
Unveiling Genomic Islands Hosting Antibiotic Resistance Genes and Virulence Genes in Foodborne Multidrug-Resistant Patho-Genic Proteus vulgaris

Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Proteus Vulgaris Genomes and Data Acquisition
2.2. Prediction and Comparison of Genomic Islands (GIs)
2.3. Antibiotic Resistance Genes (ARGs) and Virulence Genes Prediction
2.4. Phylogenetic Tree Construction
2.5. Sequence Alignments
3. Results
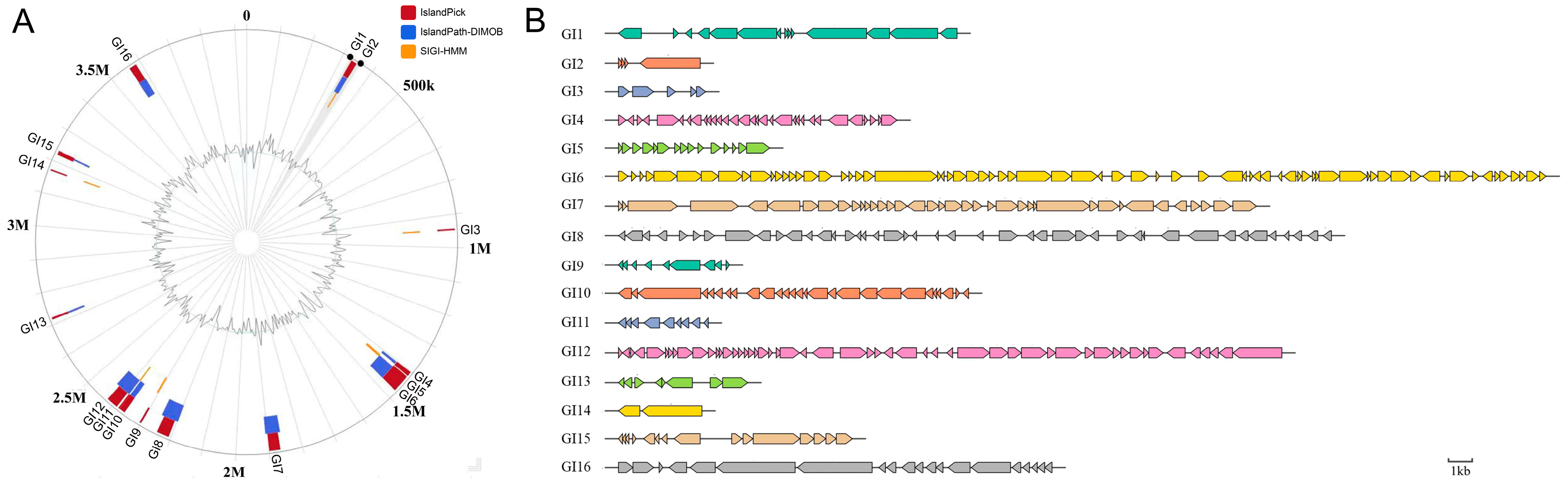
3.1. P3M Genomic Island Analysis
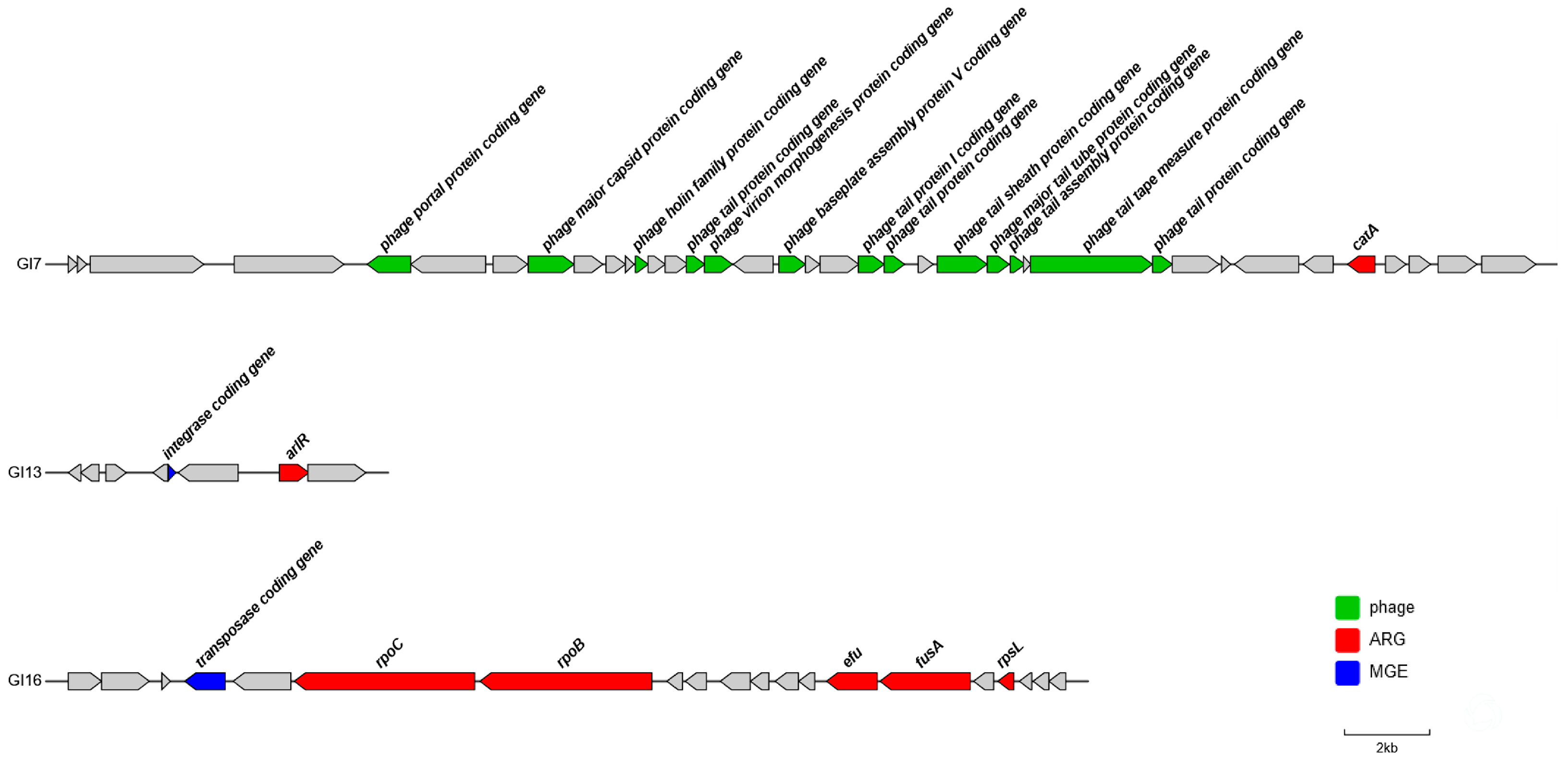
3.2. Identification of Antibiotic Resistance-Associated Genomic Islands in P3M
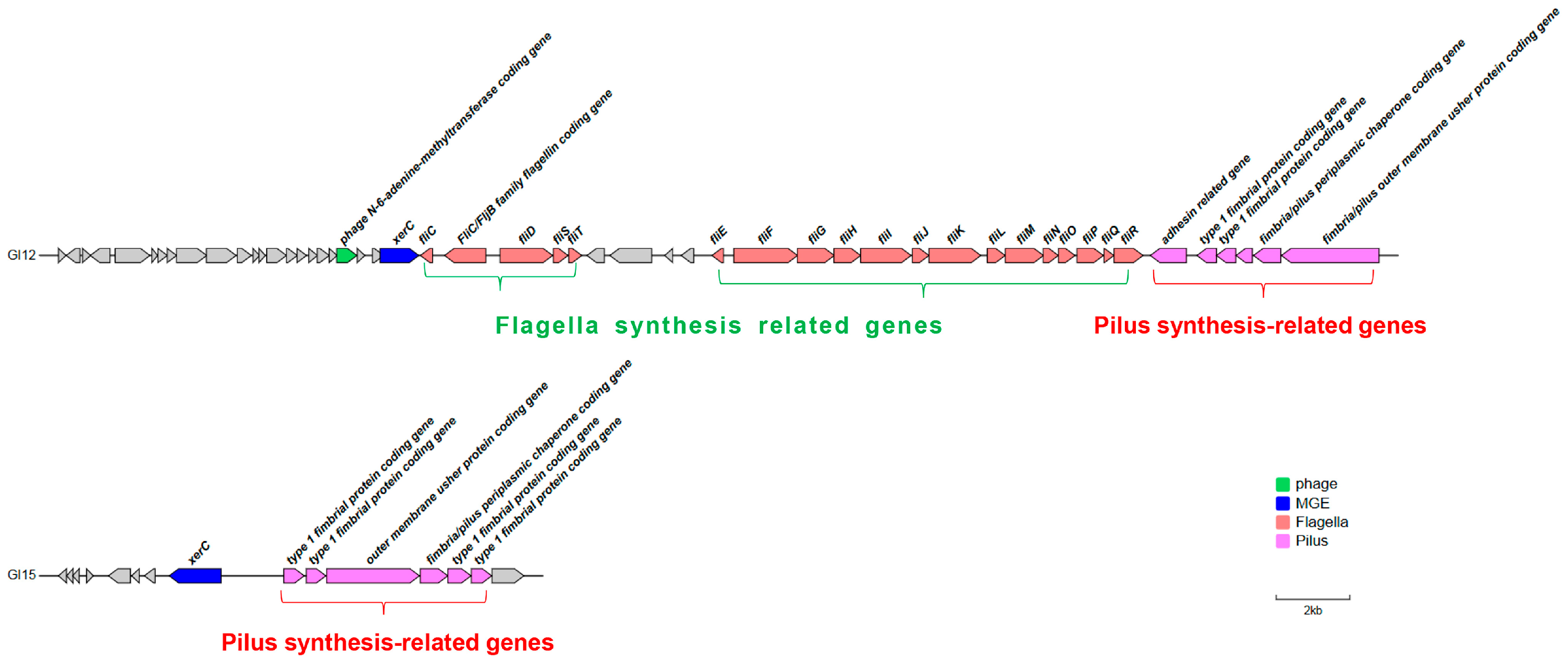
3.3. Identification Virulence-Associated Genomic Islands in P3M
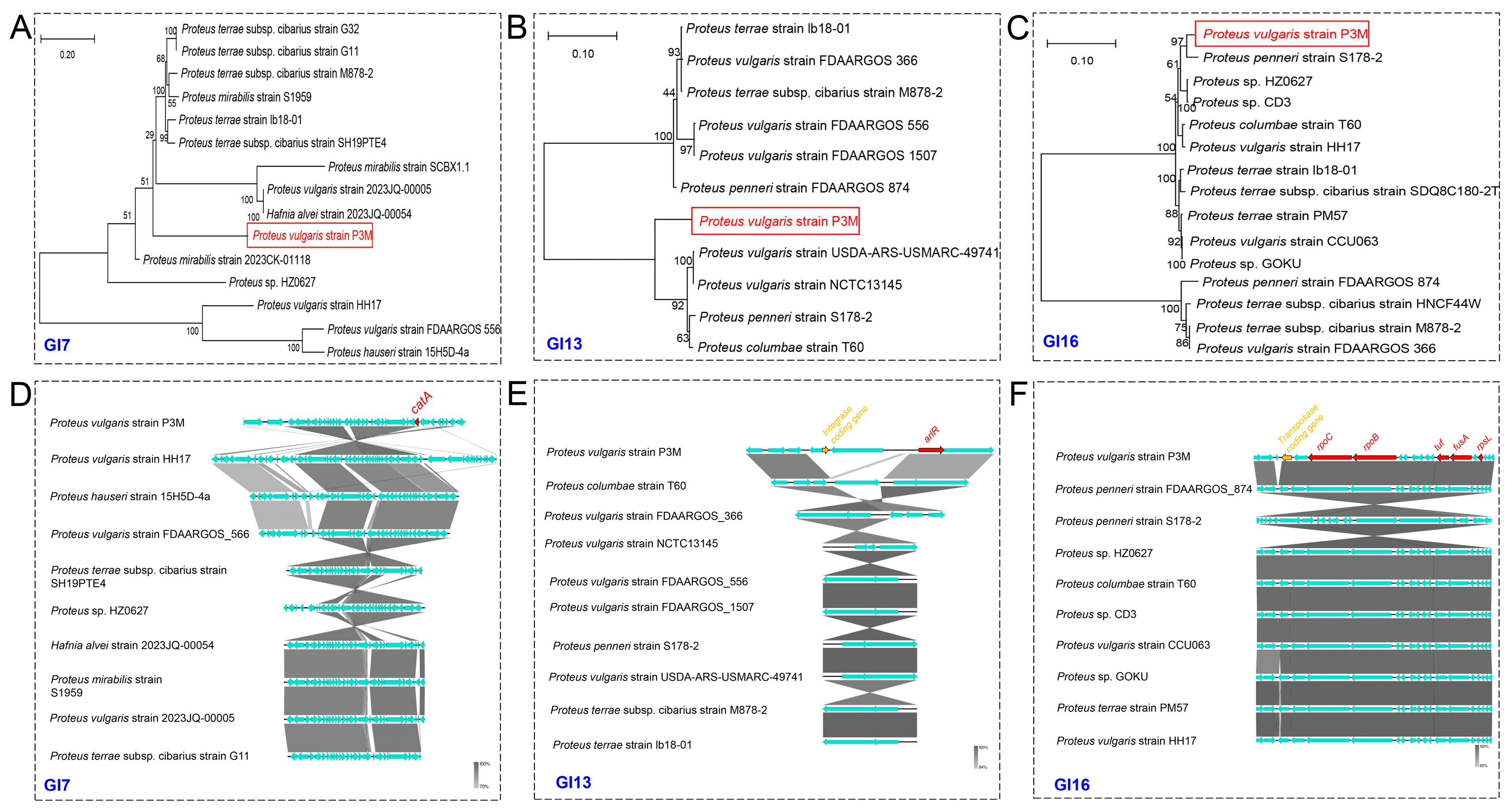
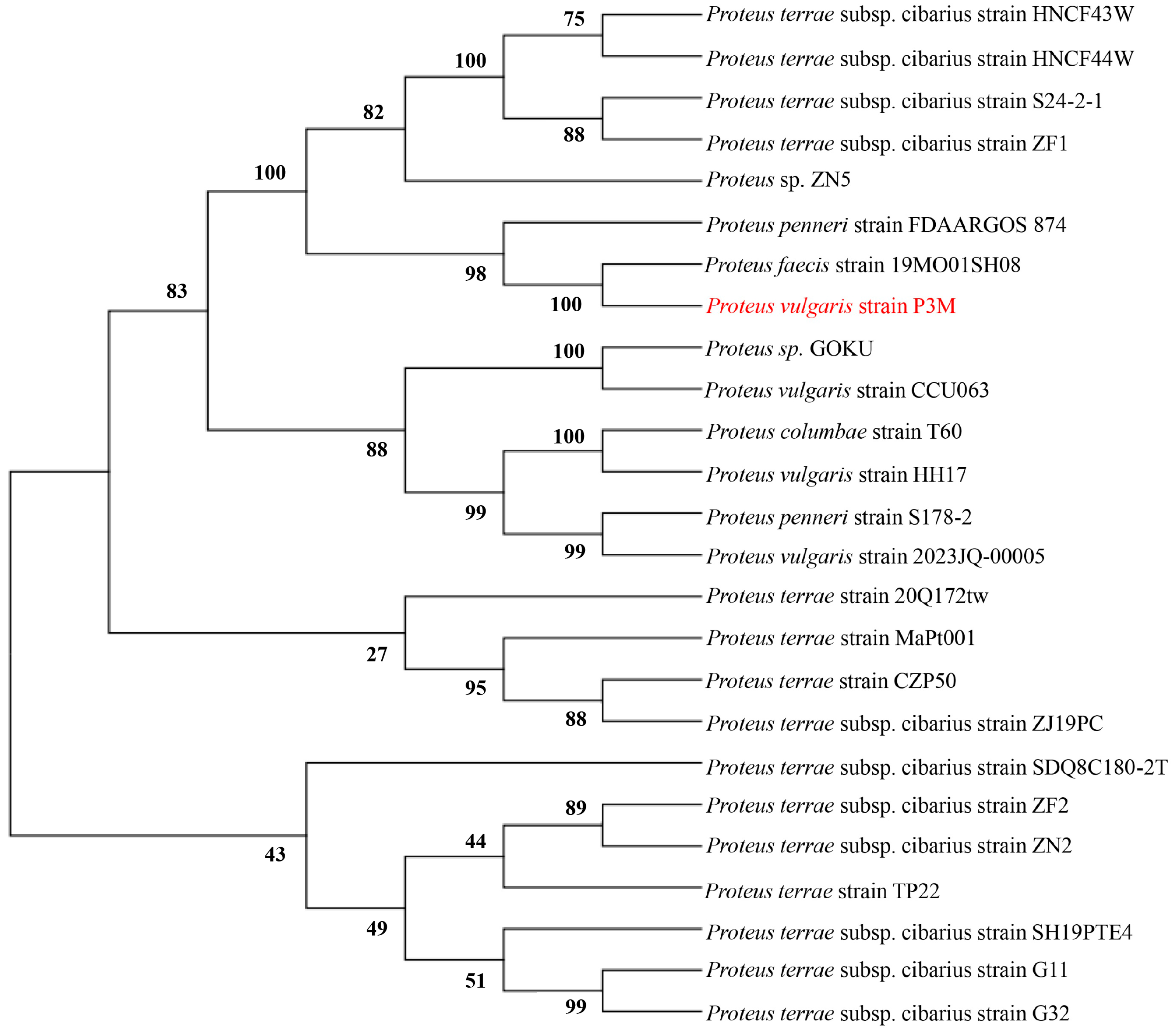
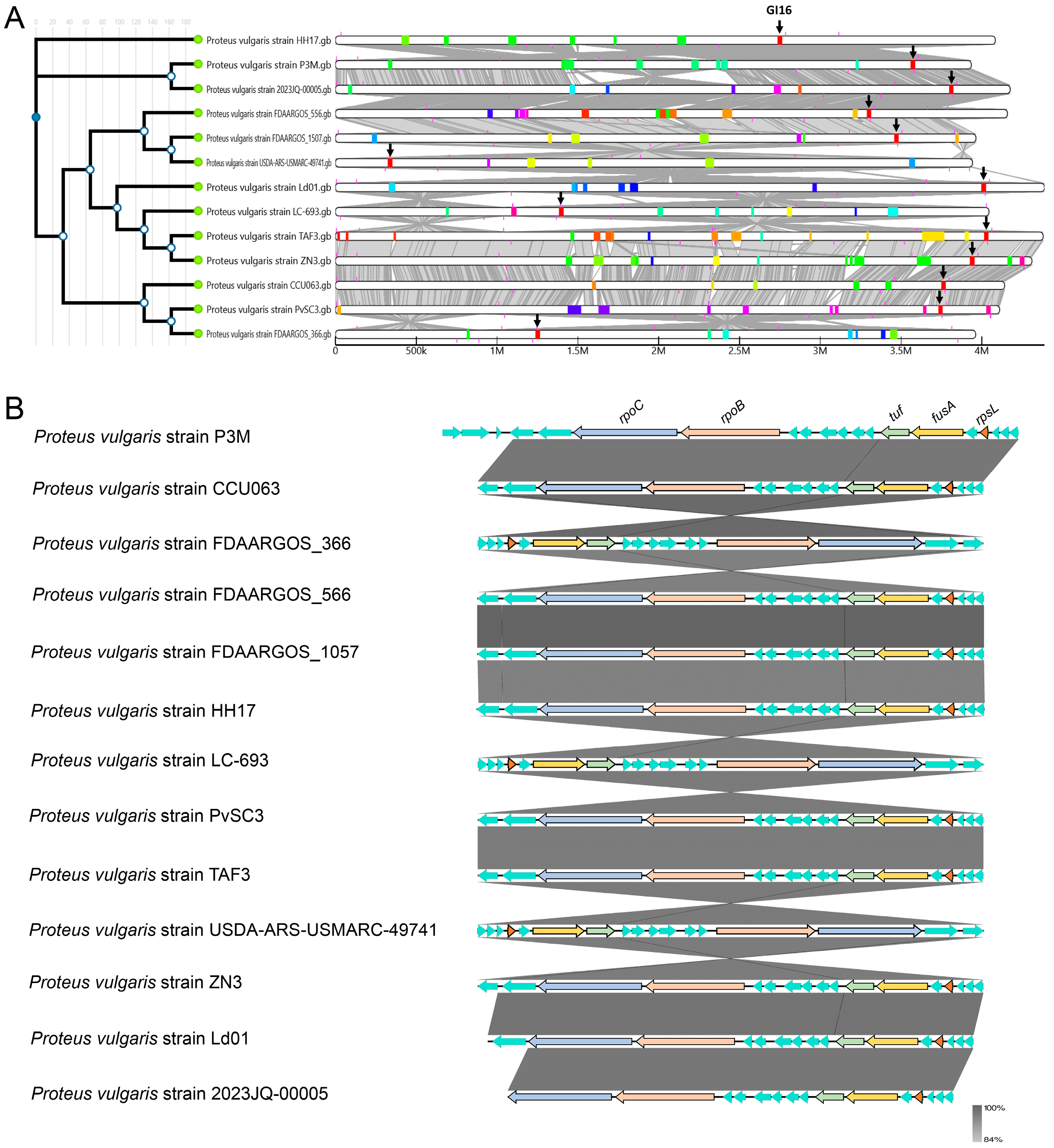
3.4. Evolutionary Conservation Analysis of Genomic Islands in P3M
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ferrari, R.G.; Rosario, D.K.A.; Cunha-Neto, A.; Mano, S.B.; Figueiredo, E.E.S.; Conte-Junior, C.A. Worldwide Epidemiology of Salmonella Serovars in Animal-Based Foods: A Meta-analysis. Appl. Environ. Microbiol. 2019, 85, e00591-19. [Google Scholar] [CrossRef]
- Babines-Orozco, L.; Balbuena-Alonso, M.G.; Barrios-Villa, E.; Lozano-Zarain, P.; Martínez-Laguna, Y.; Del Carmen Rocha-Gracia, R.; Cortés-Cortés, G. Antimicrobial resistance in food-associated Escherichia coli in Mexico and Latin America. Biosci. Microbiota Food Health 2024, 43, 4–12. [Google Scholar] [CrossRef]
- Li, R.; Zhou, M.; Lu, J.; Wei, J. Antibiofilm Effects of Epigallocatechin Gallate Against Proteus mirabilis Wild-Type and Ampicillin-Induced Strains. Foodborne Pathog. Dis. 2022, 19, 136–142. [Google Scholar] [CrossRef]
- Sergelidis, D.; Angelidis, A.S. Methicillin-resistant Staphylococcus aureus: A controversial food-borne pathogen. Lett. Appl. Microbiol. 2017, 64, 409–418. [Google Scholar] [CrossRef]
