
Comprehensive Analysis of the Complete Mitochondrial Genome of Paeonia ludlowii Reveals a Dual-Circular Structure and Extensive Inter-Organellar Gene Transfer

 , and
, and
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample and Sequencing Data Source
2.2. Mitochondrial Genome Assembly and Annotation
2.3. Repetitive Sequence Analysis
2.4. Codon Usage Preference Analysis
2.5. RNA Editing Site Prediction
2.6. Homologous Fragment Analysis of Organelle Genomes
2.7. Ka/Ks Ratio Evaluation
2.8. Calculation of Nucleotide Diversity
2.9. Comparative Analysis of Mitochondrial Genome Structure
2.10. Phylogenetic Analysis
3. Results
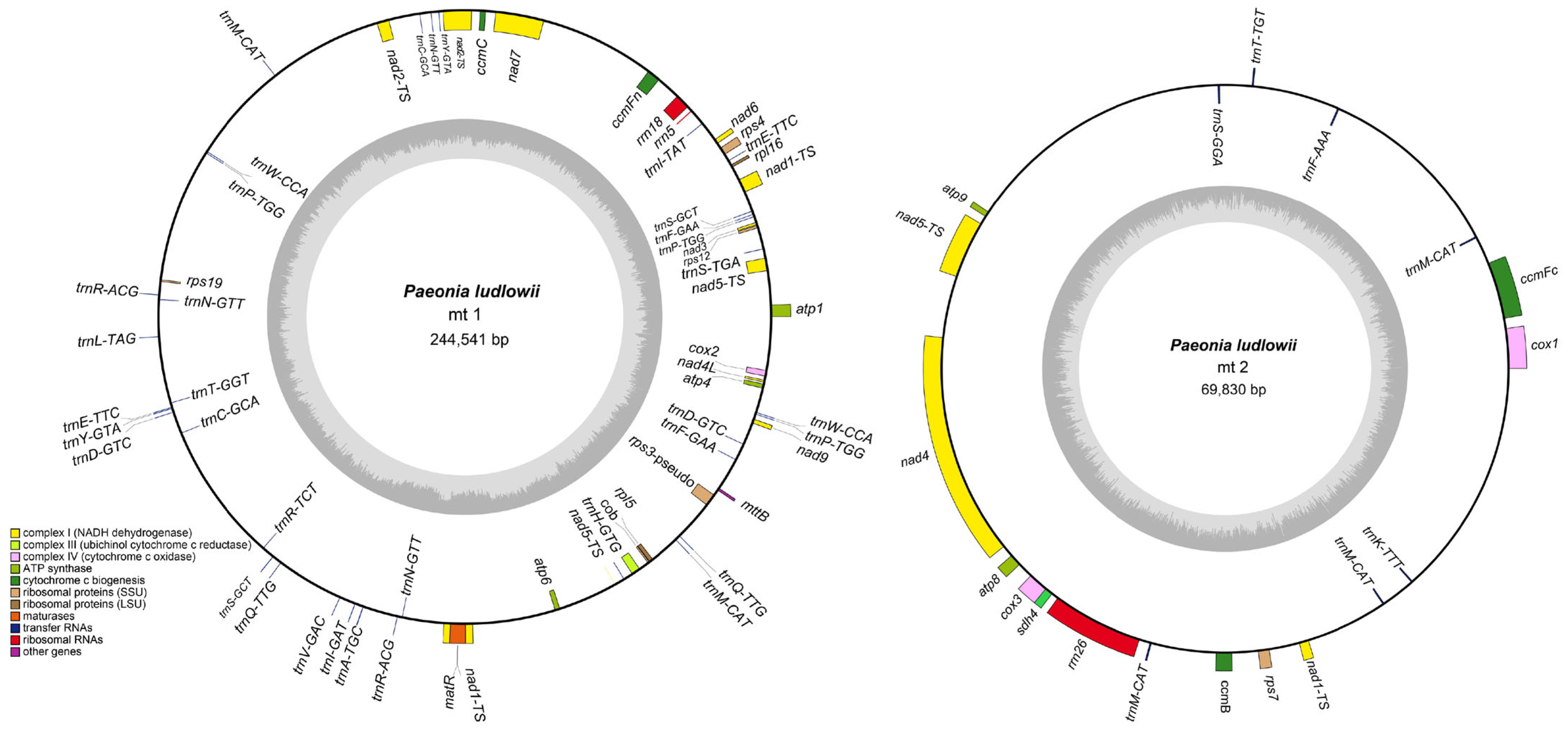
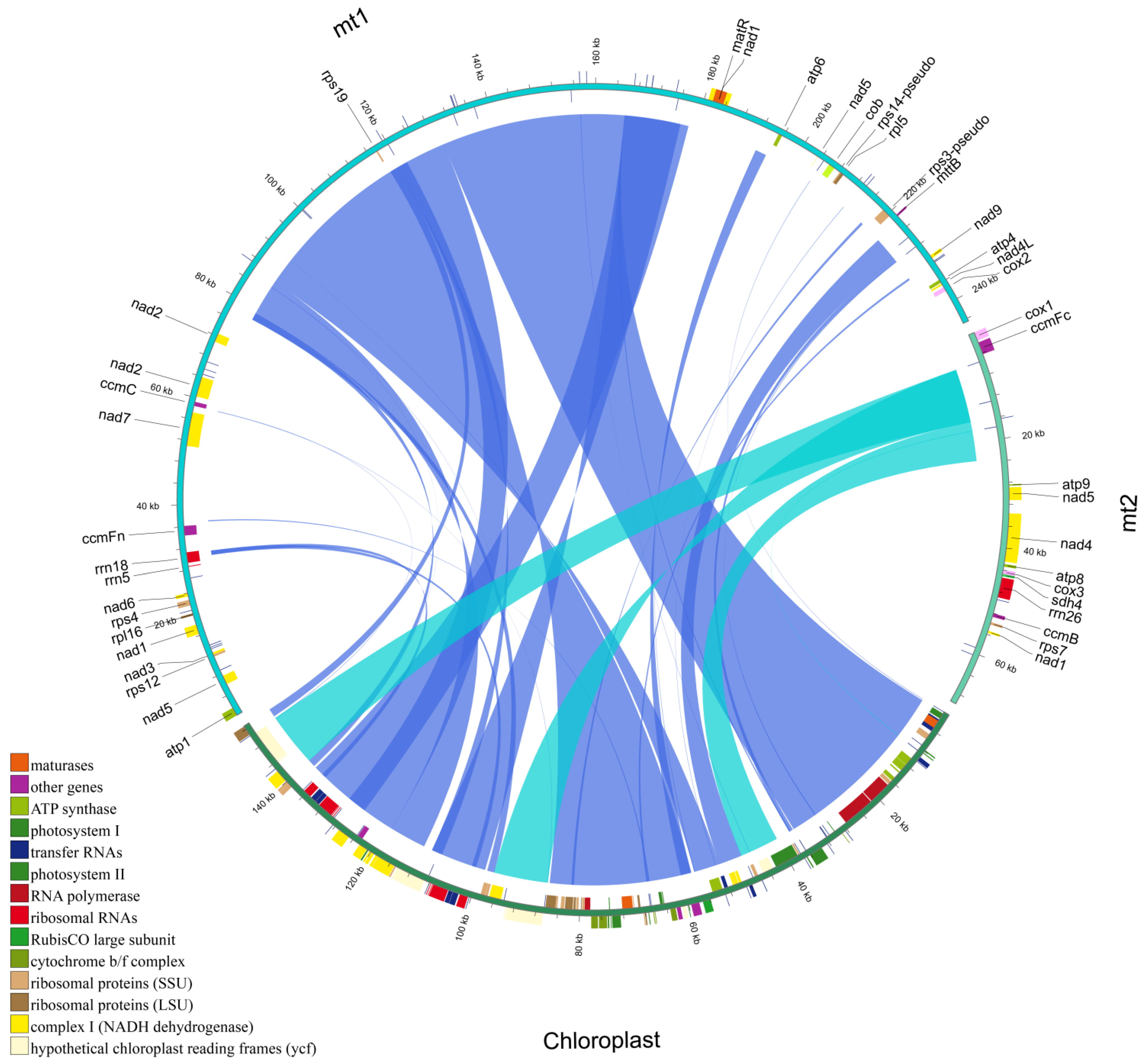
3.1. Mitochondrial Genome Structure and Content
3.2. Analysis of Repetitive Sequences
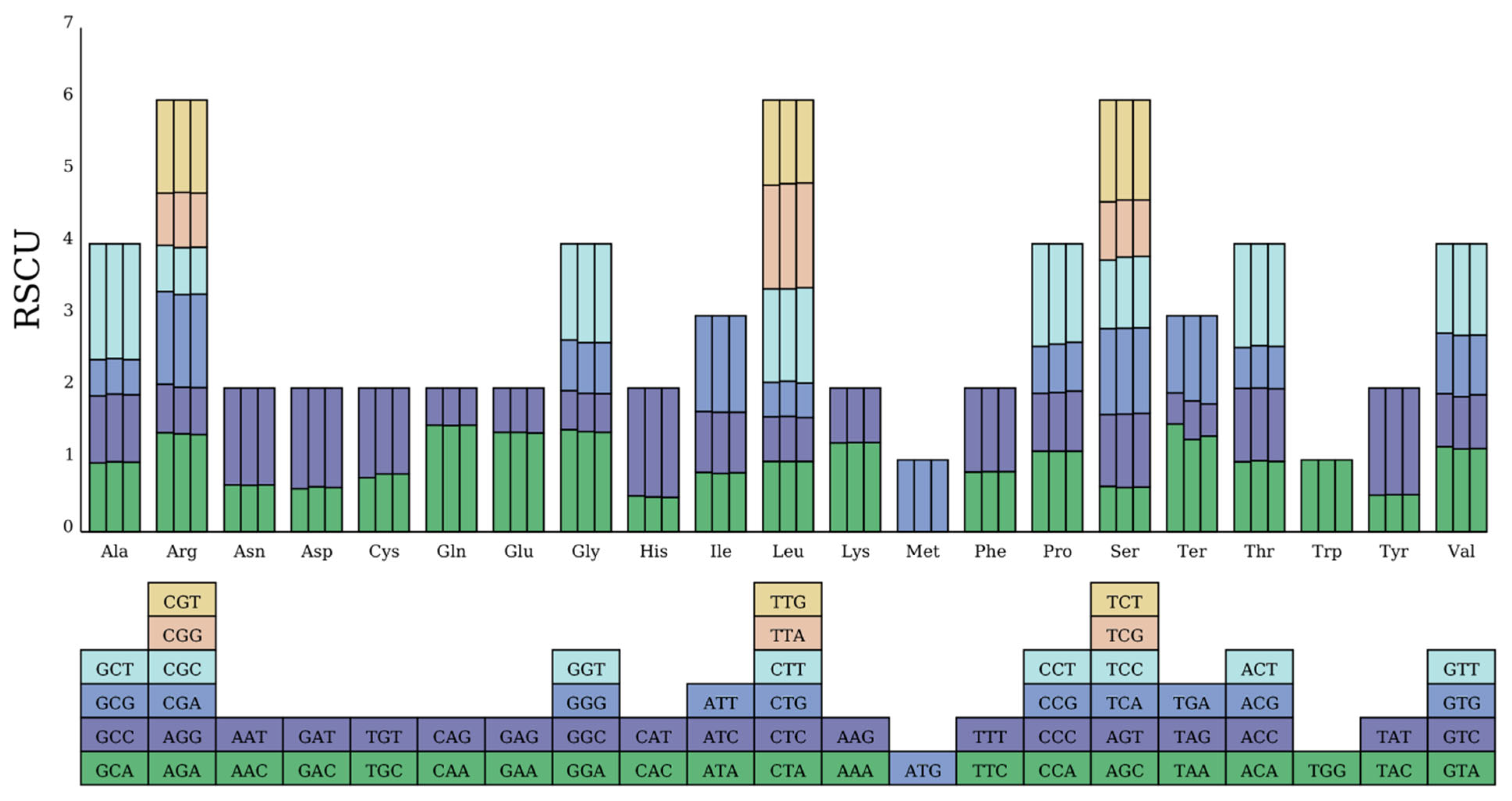
3.3. Codon Usage Analysis
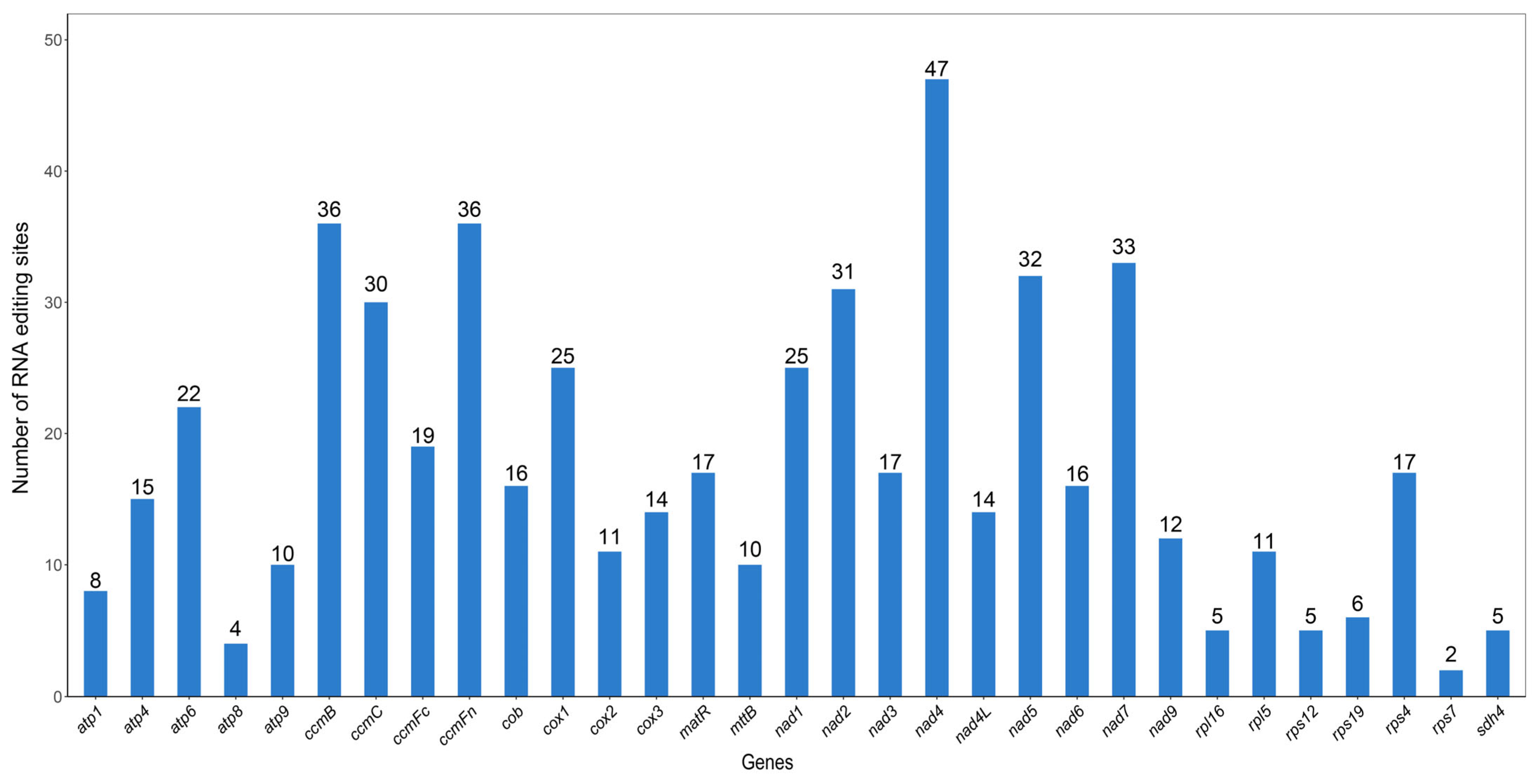
3.4. RNA Editing Prediction
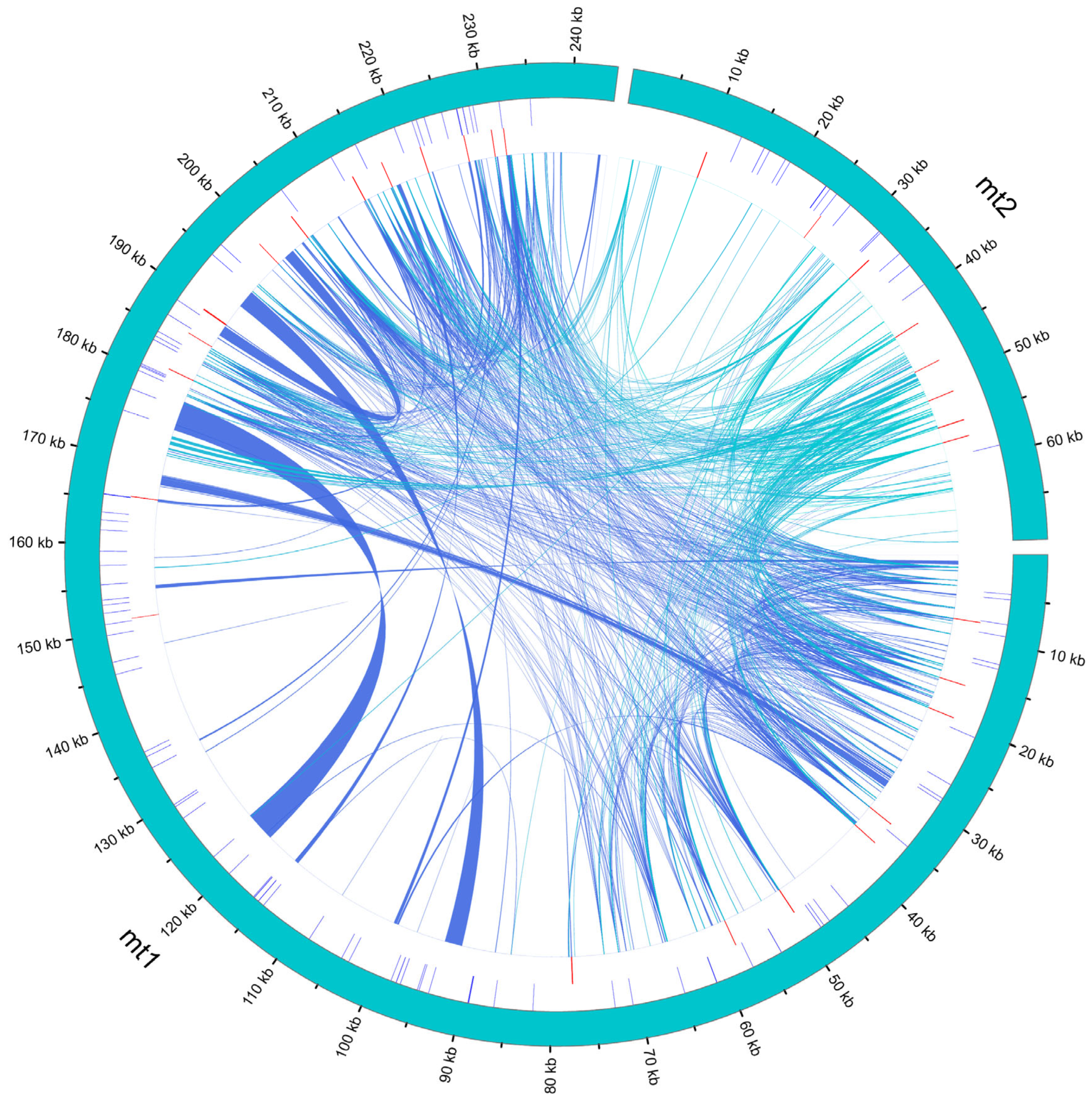
3.5. Homologous Fragment Analysis
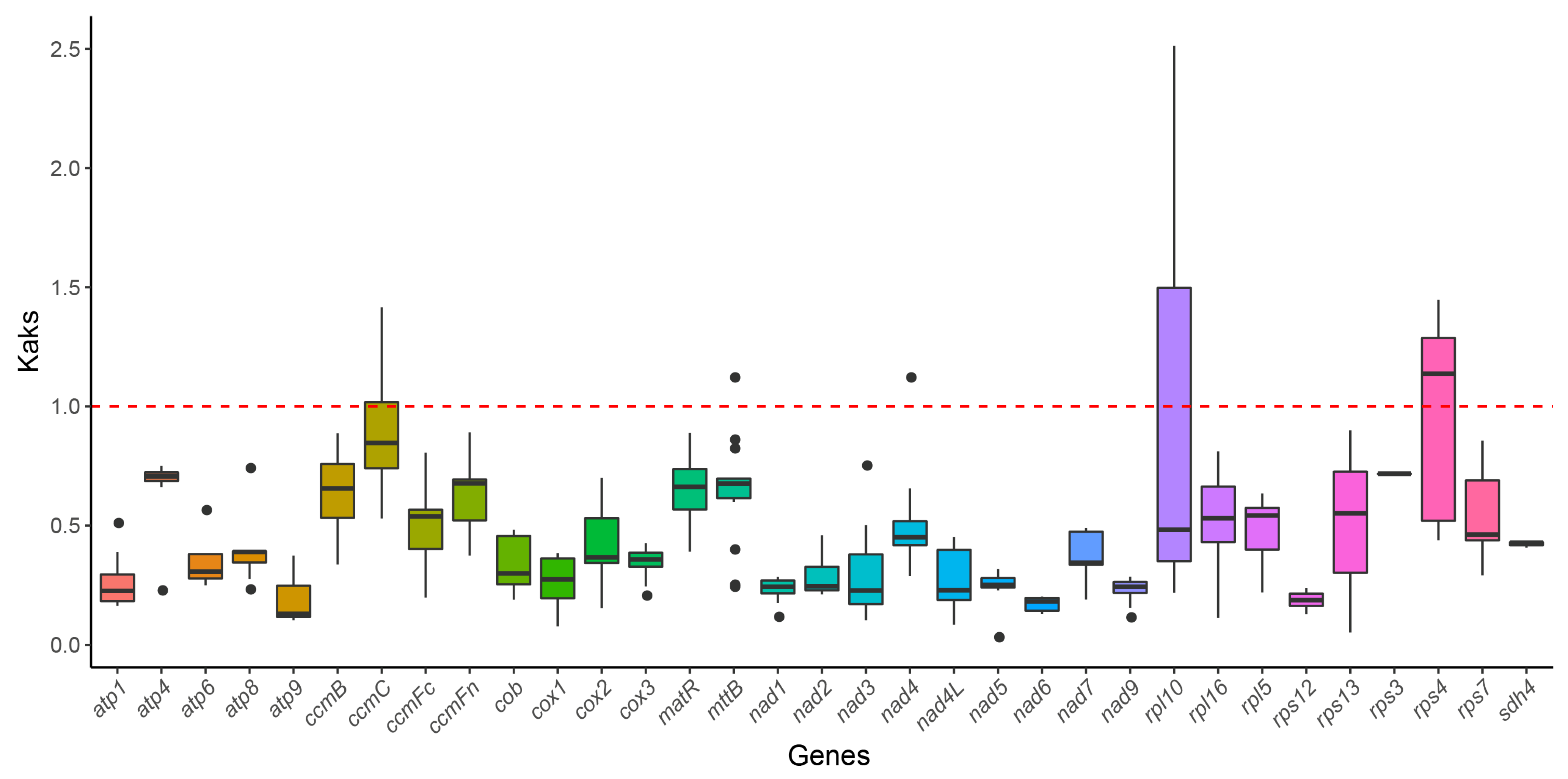
3.6. Ka/Ks Ratio Analysis
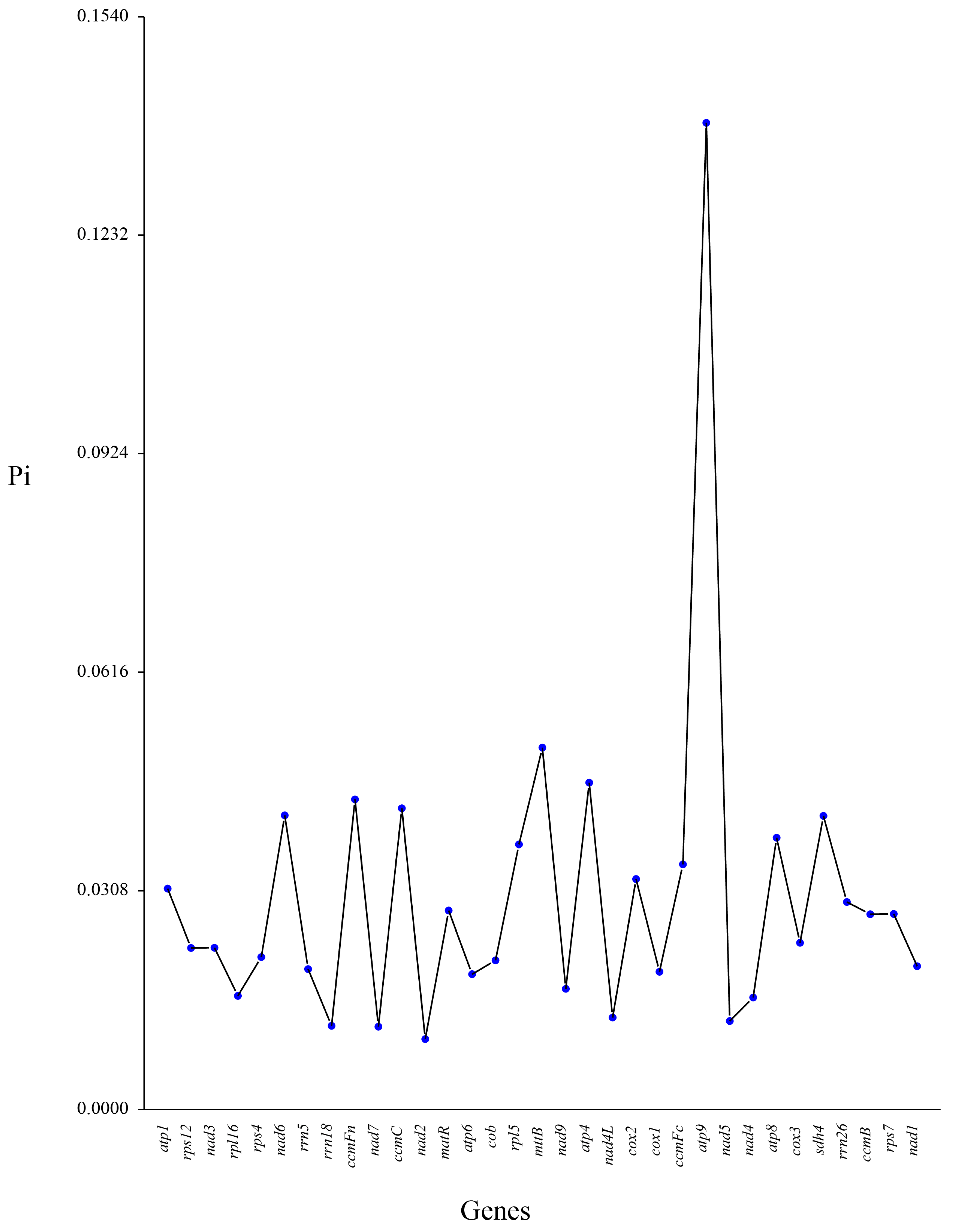
3.7. Comparative Nucleotide Diversity Analysis
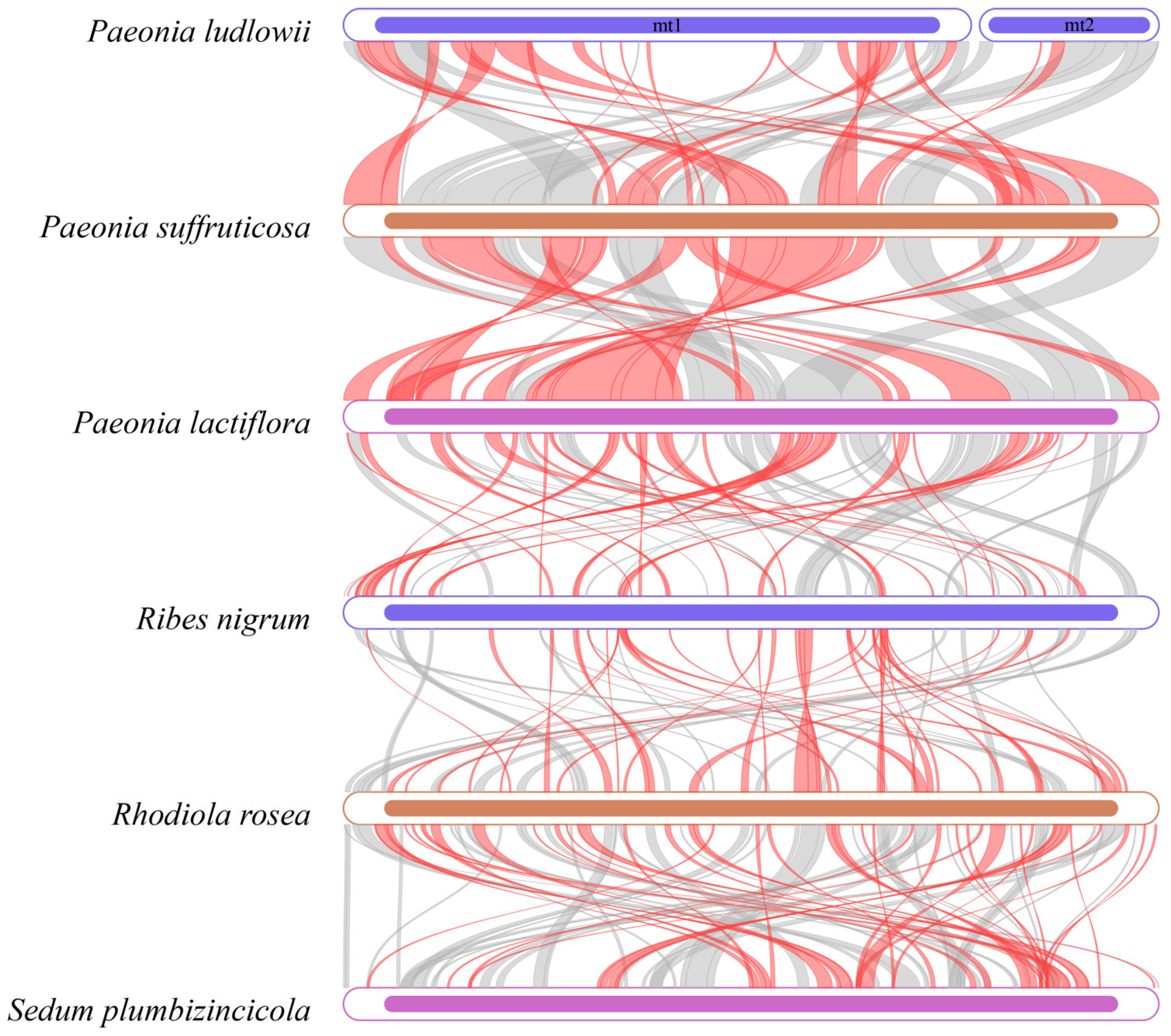
3.8. Collinearity Analysis
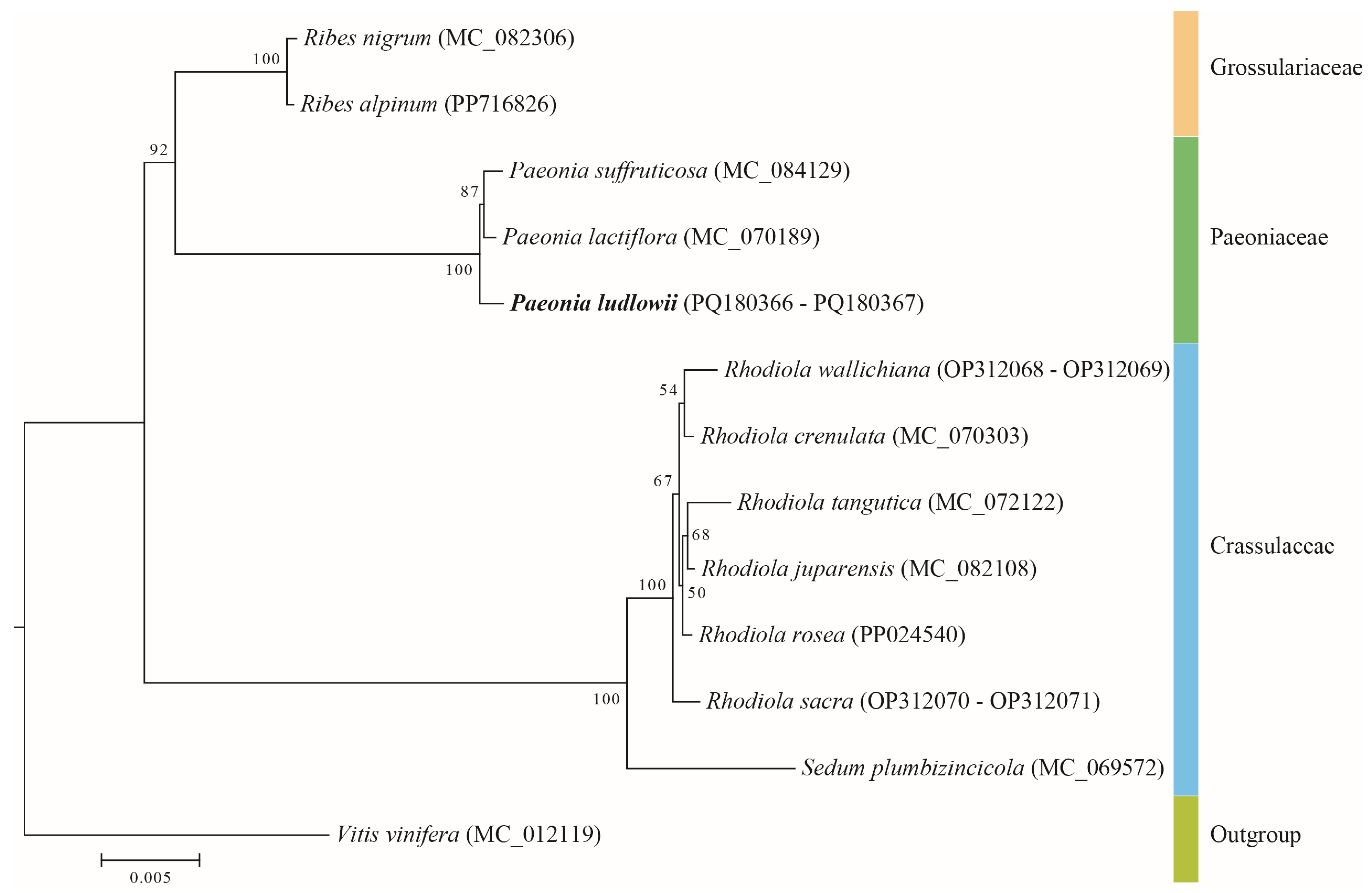
3.9. Phylogenetic Relationships
4. Discussion
4.1. Mitogenome Architecture and Gene Content
4.2. Repetitive Elements and Genome Complexity
4.3. Codon Usage Patterns and Translation Optimization
4.4. RNA Editing Site Prediction and Functional Implications
4.5. Inter-Organellar DNA Transfer and Genomic Evolution
4.6. Selective Pressures and Evolutionary Constraints
4.7. Phylogenetic Analysis and Genome Structural Evolution
4.8. Study Limitations and Future Research Directions
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mackenzie, S.; McIntosh, L. Higher plant mitochondria. Plant Cell 1999, 11, 571–585. [Google Scholar] [CrossRef]
- Barreto, P.; Koltun, A.; Nonato, J.; Yassitepe, J.; Maia, I.D.G.; Arruda, P. Metabolism and signaling of plant mitochondria in adaptation to environmental stresses. Int. J. Mol. Sci. 2022, 23, 11176. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Z.; Zhang, Z.; Tso, N.; Zhang, S.; Chen, Y.; Shu, Q.; Li, J.; Liang, Z.; Wang, R.; Wang, J.; et al. Complete mitochondrial genome of Hippophae tibetana: Insights into adaptation to high-altitude environments. Front. Plant Sci. 2024, 15, 1449606. [Google Scholar] [CrossRef]
