
Whole-Transcriptome Sequencing and Differential Expression Analysis of the Epididymis in Junggar Bactrian Camels Before and After Sexual Maturity

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Experimental Animals
2.2. RNA Extraction, Library Construction and Sequencing
2.3. Data Quality Control and Identification of mRNAs, LncRNAs, and miRNAs
2.4. Differential Expression Analysis
2.5. GO and KEGG Enrichment Analysis
2.6. Quantitative Real-Time PCR
2.7. Construction of the lncRNA-miRNA-mRNA Regulatory Network
2.8. Statistical Analysis
3. Results and Analysis
3.1. RNA-Seq Data Analysis
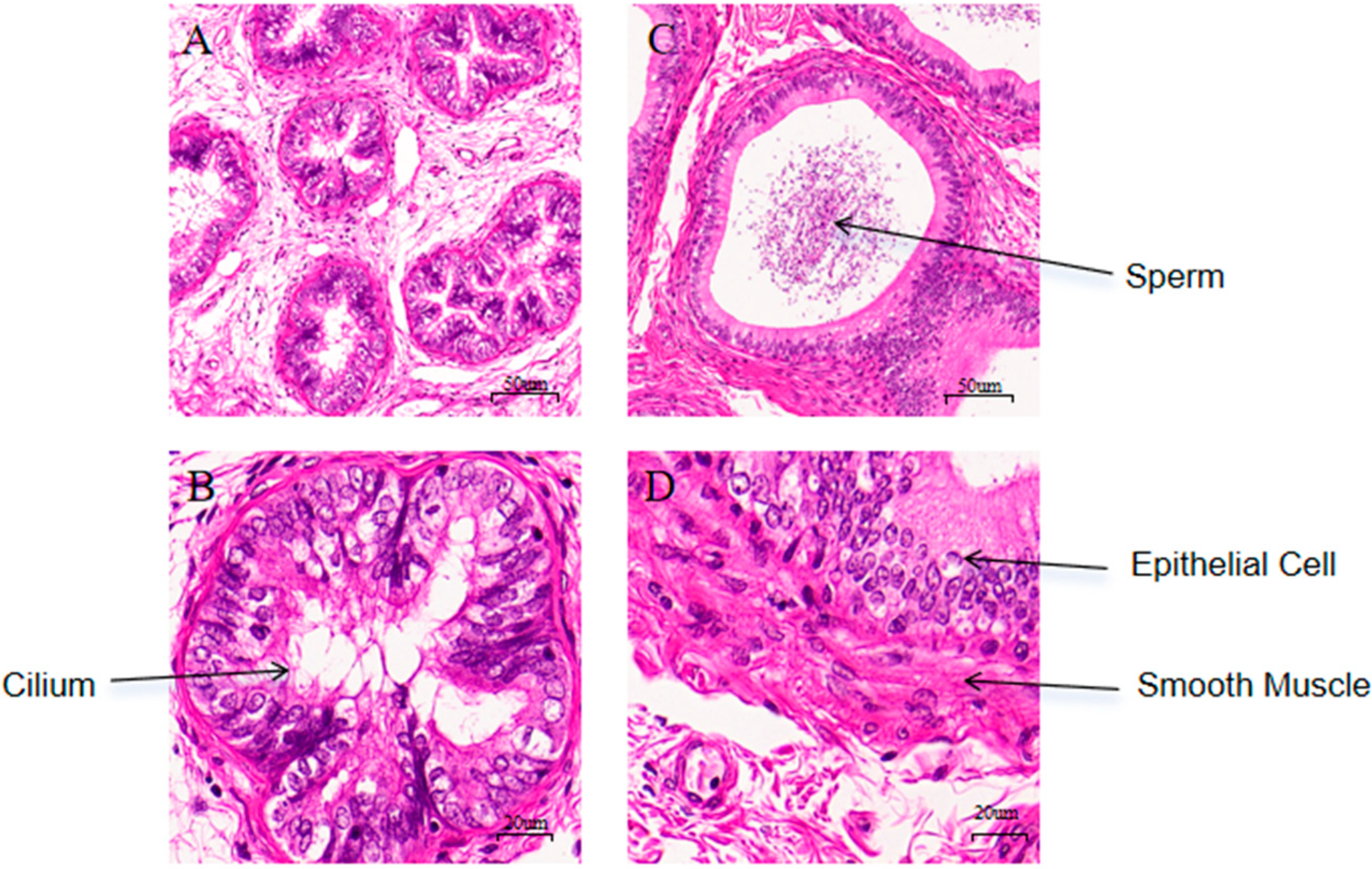
3.2. Histological Observation of Epididymal Tissues Before and After Sexual Maturity
3.3. Expression Analysis of Samples from Epididymal Tissue Before and After Sexual Maturity
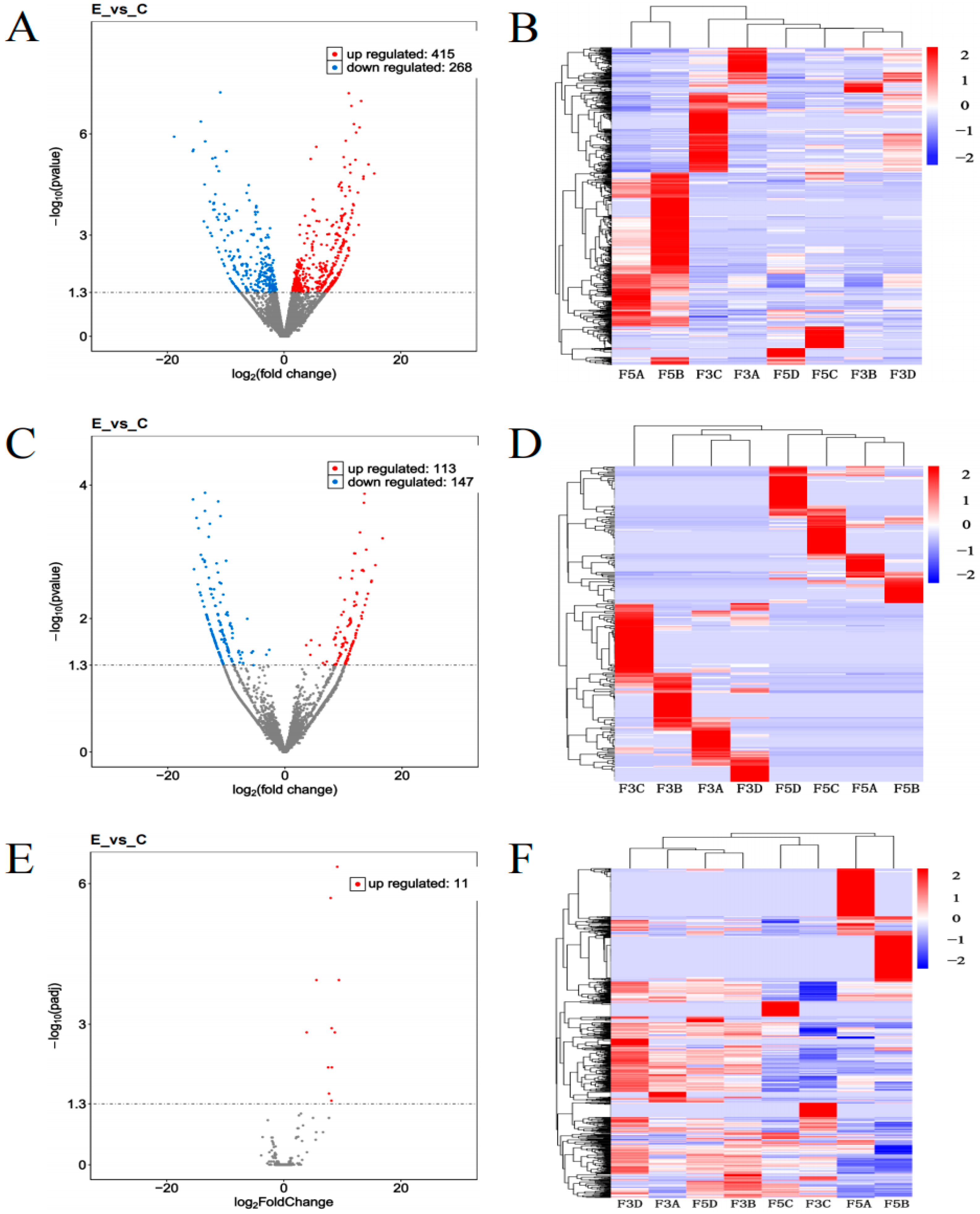
3.4. Differential Expression Analysis of Epididymal Tissues Before and After Sexual Maturity
3.5. GO Functional Annotation and KEGG Enrichment Analysis of Differentially Expressed Genes in the Epididymal Tissues of Before and After Sexual Maturity
3.5.1. GO Annotation and KEGG Enrichment of DEmRNAs
3.5.2. GO Annotation and KEGG Enrichment of DELncRNAs
3.5.3. GO Annotation and KEGG Enrichment of DEmiRNAs
3.6. RT-qPCR Validation
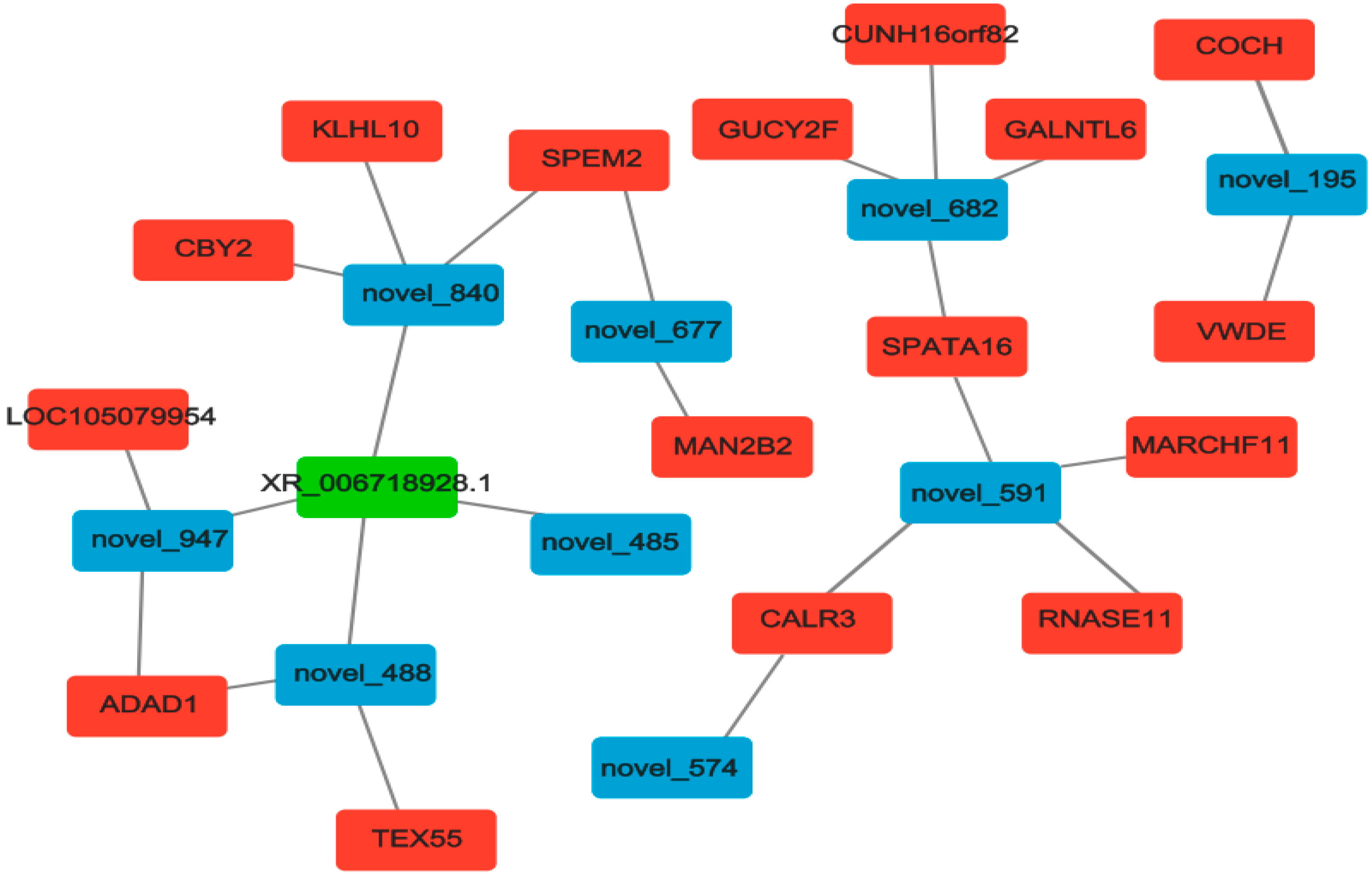
3.7. Construction and Analysis of ceRNAs Regulatory Network
4. Discussion
4.1. Differential Expression Analysis of mRNAs in the Epididymal Tissues of Bactrian Camels Before and After Sexual Maturity
4.2. Differential Expression Analysis of LncRNAs in the Epididymal Tissues of Bactrian Camels Before and After Sexual Maturity
4.3. Differential Expression Analysis of miRNAs in the Epididymal Tissues of Bactrian Camels Before and After Sexual Maturity
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Yu, Q.; Wang, T.; Li, N.; Deng, J.; Jia, J.; Yan, P. Conservation and utilization of genetic resources of Bactrian camel in Tarim, Xinjiang. Chin. Herbiv. Sci. 2025, 45, 84–87. [Google Scholar]
- Amandykova, M.; Dossybayev, K.; Mussayeva, A.; Saitou, N.; Zhunusbayeva, Z.; Bekmanov, B. A study of the genetic structure of hybrid camels in Kazakhstan. Genes 2023, 14, 1373. [Google Scholar] [CrossRef] [PubMed]