- Cangemi, J.R. Food poisoning and diarrhea: Small intestine effects. Curr. Gastroenterol. Rep. 2011, 13, 442–448. [Google Scholar] [CrossRef] [PubMed]
- Kocot, A.M.; Briers, Y.; Plotka, M. Phages and engineered lysins as an effective tool to combat Gram-negative foodborne pathogens. Compr. Rev. Food Sci. Food Saf. 2023, 22, 2235–2266. [Google Scholar] [CrossRef]
- Shakeran, Z.; Javadi-Zarnaghi, F.; Emamzadeh, R. Novel luminescent affiprobes for molecular detection of Staphylococcus aureus using flow cytometry. J. Appl. Microbiol. 2021, 130, 493–503. [Google Scholar] [CrossRef] [PubMed]
- Bakhshandeh, B.; Sorboni, S.G.; Haghighi, D.M.; Ahmadi, F.; Dehghani, Z.; Badiei, A. New analytical methods using carbon-based nanomaterials for detection of Salmonella species as a major food poisoning organism in water and soil resources. Chemosphere 2022, 287, 132243. [Google Scholar] [CrossRef] [PubMed]
- Ge, H.; Wang, Y.; Zhao, X. Research on the drug resistance mechanism of foodborne pathogens. Microb. Pathog. 2022, 162, 105306. [Google Scholar] [CrossRef]
- Asfaw, T.; Genetu, D.; Shenkute, D.; Shenkutie, T.T.; Amare, Y.E.; Yitayew, B. Foodborne Pathogens and Antimicrobial Resistance in Ethiopia: An Urgent Call for Action on “One Health”. Infect. Drug Resist. 2022, 15, 5265–5274. [Google Scholar] [CrossRef]
- Patil, A.; Banerji, R.; Kanojiya, P.; Saroj, S.D. Foodborne ESKAPE Biofilms and Antimicrobial Resistance: Lessons Learned from Clinical Isolates. Pathog. Glob. Health 2021, 115, 339–356. [Google Scholar] [CrossRef]
- Wu-Wu, J.W.F.; Guadamuz-Mayorga, C.; Oviedo-Cerdas, D.; Zamora, W.J. Antibiotic Resistance and Food Safety: Perspectives on New Technologies and Molecules for Microbial Control in the Food Industry. Antibiotics 2023, 12, 550. [Google Scholar] [CrossRef]
- Parra-Flores, J.; Holý, O.; Bustamante, F.; Lepuschitz, S.; Pietzka, A.; Contreras-Fernández, A.; Castillo, C.; Ovalle, C.; Alarcón-Lavín, M.P.; Cruz-Córdova, A.; et al. Virulence and Antibiotic Resistance Genes in Listeria monocytogenes Strains Isolated From Ready-to-Eat Foods in Chile. Front. Microbiol. 2022, 12, 796040. [Google Scholar] [CrossRef]
