, P.W. Effect of Maternal Obesity on Fetal and Postnatal Baboon (Papio Species) Early Life Phenotype. J. Med. Primatol. 2019, 48, 90–98. [Google Scholar] [CrossRef] [PubMed]
- Jaganathan, R.; Ravindran, R.; Dhanasekaran, S. Emerging Role of Adipocytokines in Type 2 Diabetes as Mediators of Insulin Resistance and Cardiovascular Disease. Can. J. Diabetes 2018, 42, 446–456.e1. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Li, Y.; Duan, Y.; Hu, C.A.A.; Tang, Y.; Yin, Y. Myokines and Adipokines: Involvement in the Crosstalk between Skeletal Muscle and Adipose Tissue. Cytokine Growth Factor Rev. 2017, 33, 73–82. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Leung, J.C.K.; Chan, L.Y.Y.; Yiu, W.H.; Tang, S.C.W. A Global Perspective on the Crosstalk between Saturated Fatty Acids and Toll-like Receptor 4 in the Etiology of Inflammation and Insulin Resistance. Prog. Lipid Res. 2020, 77, 101020. [Google Scholar] [CrossRef] [PubMed]
- Nisr, R.B.; Shah, D.S.; Ganley, I.G.; Hundal, H.S. Proinflammatory NFkB Signalling Promotes Mitochondrial Dysfunction in Skeletal Muscle in Response to Cellular Fuel Overloading. Cell. Mol. Life Sci. 2019, 76, 4887–4904. [Google Scholar] [CrossRef] [PubMed]
- Crossland, H.; Constantin-Teodosiu, D.; Greenhaff, P.L. The Regulatory Roles of Ppars in Skeletal Muscle Fuel Metabolism and Inflammation: Impact of Ppar Agonism on Muscle in Chronic Disease, Contraction and Sepsis. Int. J. Mol. Sci. 2021, 22, 9775. [Google Scholar] [CrossRef] [PubMed]
- Manickam, R.; Wahli, W. Roles of Peroxisome Proliferator-Activated Receptor β/δ in Skeletal Muscle Physiology. Biochimie 2017, 136, 42–48. [Google Scholar] [CrossRef] [PubMed]
- Schuler, M.; Ali, F.; Chambon, C.; Duteil, D.; Bornert, J.M.; Tardivel, A.; Desvergne, B.; Wahli, W.; Chambon, P.; Metzger, D. PGC1α Expression Is Controlled in Skeletal Muscles by PPARβ, Whose Ablation Results in Fiber-Type Switching, Obesity, and Type 2 Diabetes. Cell Metab. 2006, 4, 407–414. [Google Scholar] [CrossRef] [PubMed]
- Phua, W.W.T.; Wong, M.X.Y.; Liao, Z.; Tan, N.S. An Apparent Functional Consequence in Skeletal Muscle Physiology via Peroxisome Proliferator-Activated Receptors. Int. J. Mol. Sci. 2018, 19, 1425. [Google Scholar] [CrossRef] [PubMed]
- Zorzano, A.; Hernández-Alvarez, M.I.; Palacín, M.; Mingrone, G. Alterations in the Mitochondrial Regulatory Pathways Constituted by the Nuclear Co-Factors PGC-1α or PGC-1β and Mitofusin 2 in Skeletal Muscle in Type 2 Diabetes. Biochim. Biophys. Acta-Bioenerg. 2010, 1797, 1028–1033. [Google Scholar] [CrossRef] [PubMed]
- Zou, T.D.; Yu, B.; Yu, J.; Mao, X.B.; Zheng, P.; He, J.; Huang, Z.Q.; He, D.T.; Chen, D.W. Mitochondrial Biogenesis Is Decreased in Skeletal Muscle of Pig Fetuses Exposed to Maternal High-Energy Diets. Animal 2017, 11, 54–60. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.S.; Mo, J.Y.; Huang, Y.T.; Zhu, H.; Wu, H.Y.; Lin, Z.L.; Liu, R.; Liu, X.Q.; Lv, P.P.; Feng, C.; et al. Intrauterine Hyperglycaemia during Late Gestation Caused Mitochondrial Dysfunction in Skeletal Muscle of Male Offspring through CREB/PGC1A Signaling. Nutr. Diabetes 2024, 14, 56. [Google Scholar] [CrossRef] [PubMed]
- Fealy, C.E.; Mulya, A.; Axelrod, C.L.; Kirwan, J.P. Mitochondrial Dynamics in Skeletal Muscle Insulin Resistance and Type 2 Diabetes. Transl. Res. 2018, 202, 69–82. [Google Scholar] [CrossRef] [PubMed]
- Taylor, D.F.; Bishop, D.J. Transcription Factor Movement and Exercise-Induced Mitochondrial Biogenesis in Human Skeletal Muscle: Current Knowledge and Future Perspectives. Int. J. Mol. Sci. 2022, 23, 1517. [Google Scholar] [CrossRef] [PubMed]