- Burger, P.A.; Ciani, E.; Faye, B. Old World camels in a modern world—A balancing act between conservation and genetic improvement. Anim. Genet. 2019, 50, 598–612. [Google Scholar] [CrossRef]
- Xie, W.; Liu, H.; Ji, M.; Na, H.; Ha, S.; Zhang, Y.; Tserennadmid, S.; Shi, H.; Zhang, X.; Na, R. Screening and analysis of differential genes of hypothalamus and pituitary gland in Bactrian camel breeding and non-breeding season. J. Inn. Mong. Agric. Univ. (Nat. Sci. Ed.) 2025, 46, 1–11. [Google Scholar]
- Sary, R.; Khalil, K.; Sindi, R.A.; Mohamed, R.H.; Hussein, H.A.; Eid, R.A.; Samir, H.; Alkahtani, M.M.; Swelum, A.A.; Ahmed, A.E. Characteristics of ultrasound and magnetic resonance imaging of normal testes and epididymis besides angiography of testicular artery in dromedary camel. Front. Vet. Sci. 2022, 9, 899570. [Google Scholar] [CrossRef]
- Yang, D.; Yuan, L.; Ma, X.; Qi, Y.; Cheng, S.; Zhang, Y. Histological study of Bactrian camel cryptorchidism and expression of immunoglobulin λ light chain in the testicular and epididymis of cryptorchid Bactrian camel. Reprod. Domest. Anim. 2024, 59, 14512. [Google Scholar] [CrossRef]
- Cao, H. Study on the Mechanism of Palmitoylation Modification of C4BPA in Epididymis and Its Effect on Sperm Regulation; Northwest A&F University: Yangling, China, 2024. [Google Scholar]
- Lee, V.; Hinton, B.T.; Hirashima, T. Collective cell dynamics and luminal fluid flow in the epididymis: A mechanobiological perspective. Andrology 2024, 12, 939–948. [Google Scholar] [CrossRef]
- Elbashir, S.; Magdi, Y.; Rashed, A.; Henkel, R.; Agarwal, A. Epididymal contribution to male infertility: An overlooked problem. Andrologia 2021, 53, 13721. [Google Scholar] [CrossRef]
- Pan, M. Transcriptome Sequencing and SLC Gene Family Expression Analysis of Head, Body and Tail Epithelial Cells of Yak Epididymis; Southwest University of Science and Technology: Mianyang, China, 2024. [Google Scholar]
- Wang, X.; Qiu, F.; Yu, J.; Zhou, M.; Zuo, A.; Xu, X.; Sun, X.-Y.; Wang, Z. Transcriptome profiling of the initial segment and proximal caput of mouse epididymis. Front. Endocrinol. 2023, 14, 1190890. [Google Scholar] [CrossRef]