- Hartantyo, S.H.P.; Chau, M.L.; Koh, T.H.; Yap, M.; Yi, T.; Cao, D.Y.H.; GutiÉrrez, R.A.; Ng, L.C. Foodborne Klebsiella pneumoniae: Virulence Potential, Antibiotic Resistance, and Risks to Food Safety. J. Food Prot. 2020, 83, 1096–1103. [Google Scholar] [CrossRef] [PubMed]
- Tonkin-Hill, G.; Corander, J.; Parkhill, J. Challenges in prokaryote pangenomics. Microb. Genom. 2023, 9, mgen001021. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Wang, A. Genomic islands mediate environmental adaptation and the spread of antibiotic resistance in multiresistant Enterococci-evidence from genomic sequences. BMC Microbiol. 2021, 21, 55. [Google Scholar] [CrossRef] [PubMed]
- Lloyd, A.L.; Henderson, T.A.; Vigil, P.D.; Mobley, H.L. Genomic islands of uropathogenic Escherichia coli contribute to virulence. J. Bacteriol. 2009, 191, 3469–3481. [Google Scholar] [CrossRef]
- Li, B.; Chen, D.; Lin, F.; Wu, C.; Cao, L.; Chen, H.; Hu, Y.; Yin, Y. Genomic Island-Mediated Horizontal Transfer of the Erythromycin Resistance Gene erm(X) among Bifidobacteria. Appl. Environ. Microbiol. 2022, 88, e0041022. [Google Scholar] [CrossRef]
- Juhas, M.; van der Meer, J.R.; Gaillard, M.; Harding, R.M.; Hood, D.W.; Crook, D.W. Genomic islands: Tools of bacterial horizontal gene transfer and evolution. FEMS Microbiol. Rev. 2009, 33, 376–393. [Google Scholar] [CrossRef]
- Molina-Pardines, C.; Haro-Moreno, J.M.; López-Pérez, M. Phosphate-related genomic islands as drivers of environmental adaptation in the streamlined marine alphaproteobacterial HIMB59. mSystems 2023, 8, e0089823. [Google Scholar] [CrossRef]
- Chen, S.; Liu, H.; Liang, W.; Hong, L.; Zhang, B.; Huang, L.; Guo, X.; Duan, G. Insertion sequences in the CRISPR-Cas system regulate horizontal antimicrobial resistance gene transfer in Shigella strains. Int. J. Antimicrob. Agents 2019, 53, 109–115. [Google Scholar] [CrossRef] [PubMed]