- Cantó, C.; Gerhart-Hines, Z.; Feige, J.N.; Lagouge, M.; Noriega, L.; Milne, J.C.; Elliott, P.J.; Puigserver, P.; Auwerx, J. AMPK Regulates Energy Expenditure by Modulating NAD + Metabolism and SIRT1 Activity. Nature 2009, 458, 1056–1060. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.W.; Chang, S.J. Moderate Exercise Suppresses NF-ΚB Signaling and Activates the SIRT1-AMPK-PGC1α Axis to Attenuate Muscle Loss in Diabetic Db/Db Mice. Front. Physiol. 2018, 9, 636. [Google Scholar] [CrossRef] [PubMed]
- McCurdy, C.E.; Schenk, S.; Hetrick, B.; Houck, J.; Drew, B.G.; Kaye, S.; Lashbrook, M.; Bergman, B.C.; Takahashi, D.L.; Dean, T.A.; et al. Maternal Obesity Reduces Oxidative Capacity in Fetal Skeletal Muscle of Japanese Macaques. JCI Insight 2016, 1, e86612. [Google Scholar] [CrossRef] [PubMed]
- Valerio, A.; Cardile, A.; Cozzi, V.; Bracale, R.; Tedesco, L.; Pisconti, A.; Palomba, L.; Cantoni, O.; Clementi, E.; Moncada, S.; et al. TNF-α Downregulates ENOS Expression and Mitochondrial Biogenesis in Fat and Muscle of Obese Rodents. J. Clin. Investig. 2006, 116, 2791–2798. [Google Scholar] [CrossRef] [PubMed]
- Ampong, I.; Zimmerman, K.D.; Perumalla, D.S.; Wallis, K.E.; Li, G.; Huber, H.F.; Li, C.; Nathanielsz, P.W.; Cox, L.A.; Olivier, M. Maternal Obesity Alters Offspring Liver and Skeletal Muscle Metabolism in Early Post-Puberty despite Maintaining a Normal Post-Weaning Dietary Lifestyle. FASEB J. 2022, 36, e22644. [Google Scholar] [CrossRef] [PubMed]
- Pereira, S.P.; Diniz, M.S.; Tavares, L.C.; Cunha-Oliveira, T.; Li, C.; Cox, L.A.; Nijland, M.J.; Nathanielsz, P.W.; Oliveira, P.J. Characterizing Early Cardiac Metabolic Programming via 30% Maternal Nutrient Reduction during Fetal Development in a Non-Human Primate Model. Int. J. Mol. Sci. 2023, 24, 15192. [Google Scholar] [CrossRef] [PubMed]
- Ceasrine, A.M.; Devlin, B.A.; Bolton, J.L.; Green, L.A.; Jo, Y.C.; Huynh, C.; Patrick, B.; Washington, K.; Sanchez, C.L.; Joo, F.; et al. Maternal Diet Disrupts the Placenta–Brain Axis in a Sex-Specific Manner. Nat. Metab. 2022, 4, 1732–1745. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Energy Source (%) | Purina Monkey Diet 5038 | Purina 5045-6 |
|---|---|---|
| Fat | 12 | 45 |
| Glucose | 0.29 | 4.62 |
| Fructose | 0.32 | 5.64 |
| Metabolizable Energy Content (kcal/g) | 3.07 | 4.05 |
| Biscuit Basic Composition | Crude protein (≥15%), crude fat (≥5%), crude fiber (≤6%), ash (≤5%), added minerals (≤3%) | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yan, X.; Tocantins, C.; Zhu, M.-J.; Pereira, S.P.; Du, M. Maternal Nutrient Excess Induces Stress Signaling and Decreases Mitochondrial Number in Term Fetal Baboon Skeletal Muscle. Biology 2025, 14, 868. https://doi.org/10.3390/biology14070868
Yan X, Tocantins C, Zhu M-J, Pereira SP, Du M. Maternal Nutrient Excess Induces Stress Signaling and Decreases Mitochondrial Number in Term Fetal Baboon Skeletal Muscle. Biology. 2025; 14(7):868. https://doi.org/10.3390/biology14070868
Chicago/Turabian StyleYan, Xu, Carolina Tocantins, Mei-Jun Zhu, Susana P. Pereira, and Min Du. 2025. "Maternal Nutrient Excess Induces Stress Signaling and Decreases Mitochondrial Number in Term Fetal Baboon Skeletal Muscle" Biology 14, no. 7: 868. https://doi.org/10.3390/biology14070868
APA StyleYan, X., Tocantins, C., Zhu, M.-J., Pereira, S. P., & Du, M. (2025). Maternal Nutrient Excess Induces Stress Signaling and Decreases Mitochondrial Number in Term Fetal Baboon Skeletal Muscle. Biology, 14(7), 868. https://doi.org/10.3390/biology14070868