- Wynn, E.L.; Christensen, A.C. Repeats of unusual size in plant mitochondrial genomes: Identification, incidence and evolution. G3 Genes Genomes Genet. 2019, 9, 549–559. [Google Scholar] [CrossRef] [PubMed]
- Lang, B.F.; Gray, M.W.; Burger, G. Mitochondrial genome evolution and the origin of eukaryotes. Annu. Rev. Genet. 1999, 33, 351–397. [Google Scholar] [CrossRef] [PubMed]
- Turmel, M.; Otis, C.; Lemieux, C. The complete mitochondrial DNA sequence of Mesostigma viride identifies this green alga as the earliest green plant divergence and predicts a highly compact mitochondrial genome in the ancestor of all green plants. Mol. Biol. Evol. 2022, 19, 24–38. [Google Scholar] [CrossRef]
- Putintseva, Y.A.; Bondar, E.I.; Simonov, E.P.; Sharov, V.V.; Oreshkova, N.V.; Kuzmin, D.A.; Konstantinov, Y.M.; Shmakov, V.N.; Belkov, V.I.; Sadovsky, M.G.; et al. Siberian larch (Larix sibirica Ledeb.) mitochondrial genome assembled using both short and long nucleotide sequence reads is currently the largest known mitogenome. BMC Genom. 2020, 21, 1–12. [Google Scholar] [CrossRef]
- Xiong, A.S.; Peng, R.H.; Zhuang, J.; Gao, F.; Zhu, B.; Fu, X.Y.; Xue, Y.; Jin, X.F.; Tian, Y.S.; Zhao, W.; et al. Gene duplication and transfer events in plant mitochondria genome. Biochem. Biophys. Res. Commun. 2008, 376, 1–4. [Google Scholar] [CrossRef]
- Møller, I.M.; Rasmusson, A.G.; Van Aken, O. Plant mitochondria–past, present and future. Plant J. 2021, 108, 912–959. [Google Scholar] [CrossRef]
- Sloan, D.B. One ring to rule them all? Genome sequencing provides new insights into the ‘master circle’model of plant mitochondrial DNA structure. New Phytol. 2013, 200, 978–985. [Google Scholar] [CrossRef]
- Yang, Z.; Ni, Y.; Lin, Z.; Yang, L.; Chen, G.; Nijiati, N.; Hu, y.; Chen, X. De novo assembly of the complete mitochondrial genome of sweet potato (Ipomoea batatas [L.] Lam) revealed the existence of homologous conformations generated by the repeat-mediated recombination. BMC Plant Biol. 2022, 22, 285. [Google Scholar] [CrossRef] [PubMed]