- Che, Y.; Yang, Y.; Xu, X.; Břinda, K.; Polz, M.F.; Hanage, W.P.; Zhang, T. Conjugative plasmids interact with insertion sequences to shape the horizontal transfer of antimicrobial resistance genes. Proc. Natl. Acad. Sci. USA 2021, 118, e2008731118. [Google Scholar] [CrossRef] [PubMed]
- Das, B.K.; Kumar, V.; Baitha, R.; Ramteke, M.H.; Adhikari, A.; Bisai, K.; Jana, A.K.; Das, A.K. The emergence of multidrug-resistant Proteus vulgaris infection in cage reared Pangasianodon hypophthalmus: Molecular characterization and host-pathogen response. Microb. Pathog. 2024, 197, 107029. [Google Scholar] [CrossRef] [PubMed]
- Kateete, D.P.; Kabugo, U.; Baluku, H.; Nyakarahuka, L.; Kyobe, S.; Okee, M.; Najjuka, C.F.; Joloba, M.L. Prevalence and antimicrobial susceptibility patterns of bacteria from milkmen and cows with clinical mastitis in and around Kampala, Uganda. PLoS ONE 2013, 8, e63413. [Google Scholar] [CrossRef]
- Sridhar, A.; Krishnasamy, S.R.; Manikandan, D.B.; Arumugam, M.; Veeran, S.; Ramasamy, T. Activity profile of innate immune-related enzymes and bactericidal of freshwater fish epidermal mucus extract at different pH. Environ. Sci. Pollut. Res. Int. 2021, 28, 33914–33926. [Google Scholar] [CrossRef]
- Hamilton, A.L.; Kamm, M.A.; Ng, S.C.; Morrison, M. Proteus spp. as Putative Gastrointestinal Pathogens. Clin. Microbiol. Rev. 2018, 31, e00085-17. [Google Scholar] [CrossRef]
- Yu, X.; Torzewska, A.; Zhang, X.; Yin, Z.; Drzewiecka, D.; Cao, H.; Liu, B.; Knirel, Y.A.; Rozalski, A.; Wang, L. Genetic diversity of the O antigens of Proteus species and the development of a suspension array for molecular serotyping. PLoS ONE 2017, 12, e0183267. [Google Scholar] [CrossRef]
- Zhang, H.; Wang, H.; Ma, Z.; Liu, Y.; Wu, Z.; Xu, H.; Qiao, M. Characterization of Proteus vulgaris Strain P3M, a Foodborne Multidrug-Resistant Bacterium Isolated from Penaeus vannamei in China. Microb. Drug Resist. 2021, 27, 1360–1370. [Google Scholar] [CrossRef]