- Liu, M.M.; Feng, X.L.; Qi, C.; Zhang, S.E.; Zhang, G.L. The significance of single-cell transcriptome analysis in epididymis research. Front. Cell Dev. Biol. 2024, 12, 1357370. [Google Scholar] [CrossRef]
- Tang, W. Analysis of Molecular Regulation Mechanism of Testicular Development in Northern Qianbei Hemp Sheep Based on Whole Transcriptomics; Guizhou University: Guiyang, China, 2024. [Google Scholar]
- Shen, Y.; Ulaangerel, T.; Ren, H.; Liu, Q.; Davshilt, T.; Yi, M.; Dugarjaviin, M.; Bou, G. Comprehensive analysis of the whole-transcriptome landscape of the ovarian cortex from Mongolian horses that reproduce seasonally. Comp. Biochem. Physiol. Part D Genom. Proteom. 2024, 49, 101179. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Zhang, F.; Ma, R.; Xing, J.; Wang, M.; Sun, Y.; Zhang, G. Single-cell RNA sequencing unveils dynamic transcriptional profiles during the process of donkey spermatogenesis and maturation. Genomics 2025, 117, 110974. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Dong, S.; Chen, C.; Liu, X.; Zeng, X.; Gao, Y.; Zhang, X. lncRNA 1700101O22Rik and NONMMUG030480.1 are not essential for spermatogenesis in mice. Int. J. Mol. Sci. 2022, 23, 8627. [Google Scholar] [CrossRef]
- Necsulea, A.; Soumillon, M.; Warnefors, M.; Liechti, A.; Daish, T.; Zeller, U.; Baker, C.J.; Grutzner, F.; Kaessmann, H. The evolution of lncRNA repertoires and expression patterns in tetrapods. Nature 2014, 505, 635–640. [Google Scholar] [CrossRef] [PubMed]
- Saberiyan, M.; Mirfakhraie, R.; Moghni, M.; Teimori, H. Study of Linc00574 Regulatory effect on the TCTE3 expression in sperm motility. Reprod. Sci. 2021, 28, 159–165. [Google Scholar] [CrossRef]
- An, S.Y.; Liu, Z.F.; El-Samahy, M.A.; Deng, M.T.; Gao, X.X.; Liang, Y.X.; Shi, C.B.; Lei, Z.H.; Wang, F.; Zhang, G.M. LncRNA LOC102176306 plays important roles in goat testicular development. Reproduction 2021, 161, 523–537. [Google Scholar] [CrossRef]
- Hasi, G.; Sodnompil, T.; Na, H.; Liu, H.; Ji, M.; Xie, W.; Nasenochir, N. Whole transcriptome sequencing reveals core genes related to spermatogenesis in Bactrian camels. J. Anim. Sci. 2023, 101, skad115. [Google Scholar] [CrossRef]