- Christensen, A.C. Plant mitochondrial genome evolution can be explained by DNA repair mechanisms. Genome Biol. Evol. 2013, 5, 1079–1086. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Cao, D.; Li, S.; Su, A.; Geng, J.; Grover, C.E.; Hu, S.; Hua, J. The complete mitochondrial genome of Gossypium hirsutum and evolutionary analysis of higher plant mitochondrial genomes. PLoS ONE 2013, 8, e69476. [Google Scholar] [CrossRef]
- Maréchal, A.; Brisson, N. Recombination and the maintenance of plant organelle genome stability. New Phytol. 2010, 186, 299–317. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Kan, S.; Liao, X.; Zhou, J.; Tembrock, L.R.; Daniell, H.; Jin, S.; Wu, Z. Plant organellar genomes: Much done, much more to do. Trends Plant Sci. 2024, 29, 754–769. [Google Scholar] [CrossRef]
- Yuan, Y.; Bayer, P.E.; Batley, J.; Edwards, D. Improvements in genomic technologies: Application to crop genomics. Trends Biotechnol. 2017, 35, 547–558. [Google Scholar] [CrossRef]
- Lang, D.; Zhang, S.; Ren, P.; Liang, F.; Sun, Z.; Meng, G.; Tan, Y.; Li, X.; Lai, Q.; Han, L.; et al. Comparison of the two up-to-date sequencing technologies for genome assembly: HiFi reads of Pacific Biosciences Sequel II system and ultralong reads of Oxford Nanopore. Gigascience 2020, 9, giaa123. [Google Scholar] [CrossRef]
- Yu, H.; Deane, D.C.; Sui, X.; Fang, S.; Chu, C.; Liu, Y.; He, F. Testing multiple hypotheses for the high endemic plant diversity of the Tibetan Plateau. Glob. Ecol. Biogeogr. 2019, 28, 131–144. [Google Scholar] [CrossRef]
- Yang, J.; Cai, L.; Liu, D.; Chen, G.; Gratzfeld, J.; Sun, W. China’s conservation program on plant species with extremely small populations (PSESP): Progress and perspectives. Biol. Conserv. 2020, 244, 108535. [Google Scholar] [CrossRef]
- Hong, D.Y.; Zhou, S.L.; He, X.J.; Yuan, J.H.; Zhang, Y.L.; Cheng, F.Y.; Zeng, X.L.; Wang, Y.; Zhang, X.X. Current status of wild tree peony species with special reference to conservation. Biodivers. Sci. 2017, 25, 781. [Google Scholar] [CrossRef]
- Xiao, P.X.; Li, Y.; Lu, J.; Zuo, H.; Pingcuo, G.; Ying, H.; Zhao, F.; Xu, Q.; Zeng, X.; Jiao, W.B. High-quality assembly and methylome of a Tibetan wild tree peony genome (Paeonia ludlowii) reveal the evolution of giant genome architecture. Hortic. Res. 2023, 10, uhad241. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.J.; Yin, G.S.; Gong, X. RAD-sequencing improves the genetic characterization of a threatened tree peony (Paeonia ludlowii) endemic to China: Implications for conservation. Plant Divers. 2023, 45, 513–522. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Du, H.; Wang, L.; Shu, Q.; Zheng, Y.; Xu, Y.; Zhang, J.; Zhang, J.; Yang, R.; Ge, Y. Flavonoid composition and antioxidant activity of tree peony (Paeonia section Moutan) yellow flowers. J. Agric. Food Chem. 2009, 57, 8496–8503. [Google Scholar] [CrossRef]