- Baron, S.; Leulmi, Z.; Villard, C.; Olaitan, A.O.; Telke, A.A.; Rolain, J.M. Inactivation of the arn operon and loss of aminoarabinose on lipopolysaccharide as the cause of susceptibility to colistin in an atypical clinical isolate of Proteus vulgaris. Int. J. Antimicrob. Agents 2018, 51, 50–457. [Google Scholar] [CrossRef]
- Sheng, H.; Zhao, L.; Suo, J.; Yang, Q.; Cao, C.; Chen, J.; Cui, G.; Fan, Y.; Ma, Y.; Huo, S.; et al. Niche-specific evolution and gene exchange of Salmonella in retail pork and chicken. Food Res. Int. 2024, 197, 115299. [Google Scholar] [CrossRef]
- Su, H.; Xia, T.; Xu, W.; Hu, X.; Xu, Y.; Wen, G.; Cao, Y. Temporal variations, distribution, and dissemination of antibiotic resistance genes and changes of bacterial communities in a biofloc-based zero-water-exchange mariculture system. Ecotoxicol. Environ. Saf. 2023, 256, 114904. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Lei, C.W.; Wang, H.N. Identification of a novel conjugative plasmid carrying the multiresistance gene cfr in Proteus vulgaris isolated from swine origin in China. Plasmid 2019, 105, 102440. [Google Scholar] [CrossRef] [PubMed]
- Drzewiecka, D. Significance and roles of Proteus spp. bacteria in natural environments. Microb. Ecol. 2016, 72, 741–758. [Google Scholar] [CrossRef] [PubMed]
- Beutlich, J.; Jahn, S.; Malorny, B.; Hauser, E.; Hühn, S.; Schroeter, A.; Rodicio, M.R.; Appel, B.; Threlfall, J.; Mevius, D.; et al. Med-Vet-Net WP21 Project Group. Antimicrobial resistance and virulence determinants in European Salmonella genomic island 1-positive Salmonella enterica isolates from different origins. Appl. Environ. Microbiol. 2011, 77, 5655–5664. [Google Scholar] [CrossRef]
- Hall, R.M. Salmonella genomic islands and antibiotic resistance in Salmonella enterica. Future Microbiol. 2010, 5, 1525–1538. [Google Scholar] [CrossRef]
- Yang, D.; Yang, Y.; Qiao, P.; Jiang, F.; Zhang, X.; Zhao, Z.; Cai, T.; Li, G.; Cai, W. Genomic Island-Encoded Histidine Kinase and Response Regulator Coordinate Mannose Utilization with Virulence in Enterohemorrhagic Escherichia coli. mBio 2023, 14, e0315222. [Google Scholar] [CrossRef]