- Gao, Y. Transcriptomics Comparison of Testicular Development in Hornless and Horned Sheep; Northwest A&F University: Yangling, China, 2024. [Google Scholar]
- Zhu, Y. Exploration of Key Regulators of Back Fat Deposition in Dahe Pigs Based on Whole Transcriptome Sequencing; Yunnan Agricultural University: Kunming, China, 2024. [Google Scholar]
- Ibrahim, Z.H.; Al-Kheraije, K.A.; Singh, S.K. Morphological and histochemical changes in the dromedary camel epididymis in relation to reproductive activity. Histol. Histopathol. 2021, 36, 485–504. [Google Scholar]
- James, E.R.; Carrell, D.T.; Aston, K.I.; Jenkins, T.G.; Yeste, M.; Huetos, A.S. The role of the epididymis and the contribution of epididymosomes to mammalian reproduction. Int. J. Mol. Sci. 2020, 21, 5377. [Google Scholar] [CrossRef]
- Saitou, M.; Miyauchi, H. Gametogenesis from pluripotent stem cells. Cell Stem Cell 2016, 18, 721–735. [Google Scholar] [CrossRef]
- Esfahani, S.N.; Zheng, Y.; Arabpour, A.; Lrizarry, A.M.; Kobayashi, N.; Xue, X.; Shao, Y.; Zhao, C.; Agranonik, N.L.; Sparrow, M.; et al. Derivation of human primordial germ cell-like cells in an embryonic-like culture. Nat. Commun. 2024, 15, 167. [Google Scholar] [CrossRef] [PubMed]
- Teves, M.E.; Roldan, E.R.S. Sperm bauplan and function and underlying processes of sperm formation and selection. Physiol. Rev. 2022, 102, 7–60. [Google Scholar] [CrossRef] [PubMed]
- Meinhardt, A.; Middendorff, R.; Berger, T. Editorial on the Special Issue of Andrology “The clinic and biology of the epididymis”. Andrology 2024, 12, 937–938. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Yuan, L.; Chen, S.; Zhang, Y.; Ma, X.; Xing, Y.; Song, J. Morphological and histochemical identification of telocytes in adult yak epididymis. Sci. Rep. 2023, 13, 5295. [Google Scholar] [CrossRef]
- Pan, M.; Luo, X.; Zhang, Z.; Li, J.; Shahzad, K.; Danba, Z.; Caiwang, G.; Chilie, W.; Chen, X.; Zhao, W. The expression spectrum of yak epididymal epithelial cells reveals the functional diversity of caput, corpus and cauda regions. Genomics 2024, 116, 110912. [Google Scholar] [CrossRef]