- Zhang, C.Q.; Xu, Y.J.; Lu, Y.Z.; Li, L.Q.; Lan, X.Z.; Zhong, Z.C. Study on the fatty acids, aromatic compounds and shelf life of Paeonia ludlowii kernel oil. J. Oleo Sci. 2020, 69, 1001–1009. [Google Scholar] [CrossRef]
- Zhang, X.X.; Shi, Q.Q.; Ji, D.; Niu, L.X.; Zhang, Y.L. Determination of the phenolic content, profile, and antioxidant activity of seeds from nine tree peony (Paeonia section Moutan DC) species native to China. Food Res. Int. 2017, 97, 141–148. [Google Scholar] [CrossRef] [PubMed]
- Li, J.J.; Chen, D.Z.; Yu, L.; He, L.X.; Chen, X.L. A study on taxonomic position of Paeonia ludlowii. Bull. Bot. Res. 1998, 18, 152. [Google Scholar]
- Cai, H.; Xu, R.; Tian, P.; Zhang, M.; Zhu, L.; Yin, T.; Zhang, H.; Liu, X. Complete Chloroplast Genomes and the Phylogenetic Analysis of Three Native Species of Paeoniaceae from the Sino-Himalayan Flora Subkingdom. Int. J. Mol. Sci. 2023, 25, 257. [Google Scholar] [CrossRef]
- Tang, P.; Ni, Y.; Li, J.; Lu, Q.; Liu, C.; Guo, J. The Complete Mitochondrial Genome of Paeonia lactiflora Pall. (Saxifragales: Paeoniaceae): Evidence of Gene Transfer from Chloroplast to Mitochondrial Genome. Genes 2024, 15, 239. [Google Scholar]
- Bi, C.; Shen, F.; Han, F.; Qu, Y.; Hou, J.; Xu, K.; Xu, L.A.; He, W.; Wu, Z.; Yin, T. PMAT: An efficient plant mitogenome assembly toolkit using low-coverage HiFi sequencing data. Hortic. Res. 2024, 11, uhae023. [Google Scholar] [CrossRef]
- Wick, R.R.; Schultz, M.B.; Zobel, J.; Holt, K.E. Bandage: Interactive visualization of de novo genome assemblies. Bioinformatics 2015, 31, 3350–3352. [Google Scholar] [CrossRef]
- Chen, Y.; Ye, W.; Zhang, Y.; Xu, Y. High speed BLASTN: An accelerated MegaBLAST search tool. Nucleic Acids Res. 2015, 43, 7762–7768. [Google Scholar] [CrossRef]
- Tillich, M.; Lehwark, P.; Pellizzer, T.; Ulbricht-Jones, E.S.; Fischer, A.; Bock, R.; Greiner, S. GeSeq–versatile and accurate annotation of organelle genomes. Nucleic Acids Res. 2017, 45, W6–W11. [Google Scholar] [CrossRef] [PubMed]