- Zhou, Y.; Li, J.; Huang, F.; Ai, H.; Gao, J.; Chen, C.; Huang, L. Characterization of the pig lower respiratory tract antibiotic resistome. Nat. Commun. 2023, 14, 4868. [Google Scholar] [CrossRef]
- Turner, L.; Zhang, R.; Darnton, N.C.; Berg, H.C. Visualization of Flagella during bacterial Swarming. J. Bacteriol. 2010, 192, 3259–3267. [Google Scholar] [CrossRef]
- Chahales, P.; Thanassi, D.G. Structure, Function, and Assembly of Adhesive Organelles by Uropathogenic Bacteria. Microbiol. Spectr. 2015, 3, 39. [Google Scholar] [CrossRef]
- Lou, L.; Zhang, P.; Piao, R.; Wang, Y. Salmonella Pathogenicity Island 1 (SPI-1) and Its Complex Regulatory Network. Front. Cell. Infect. Microbiol. 2019, 9, 270. [Google Scholar] [CrossRef]
- Desvaux, M.; Dalmasso, G.; Beyrouthy, R.; Barnich, N.; Delmas, J.; Bonnet, R. Pathogenicity Factors of Genomic Islands in Intestinal and Extraintestinal Escherichia coli. Front. Microbiol. 2020, 11, 2065. [Google Scholar] [CrossRef]
- Alibayov, B.; Baba-Moussa, L.; Sina, H.; Zdeňková, K.; Demnerová, K. Staphylococcus aureus mobile genetic elements. Mol. Biol. Rep. 2014, 41, 5005–5018. [Google Scholar] [CrossRef]
- Bertelli, C.; Laird, M.R.; Williams, K.P.; Simon Fraser University Research Computing Group; Lau, B.Y.; Hoad, G.; Winsor, G.L.; Brinkman, F.S.L. IslandViewer 4: Expanded prediction of genomic islands for larger-scale datasets. Nucleic Acids Res. 2017, 45, W30–W35. [Google Scholar] [CrossRef]
- Bertelli, C.; Gray, K.L.; Woods, N.; Lim, A.C.; Tilley, K.E.; Winsor, G.L.; Hoad, G.R.; Roudgar, A.; Spencer, A.; Peltier, J.; et al. Enabling genomic island prediction and comparison in multiple genomes to investigate bacterial evolution and outbreaks. Microb. Genom. 2022, 8, mgen000818. [Google Scholar] [CrossRef] [PubMed]