- Wang, C.; Hussain Solangi, T.; Wang, H.; Yang, L.; Adjei, M.; Ahmed, S.; Shahzad, K.; Zhao, W.; Lang, X. High-throughput sequencing reveals differential expression of miRNAs in yak and cattleyak epididymis. Reprod. Domest. Anim. 2022, 57, 125–140. [Google Scholar] [CrossRef]
- Wang, P.; Zhang, X.; Huo, H.; Li, W.; Liu, Z.; Wang, L.; Li, L.; Sun, Y.H.; Hou, J. Transcriptomic analysis of testis and epididymis tissues from Banna mini-pig inbred line boars with single-molecule long-read sequencing. Biol. Reprod. 2023, 108, 465–478. [Google Scholar] [CrossRef]
- Zhao, Y.; Qin, J.; Sun, J.; He, J.; Sun, Y.; Yuan, R.; Li, Z. Motility-related microRNAs identified in pig seminal plasma exosomes by high-throughput small RNA sequencing. Theriogenology 2024, 215, 351–360. [Google Scholar] [CrossRef]
- Wu, C.; Wang, C.; Zhai, B.; Zhao, Y.; Zhao, Z.; Yuan, Z.; Fu, X.; Zhang, M. Study on the region-specific expression of epididymis mRNA in the rams. PLoS ONE 2021, 16, 0245933. [Google Scholar] [CrossRef]
- Guo, S.; Cong, B.; Zhu, L.; Zhang, Y.; Yang, Y.; Qi, X.; Wang, X.; Xiao, L.; Long, C.; Xu, Y.; et al. Whole transcriptome sequencing of testis and epididymis reveals genes associated with sperm development in roosters. BMC Genom. 2024, 25, 1029. [Google Scholar] [CrossRef]
- Zhang, M.; Nii, T.; Isobe, N.; Yoshimura, Y. Expression of Toll-like receptors and effects of lipopolysaccharide on the expression of proinflammatory cytokines and chemokine in the testis and epididymis of roosters. Poult. Sci. 2012, 91, 1997–2003. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Zhang, X.; Yan, J.; Gou, J.; Zhang, F.; Zhu, K.; Liu, S.; Sun, Y.; Shen, W.; Wang, J. Transcriptional specificity analysis of testis and epididymis tissues in donkey. Genes 2022, 13, 2339. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Alves, M.B.R.; Belleannée, C. Contribution of epididymal epithelial cell functions to sperm epigenetic changes and the health of progeny. Hum. Reprod. Update 2022, 28, 51–66. [Google Scholar] [CrossRef] [PubMed]
- Du, W. Biological Characteristics and Gene Expression of Head, Body and Tail Epithelial Cells of Sheep Epididymis; Inner Mongolia Agricultural University: Hohhot, China, 2021. [Google Scholar]