- Chan, P.P.; Lin, B.Y.; Mak, A.J.; Lowe, T.M. tRNAscan-SE 2.0: Improved detection and functional classification of transfer RNA genes. Nucleic Acids Res. 2021, 49, 9077–9096. [Google Scholar] [CrossRef]
- Lee, E.; Harris, N.; Gibson, M.; Chetty, R.; Lewis, S. Apollo: A community resource for genome annotation editing. Bioinformatics 2009, 25, 1836–1837. [Google Scholar] [CrossRef]
- Shi, L.; Chen, H.; Jiang, M.; Wang, L.; Wu, X.; Huang, L.; Liu, C. CPGAVAS2, an integrated plastome sequence annotator and analyzer. Nucleic Acids Res. 2019, 47, W65–W73. [Google Scholar] [CrossRef]
- Beier, S.; Thiel, T.; Münch, T.; Scholz, U.; Mascher, M. MISA-web: A web server for microsatellite prediction. Bioinformatics 2017, 33, 2583–2585. [Google Scholar] [CrossRef] [PubMed]
- Benson, G. Tandem repeats finder: A program to analyze DNA sequences. Nucleic Acids Res. 1999, 27, 573–580. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Gao, F.; Jakovlić, I.; Zou, H.; Zhang, J.; Li, W.X.; Wang, G.T. PhyloSuite: An integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Mol. Ecol. Resour. 2020, 20, 348–355. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Wang, D.; Zhang, Y.; Zhang, Z.; Zhu, J.; Yu, J. KaKs_Calculator 2.0: A toolkit incorporating gamma-series methods and sliding window strategies. GPB 2010, 8, 77–80. [Google Scholar] [CrossRef]
- Rozas, J.; Ferrer-Mata, A.; Sánchez-DelBarrio, J.C.; Guirao-Rico, S.; Librado, P.; Ramos-Onsins, S.E.; Sánchez-Gracia, A. DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol. Biol. Evol. 2017, 34, 3299–3302. [Google Scholar] [CrossRef]
- Marçais, G.; Delcher, A.L.; Phillippy, A.M.; Coston, R.; Salzberg, S.L.; MUMmer, A.Z. A fast and versatile genome alignment system. PLoS Comput. Biol. 2018, 14, e1005944. [Google Scholar] [CrossRef]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. jModelTest 2: More models, new heuristics and high-performance computing. Nat. Methods 2012, 9, 772. [Google Scholar] [CrossRef]
- Nguyen, L.T.; Schmidt, H.A.; Von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Mehrotra, S.; Goyal, V. Repetitive sequences in plant nuclear DNA: Types, distribution, evolution and function. GPB 2014, 12, 164–171. [Google Scholar] [CrossRef]
- Gualberto, J.M.; Mileshina, D.; Wallet, C.; Niazi, A.K.; Weber-Lotfi, F.; Dietrich, A. The plant mitochondrial genome: Dynamics and maintenance. Biochimie 2014, 100, 107–120. [Google Scholar] [CrossRef]