- Alcock, B.P.; Huynh, W.; Chalil, R.; Smith, K.W.; Raphenya, A.R.; Wlodarski, M.A.; Edalatmand, A.; Petkau, A.; Syed, S.A.; Tsang, K.K.; et al. CARD 2023: Expanded curation, support for machine learning, and resistome prediction at the Comprehensive Antibiotic Resistance Database. Nucleic Acids Res. 2023, 51, D690–D699. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Nei, M.; Kumar, S. Prospects for inferring very large phylogenies by using the neighbor-joining method. Proc. Natl. Acad. Sci. USA 2004, 101, 11030–11035. [Google Scholar] [CrossRef]
- Sullivan, M.J.; Petty, N.K.; Beatson, S.A. Easyfig: A genome comparison visualizer. Bioinformatics 2011, 27, 1009–1010. [Google Scholar] [CrossRef] [PubMed]
- Tumapa, S.; Holden, M.T.; Vesaratchavest, M.; Wuthiekanun, V.; Limmathurotsakul, D.; Chierakul, W.; Feil, E.J.; Currie, B.J.; Day, N.P.; Nierman, W.C.; et al. Burkholderia pseudomallei genome plasticity associated with genomic island variation. BMC Genom. 2008, 9, 190. [Google Scholar] [CrossRef]
- Jorquera, M.A.; Acuña, J.J.; Huerta, N.; Bai, J.; Zhang, L.; Xiao, R.; Sadowsky, M.J. Multiple antibiotic resistance and herbicide catabolic profiles of bacteria isolated from Lake Villarrica surface sediments (Chile). Environ. Pollut. 2024, 358, 124538. [Google Scholar] [CrossRef]
- Letchumanan, V.; Yin, W.F.; Lee, L.H.; Chan, K.G. Prevalence and antimicrobial susceptibility of Vibrio parahaemolyticus isolated from retail shrimps in Malaysia. Front. Microbiol. 2015, 6, 33. [Google Scholar] [CrossRef]
- Gu, W.; Li, W.; Jia, S.; Zhou, Y.; Yin, J.; Wu, Y.; Fu, X. Antibiotic resistance and genomic features of Clostridioides difficile in southwest China. PeerJ 2022, 10, e14016. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Chang, M.; Zhang, X.; Cai, P.; Dai, Y.; Song, T.; Wu, Z.; Xu, H.; Qiao, M. Functional Identification and Evolutionary Analysis of Two Novel Plasmids Mediating Quinolone Resistance in Proteus vulgaris. Microorganisms 2020, 8, 1074. [Google Scholar] [CrossRef] [PubMed]