- Battistone, M.A.; Merkulova, M.; Park, Y.J.; Peralta, M.A.; Gombar, F.; Brown, D.; Breton, S. Unravelling purinergic regulation in the epididymis: Activation of V-ATPase-dependent acidification by luminal ATP and adenosine. J. Physiol. 2019, 597, 1957–1973. [Google Scholar] [CrossRef]
- Cajas, D.; Guajardo, E.; Jara-Rosales, S.; Nunez, C.; Vargas, R.; Carriel, V.; Campos, A.; Milla, L.; Orihuela, P.; Guzman, C.G. Molecules involved in the sperm interaction in the human uterine tube: A histochemical and immunohistochemical approach. Eur. J. Histochem. 2023, 67, 3513. [Google Scholar] [CrossRef]
- Nguyen, H.T.; Martin, L.J. Classical cadherins in the testis: How are they regulated? Reprod. Fertil. Dev. 2023, 35, 641–660. [Google Scholar] [CrossRef]
- Logvinov, A.S.; Nefedova, V.V.; Yampolskaya, D.S.; Kleymenov, S.Y.; Levitsky, D.I.; Matyushenko, A.M. Structural and Functional Properties of Tropomyosin Isoforms Tpm4.1 and Tpm2.1. Biochemistry 2023, 88, 801–809. [Google Scholar] [CrossRef]
- Wallgren-Pettersson, C.; Jokela, M.; Lehtokari, V.L.; Tyynismaa, H.; Sainio, M.T.; Ylikallio, E.; Tynninen, O.; Pelin, K.; Auranen, M. Variants in tropomyosins TPM2 and TPM3 causing muscle hypertonia. Neuromuscul. Disord. 2024, 35, 29–32. [Google Scholar] [CrossRef]
- Mele, V.; Basso, C.; Governa, V.; Garzon, J.F.; Muraro, M.G.; Daster, S.; Nebiker, C.A.; Mechera, R.; Bolli, M.; Schmidt, A.; et al. Identification of TPM2 and CNN1 as novel prognostic markers in functionally characterized human colon cancer-associated stromal cells. Cancers 2022, 14, 2024. [Google Scholar] [CrossRef]
- Joshi, M.; Rajender, S. Long non-coding RNAs (LncRNAs) in spermatogenesis and male infertility. Reprod. Biol. Endocrinol. 2020, 18, 103. [Google Scholar] [CrossRef]
- Wu, Z.; Ge, L.; Ma, L.; Lu, M.; Song, Y.; Deng, S.; Duan, P.; Du, T.; Wu, Y.; Zhang, Z.; et al. TPM2 attenuates progression of prostate cancer by blocking PDLIM7-mediated nuclear translocation of YAP1. Cell Biosci. 2023, 13, 39. [Google Scholar] [CrossRef]