- Li, X.; Han, Q.; Li, M.; Luo, Q.; Zhu, S.; Zheng, Y.; Tan, G. Complete Mitochondrial Genome Sequence, Characteristics, and Phylogenetic Analysis of Oenanthe javanica. Agronomy 2023, 13, 2103. [Google Scholar] [CrossRef]
- Small, I.D.; Schallenberg-Rüdinger, M.; Takenaka, M.; Mireau, H.; Ostersetzer-Biran, O. Plant organellar RNA editing: What 30 years of research has revealed. Plant J. 2020, 101, 1040–1056. [Google Scholar] [CrossRef]
- Knoop, V. C-to-U and U-to-C: RNA editing in plant organelles and beyond. J. Exp. Bot. 2023, 74, 2273–2294. [Google Scholar] [CrossRef]
- Hurst, L.D. The Ka/Ks ratio: Diagnosing the form of sequence evolution. Trends Genet. 2002, 18, 486–487. [Google Scholar] [CrossRef] [PubMed]
- Zhou, P.; Zhang, Q.; Li, F.; Huang, J.; Zhang, M. Assembly and comparative analysis of the complete mitochondrial genome of Ilex metabaptista (Aquifoliaceae), a Chinese endemic species with a narrow distribution. BMC Plant Biol. 2023, 23, 393. [Google Scholar] [CrossRef] [PubMed]
- Dwiningsih, Y.; Rahmaningsih, M.; Alkahtani, J. Development of single nucleotide polymorphism (SNP) markers in tropical crops. Adv. Sustain. Sci. Eng. Technol. 2020, 2, 343558. [Google Scholar] [CrossRef]
- Angiosperm Phylogeny Group; Chase, M.W.; Christenhusz, M.J.; Fay, M.F.; Byng, J.W.; Judd, W.S.; Soltis, D.E.; Mabberley, D.J.; Sennikov, A.N.; Soltis, P.S.; et al. An update of the Angiosperm Phylogeny Group classification for the orders and families of flowering plants: APG IV. Bot. J. Linn. Soc. 2016, 181, 1–20. [Google Scholar]
- Yang, H.; Chen, H.; Ni, Y.; Li, J.; Cai, Y.; Wang, J.; Liu, C. Mitochondrial genome sequence of Salvia officinalis (Lamiales: Lamiaceae) suggests diverse genome structures in cogeneric species and finds the stop gain of genes through RNA editing events. Int. J. Mol. Sci. 2023, 24, 5372. [Google Scholar] [CrossRef]
- Yu, X.; Wei, P.; Chen, Z.; Li, X.; Zhang, W.; Yang, Y.; Liu, C.; Zhao, S.; Li, X.; Liu, X. Comparative analysis of the organelle genomes of three Rhodiola species provide insights into their structural dynamics and sequence divergences. BMC Plant Biol. 2023, 23, 156. [Google Scholar] [CrossRef]
- Zhang, Y.; Ma, Y.; Yu, H.; Han, Y.; Yu, T. Deciphering Codon Usage Patterns in the Mitochondrial Genome of the Oryza Species. Agronomy 2024, 14, 2722. [Google Scholar] [CrossRef]