- Marques, P.H.; Prado, L.C.D.S.; Felice, A.G.; Rodrigues, T.C.V.; Pereira, U.P.; Jaiswal, A.K.; Azevedo, V.; Oliveira, C.J.F.; Soares, S. Insights into the Vibrio Genus: A One Health Perspective from Host Adaptability and Antibiotic Resistance to In Silico Identification of Drug Targets. Antibiotics 2022, 11, 1399. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genomic Island (GI) | Start | End | Size |
|---|---|---|---|
| GI1 | 330,090 | 348,336 | 18,246 |
| GI2 | 339,053 | 343,460 | 4407 |
| GI3 | 943,652 | 948,353 | 4701 |
| GI4 | 1,401,531 | 1,415,713 | 14,182 |
| GI5 | 1,418,845 | 1,426,992 | 8147 |
| GI6 | 1,423,836 | 1,473,830 | 49,994 |
| GI7 | 1,865,071 | 1,899,237 | 34,166 |
| GI8 | 2,206,196 | 2,244,624 | 38,428 |
| GI9 | 2,299,849 | 2,305,598 | 5749 |
| GI10 | 2,358,993 | 2,377,861 | 18,868 |
| GI11 | 2,378,798 | 2,383,241 | 4443 |
| GI12 | 2,388,800 | 2,424,554 | 35,754 |
| GI13 | 2,714,880 | 2,721,564 | 6684 |
| GI14 | 3,170,960 | 3,175,456 | 4496 |
| GI15 | 3,221,960 | 3,234,561 | 12,601 |
| GI16 | 3,561,510 | 3,584,877 | 23,367 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, H.; Wu, T.; Ruan, H. Unveiling Genomic Islands Hosting Antibiotic Resistance Genes and Virulence Genes in Foodborne Multidrug-Resistant Patho-Genic Proteus vulgaris. Biology 2025, 14, 858. https://doi.org/10.3390/biology14070858
Zhang H, Wu T, Ruan H. Unveiling Genomic Islands Hosting Antibiotic Resistance Genes and Virulence Genes in Foodborne Multidrug-Resistant Patho-Genic Proteus vulgaris. Biology. 2025; 14(7):858. https://doi.org/10.3390/biology14070858
Chicago/Turabian StyleZhang, Hongyang, Tao Wu, and Haihua Ruan. 2025. "Unveiling Genomic Islands Hosting Antibiotic Resistance Genes and Virulence Genes in Foodborne Multidrug-Resistant Patho-Genic Proteus vulgaris" Biology 14, no. 7: 858. https://doi.org/10.3390/biology14070858
APA StyleZhang, H., Wu, T., & Ruan, H. (2025). Unveiling Genomic Islands Hosting Antibiotic Resistance Genes and Virulence Genes in Foodborne Multidrug-Resistant Patho-Genic Proteus vulgaris. Biology, 14(7), 858. https://doi.org/10.3390/biology14070858