- Kulus, J.; Kranc, W.; Kulus, M.; Dziegiel, P.; Bukowska, D.; Mozdziak, P.; Kempisty, B.; Antosik, P. Expression of genes regulating cell division in porcine follicular granulosa cells. Cell Div. 2023, 18, 12. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Yang, S.; Hao, C.; Chen, J.; Wang, J.; Xu, L. DDIT4 is essential for DINP-induced autophagy of ovarian granulosa cells. Ecotoxicol. Environ. Saf. 2023, 268, 115686. [Google Scholar] [CrossRef] [PubMed]
- Kong, Z.; Lu, Y.; Yang, Y.; Chang, K.; Lin, Y.; Huang, Y.; Wang, C.; Zhang, L.; Xu, W.; Zhao, S.; et al. m6A-Mediated biogenesis of circDDIT4 inhibits prostate cancer progression by sequestrating ELAVL1/HuR. Mol. Cancer Res. 2023, 21, 1342–1355. [Google Scholar] [CrossRef] [PubMed]
- Lewandowski, J.P.; Dumbović, G.; Watson, A.R.; Hwang, T.; Palmer, E.J.; Chang, N.; Much, C.; Turner, K.M.; Kirby, C.; Rubinstein, N.D.; et al. The Tug1 LncRNA locus is essential for male fertility. Genome Biol. 2020, 21, 237. [Google Scholar] [CrossRef]
- Hitit, M.; Kaya, A.; Memili, E. Sperm long non-coding RNAs as markers for ram fertility. Front. Vet. Sci. 2024, 11, 1337939. [Google Scholar] [CrossRef]
- O’Callaghan, E.; Sánchez, J.M.; Rabaglino, M.B.; McDonald, M.; Liu, H.; Spencer, T.E.; Fair, S.; Kenny, D.A.; Lonergan, P. Influence of sire fertility status on conceptus-induced transcriptomic response of the bovine endometrium. Front. Cell Dev. Biol. 2022, 10, 950443. [Google Scholar] [CrossRef]
- Chen, T.; Yao, L.; Liu, W.; Luan, J.; Wang, Y.; Yang, C.; Zhou, X.; Ji, C.; Gou, X.; Wang, Z.; et al. Epididymal segment-specific miRNA and mRNA regulatory network at the single cell level. Cell Cycle 2023, 22, 2194–2209. [Google Scholar] [CrossRef]
- Karuthadurai, T.; Das, D.N.; Kumaresan, A.; Sinha, M.; Kamaraj, E.; Nag, P.; King, J.P.E.S.; Datta, T.K.; Manimaran, A.; Jeyakumar, S.; et al. Sperm transcripts associated with odorant binding and olfactory transduction pathways are altered in breeding bulls producing poor-quality semen. Front. Vet. Sci. 2022, 9, 799386. [Google Scholar] [CrossRef]
- Wu, H.; Gao, J.; Wang, X.; Leung, T.Y.; Duan, Y.G.; Chiu, P.C.N. Platelet-activating factor induces acrosome reaction via the activation of extracellular signal-regulated kinase in human spermatozoa. Andrologia 2020, 52, 13565. [Google Scholar] [CrossRef]