- Dang, J.; Liu, Z.; Luo, X.; Jiang, Y.; Zhang, Z.; Abdullah, S.; Yusop, M.R. Codon Usage Characteristics and Evolutionary Analysis of Mitochondrial Genome of Winter Squash (Cucurbita maxima Duch.). J. Biobased Mater. Bioenergy 2024, 18, 428–443. [Google Scholar] [CrossRef]
- Zheng, P.; Wang, D.; Huang, Y.; Chen, H.; Du, H.; Tu, J. Detection and analysis of C-to-U RNA editing in rice mitochondria-encoded ORFs. Plants 2020, 9, 1277. [Google Scholar] [CrossRef]
- Gualberto, J.M.; Lamattina, L.; Bonnard, G.; Weil, J.H.; Grienenberger, J.M. RNA editing in wheat mitochondria results in the conservation of protein sequences. Nature 1989, 341, 660–662. [Google Scholar] [CrossRef]
- Wang, L.; Liu, X.; Xu, Y.; Zhang, Z.; Wei, Y.; Hu, Y.; Zheng, C.; Qu, X. Assembly and comparative analysis of the first complete mitochondrial genome of a traditional Chinese medicine Angelica biserrata (Shan et Yuan) Yuan et Shan. Int. J. Biol. Macromol. 2024, 257, 128571. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Z.; Mao, C.; Shang, Z.; Norbu, N.; Bonjor, N.; Jia, X.; Li, W.; Zhang, W.; Wang, J.; Qiong, L. Assembly and Comparative Analysis of the Complete Mitochondrial Genome of Hippophae salicifolia. Biology 2025, 14, 448. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Yue, C.; Li, H. Mitochondrial Genome Characteristics and Comparative Genomic Analysis of Spartina alterniflora. Curr. Issues Mol. Biol. 2025, 47, 107. [Google Scholar] [CrossRef] [PubMed]
- Androsiuk, P.; Paukszto, Ł.; Jastrzębski, J.P.; Milarska, S.E.; Okorski, A.; Pszczółkowska, A. Molecular diversity and phylogeny reconstruction of genus Colobanthus (Caryophyllaceae) based on mitochondrial gene sequences. Genes 2022, 13, 1060. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genome | Genome Type | Genome Length | Genome GC (%) | CDS | tRNA | rRNA | Pseudo Gene |
|---|---|---|---|---|---|---|---|
| mt1 | circular | 244,541 | 41.72 | 22 | 35 | 2 | 2 |
| mt2 | circular | 69,830 | 44.33 | 9 | 7 | 1 | 0 |
| Total | - | 314,371 | 42.3 | 31 | 42 | 3 | 2 |
| Type | RNA Editing | Number | Percentage |
|---|---|---|---|
| hydrophilic–hydrophilic | CAC (H) ⇒ TAC (Y) | 7 | |
| CAT (H) ⇒ TAT (Y) | 18 | ||
| CGC (R) ⇒ TGC (C) | 13 | ||
| CGT (R) ⇒ TGT (C) | 28 | ||
| Total | 66 | 11.98% | |
| hydrophilic–hydrophobic | ACA (T) ⇒ ATA (I) | 5 | |
| ACC (T) ⇒ ATC (I) | 1 | ||
| ACG (T) ⇒ ATG (M) | 6 | ||
| ACT (T) ⇒ ATT (I) | 6 | ||
| CGG (R) ⇒ TGG (W) | 29 | ||
| TCA (S) ⇒ TTA (L) | 78 | ||
| TCC (S) ⇒ TTC (F) | 35 | ||
| TCG (S) ⇒ TTG (L) | 42 | ||
| TCT (S) ⇒ TTT (F) | 54 | ||
| Total | 256 | 46.46% | |
| hydrophilic–stop | CAG (Q) ⇒ TAG (X) | 1 | |
| CGA (R) ⇒ TGA (X) | 3 | ||
| Total | 4 | 0.73% | |
| hydrophobic–hydrophilic | CCA (P) ⇒ TCA (S) | 9 | |
| CCC (P) ⇒ TCC (S) | 12 | ||
| CCG (P) ⇒ TCG (S) | 5 | ||
| CCT (P) ⇒ TCT (S) | 18 | ||
| Total | 44 | 7.99% | |
| hydrophobic–hydrophobic | CCA (P) ⇒ CTA (L) | 51 | |
| CCC (P) ⇒ CTC (L) | 12 | ||
| CCC (P) ⇒ TTC (F) | 8 | ||
| CCG (P) ⇒ CTG (L) | 39 | ||
| CCT (P) ⇒ CTT (L) | 27 | ||
| CCT (P) ⇒ TTT (F) | 14 | ||
| CTC (L) ⇒ TTC (F) | 8 | ||
| CTT (L) ⇒ TTT (F) | 14 | ||
| GCC (A) ⇒ GTC (V) | 1 | ||
| GCG (A) ⇒ GTG (V) | 5 | ||
| GCT (A) ⇒ GTT (V) | 2 | ||
| Total | 181 | 32.85% | |
| All | 551 | 100% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zeng, Z.; Zhang, Z.; Norbu, N.; Bonjor, N.; Tan, X.; Zhang, S.; Tso, N.; Wang, J.; Qiong, L. Comprehensive Analysis of the Complete Mitochondrial Genome of Paeonia ludlowii Reveals a Dual-Circular Structure and Extensive Inter-Organellar Gene Transfer. Biology 2025, 14, 854. https://doi.org/10.3390/biology14070854
Zeng Z, Zhang Z, Norbu N, Bonjor N, Tan X, Zhang S, Tso N, Wang J, Qiong L. Comprehensive Analysis of the Complete Mitochondrial Genome of Paeonia ludlowii Reveals a Dual-Circular Structure and Extensive Inter-Organellar Gene Transfer. Biology. 2025; 14(7):854. https://doi.org/10.3390/biology14070854
Chicago/Turabian StyleZeng, Zhefei, Zhengyan Zhang, Ngawang Norbu, Ngawang Bonjor, Xin Tan, Shutong Zhang, Norzin Tso, Junwei Wang, and La Qiong. 2025. "Comprehensive Analysis of the Complete Mitochondrial Genome of Paeonia ludlowii Reveals a Dual-Circular Structure and Extensive Inter-Organellar Gene Transfer" Biology 14, no. 7: 854. https://doi.org/10.3390/biology14070854
APA StyleZeng, Z., Zhang, Z., Norbu, N., Bonjor, N., Tan, X., Zhang, S., Tso, N., Wang, J., & Qiong, L. (2025). Comprehensive Analysis of the Complete Mitochondrial Genome of Paeonia ludlowii Reveals a Dual-Circular Structure and Extensive Inter-Organellar Gene Transfer. Biology, 14(7), 854. https://doi.org/10.3390/biology14070854