- Ji, K.; Wei, J.; Fan, Z.; Zhu, M.; Yuan, X.; Zhang, S.; Li, S.; Xu, H.; Ling, Y. Preservative effects of curcumin on semen of Hu sheep. Animals 2024, 14, 947. [Google Scholar] [CrossRef]
- Li, C.; Shen, C.; Xiong, W.; Ge, H.; Shen, Y.; Chi, J.; Zhang, H.; Tang, L.; Lu, S.; Wang, J.; et al. Spem2, a novel testis-enriched gene, is required for spermiogenesis and fertilization in mice. Cell. Mol. Life Sci. 2024, 81, 108. [Google Scholar] [CrossRef] [PubMed]
- Jamin, S.P.; Petit, F.G.; Demini, L.; Priming, M. Tex55 encodes a conserved putative A-kinase anchoring protein dispensable for male fertility in the mouse. Biol. Reprod. 2021, 104, 731–733. [Google Scholar] [CrossRef] [PubMed]
- Gangwar, M.; Kumar, S.; Ahmad, S.F.; Singh, A.; Agrawal, S.; Anitta, P.L.; Kumar, A. Identification of genetic variants affecting reproduction traits in Vrindavani cattle. Mamm. Genome 2024, 35, 99–111. [Google Scholar] [CrossRef] [PubMed]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, J.; Yao, X.; Meng, J.; Wang, J.; Zeng, Y.; Li, L.; Ren, W. Whole-Transcriptome Sequencing and Differential Expression Analysis of the Epididymis in Junggar Bactrian Camels Before and After Sexual Maturity. Biology 2025, 14, 760. https://doi.org/10.3390/biology14070760
Liu J, Yao X, Meng J, Wang J, Zeng Y, Li L, Ren W. Whole-Transcriptome Sequencing and Differential Expression Analysis of the Epididymis in Junggar Bactrian Camels Before and After Sexual Maturity. Biology. 2025; 14(7):760. https://doi.org/10.3390/biology14070760
Chicago/Turabian StyleLiu, Jiahao, Xinkui Yao, Jun Meng, Jianwen Wang, Yaqi Zeng, Linling Li, and Wanlu Ren. 2025. "Whole-Transcriptome Sequencing and Differential Expression Analysis of the Epididymis in Junggar Bactrian Camels Before and After Sexual Maturity" Biology 14, no. 7: 760. https://doi.org/10.3390/biology14070760
APA StyleLiu, J., Yao, X., Meng, J., Wang, J., Zeng, Y., Li, L., & Ren, W. (2025). Whole-Transcriptome Sequencing and Differential Expression Analysis of the Epididymis in Junggar Bactrian Camels Before and After Sexual Maturity. Biology, 14(7), 760. https://doi.org/10.3390/biology14070760